
Abstract
The waxy cuticle on plant surfaces, comprising a cutin polymer matrix and complex very long chain fatty acids, figure in protective barrier against water loss and environmental damage. In this study, we employed a F2 population (comprising of 237 individual plants) constructed by the crossing of HX128 (female parent without cuticular wax) with HX027 (male parent with significant cuticular wax). Two extreme phenotypic bulks were constructed according to the variation of wax trait in F2 population, and bulked segregant RNA-seq (BSR-seq) was implemented to decipher the molecular underpinnings of waxiness trait in durum wheat. The QTL related to waxiness was mapped between 0.8 and 23.4 Mb on chromosome 2B. According to the differential sequence information of target interval between the parents, polymorphic primers were designed to screen F2 population. The QTL of waxiness trait was further narrowed down between 6.8 and 10.1 Mb using the composite interval mapping tool. The LOD value was 129.53, with phenotypic variance explained (PVE) up to 44.65%. The annotation and expression analysis of the genes in the QTL region entailed selection of 15 potential candidate genes. Among which, eleven new genes with four previously reported genes were found that seem to be a gene cluster mediating wax synthesis at W1 loci. This study provides important insights into fine mapping and cloning of genes involved in wax synthesis with improved efficacy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Durum wheat (Triticum turgidum L. ssp. durum (Desf.), AABB, 2n = 4x = 28) is cultivated worldwide for traditional and high-value food preparations due to its capacity to produce ultra hard endosperm (Kidane et al. 2019). Durum wheat is renowned for its superior nutritional content, characterized by high protein levels and a distinct nutty flavor, particularly well-suited for making pasta, semolina, couscous, and bulgur (Arif et al. 2020; Bassi and Sanchez-Garcia 2017). Durum wheat encompasses significant economic and agricultural value globally, making it an important cereal crop in various regions.
Epicuticular wax covers the leaves and stems of plants, serving as a protective barrier against the external environment. Its primary functions include mitigating water loss and augmenting photosynthesis, thereby protecting plants from detrimental high temperatures and intense ultraviolet radiation (Reina-Pinto and Yephremov 2009; Yeats et al. 2012). The significance of plant epicuticular wax extends across all phases of plant growth and development, significantly contributing to plant’s resilience against bacterial and fungal infestation, non-stomatal water loss, thus contributing to drought tolerance (Bernard and Joubès 2013). The leaves and stems of durum wheat also possess the wax trait; however, there is notable variation in waxiness in different cultivars. Therefore, a comprehensive analysis of the genes and regulatory mechanisms responsible for governing wax production in wheat stems and leaves bear paramount importance.
The glaucous appearance observed in wheat (Triticum aestivum) manifests as the light bluish-gray hue on the surfaces of stems and spikes, resulting from the deposition of cuticular β-diketone wax. This phenotype exhibits a notable association with increased yields in drought-prone conditions (Sun et al. 2023). In the mid-twentieth century, researchers commenced investigations into the synthesis and regulatory mechanisms of wax production. However, due to the intricate nature of wax components and their synthesis in wheat epicuticle, much of the research primarily revolved around the composition and structural aspects of epicuticular wax (Hen-Avivi et al. 2016). Numerous promising genes pertinent to wax synthesis and regulation had been identified in model crops such as Arabidopsis and rice (Rowland et al. 2006; Shaheenuzzamn et al. 2019). Notably, alkanes represented vital components within the wax synthesis process in wheat epicuticle. Their accumulation predominantly occurs during the later stages of wheat development, particularly within the leaves, stems, and spikes. The intricate biosynthesis of wax underscores the importance of comprehensive understanding of the mechanism (Wang et al. 2015).
Several studies have reported potential biosynthesis and gene identification of wheat wax trait. Chai et al. (2018) successfully cloned TaFARs, gene encoding fatty acyl coenzyme A reductase and involved in the synthesis of primary alcohols in common wheat which plays a crucial role in wax synthesis. Moreover, Li et al. (2019) found that TaCER1-1A is a key player in this process and is responsive to environmental stresses, particularly drought conditions. Kong et al. (2020) demonstrated the existence of a TaEPBM1-TaADA2-TaGCN5 ternary protein complex. This complex activates TaECR transcription by enhancing histone acetylation, thus promoting the enrichment of RNA polymerase II at TaECR genes. These mechanisms contribute significantly to the biosynthesis of wheat cuticular wax. Hen-Avivi et al. (2016) identified a cluster of metabolic genes at the W1 locus in wheat responsible for mediating β-diketone biosynthesis. These genes include those encoding several protein families, such as type III polyketide synthases, α/β hydrolases, chalcone synthases, O-acyltransferases, and cytochrome P450 associated with known fatty acid hydroxylases. Conversely, Li et al. (2022) discovered an epicuticular wax-deficient mutant derived from the high-quality wheat variety, Jimai22. This mutant was found to be under the control of an incomplete dominant gene on the long arm of chromosome 7D, distinct from the known wax loci W1–W4. This gene was consequently named W5 and was shown to impede the biosynthesis of β-diketones, thereby inhibiting wax synthesis. Huggins et al. (2014) identified high-impact quantitative trait loci (QTL) responsible for epicuticular waxiness on chromosomes 2A, 2B, 3A, 6B, and 7A, with the QTL on chromosome 2B having the most substantial influence on epicuticular waxiness. More recently, Arif et al. (2021) reported 19 QTLs for stem waxiness, 16 QTLs for leaf waxiness and 17 QTLs for spike waxiness in hexaploid wheat where the variation explained was as much as 70.5% by individual QTL. Their distribution was also throughout the wheat genome. Understanding the regulatory mechanisms of wax-related genes in wheat is impeded by the intricate nature of the wheat genome and ploidy level, the involvement of multiple genes with homologous sequences, and the intricate nature of wax alkane biosynthesis.
In this study, we employed the F2 population, comprising of 237 individuals, derived from a cross between the HX027 (waxy trait) and HX128 (non-waxy trait) cultivars. BSR-seq and molecular marker tools were employed for QTL mapping of stem and leaf wax traits of durum wheat. Based on the functional annotations of genes in the mapping intervals, the related candidate genes were analyzed together with the transcriptomic gene expression data, which provided a reference for the identification of genes of stem and leaf waxy traits in wheat.
Material and Methods
Materials and Field Experiments
An F2 population with 237 lines generated from a cross between HX128 (maternal, non-waxy/glossy) and HX027 (paternal, waxy/glaucous) was used in this study. The two parents and F2 population materials were planted in November 2021 in the Agricultural Crops Garden of Zhejiang Agriculture and Forestry University (Hangzhou, China). HX027 and HX128 were each sown in a single row, 1.5-m long, with a row spacing of 15 cm, and 6–10 plants per row, spaced 10 cm apart. The F2 population was sown in two rows, with a row spacing of 15 cm, and plant-to-plant spacing of 10 cm, in a row length of 20 m. At the jointing stage, each F2 individual plant was tagged and given a consecutive numerical code to facilitate late trait investigation and sampling. Waxy traits of each individual plant in the F2 material were examined at the heading stage.
Waxiness Trait Analysis
The morphological observations of F2 population along with their parents were recorded for wax traits during the heading stage. The waxy phenotype on the stem, leaf, and spike showed similar. The male parent HX027 showed obvious waxiness on stem, leaf, and spike, whereas female parent HX128 showed no waxiness on those tissues. The segregation of phenotypic variations in the F2 population such as waxy, non-waxy, and intermediate phenotypes were observed in fried according to comparing with the two parental phenotypes. Waxy phenotypes of the F2 population were scored for subsequent analyses according to the presence or absence of wax on stem, leaf, and spike: Materials with a non-waxy phenotype were scored as “1,” those with an intermediate type were scored as “2,” and those with a waxy phenotype was scored as “3.”
DNA Extraction and BSR-Seq Analysis
The leaves of the (HX128 × HX027) F2 population and their parents were sampled at booting stage and rapidly frozen in liquid nitrogen and stored at − 80 °C. DNA of each sample was extracted using the CTAB method (Sharp et al. 1989). Nanodrop 2000 and electrophoresis 1.0% agarose gels were used to check and purify the DNA.
BSR-seq (Bulked segregant RNA sequencing) is a BSA (Bulked segregant analysis) combined with RNA-seq sequencing (Sanzhen et al. 2012). The leaves of 30 plants of (HX128 × HX027) F2 population extreme phenotypes were taken separately with a perforator to construct waxy trait extreme mixed pools to form waxy and non-waxy pools. The leaves were frozen in liquid nitrogen at − 80 °C. RNA extraction, quality control, and library construction, as well as RNA sequencing with 150 base pair-end reads, were performed at Chengdu Tiancheng Future Technology Co., Ltd.
The raw reads quality control was performed using fastp (v. 0.19.5) (Chen et al. 2018) to remove the adapter sequences and low-quality reads to obtain high quality clean reads. Subsequently, the paired-end clean reads of two sequencing pools were individually aligned to the durum wheat (Svevo1.0) reference genome (Maccaferri et al. 2019) using Hisat2 (v. 2.2.1) software (Kim et al. 2019) with default parameters. Samtools (v. 1.3) (Li et al. 2009) was used to sort and selected the unique mapping reads. PCR replicates were annotated, and SNP calling was performed with the GATK (v.4.0.9) (McKenna et al. 2010). SNPs from both pools were integrated using the GenetypeGVCFs tool of GATK (v.4.0.9). Finally, the obtained “vcf” file was submitted to a wheat BSR online analysis website PLANT GMAP (http://183.221.124.252:4100/) for analysis (Zhang et al. 2021). The wax and non-wax mixed-pool SNP variation data were analyzed using the Euclidean Distance (ED) (Hill et al. 2013) and Gprime methods (Magwene et al. 2011) of its QTLseqr/ED functional module of PLANT GMAP (http://183.221.124.252:4100/). This analysis was used to predict the location of waxing QTLs on chromosomes.
Parental RNA-Seq Sequencing
To observe the development of parental stem waxes, the stem epicuticle of two parental materials (HX027 and HX128) of durum wheat were sampled and rapidly frozen in liquid nitrogen, and three biological replicates were taken for each of the experiments. RNA extraction, library construction and sequencing, and alignment methods employed were the same as above. Raw counts were counted by featureCounts (v. 2.0.1) (Liao et al. 2013), and the mean expression value in transcripts per million (TPM) of each gene was calculated based on its exon length.
Primer Design
According to the Indel information of the localized intervals obtained by BSR-seq, we designed primers for the differential sequences using the online tool “PrimerServer” (Design & Check) of the WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/PrimerServer/) (Ma et al. 2021). The specificity of these Indel primers were checked using the “Specificity Check Only” function on the same website. All the primers were synthesized by Zhejiang Shangya Biotechnology Co., Ltd.
Primer Polymorphism Screening
PCR amplification of DNA from the parents and four randomly selected F2 progeny materials was carried out using previously designed primers to identify polymorphisms of Indel primer-amplified fragments between the parents and in the F2 population by polyacrylamide gel electrophoresis (PAGE). PCR was performed in a 15 μL reaction solution containing 20 ng genomic DNA, 0.4 μM each primer, 0.2 mM dNTP, 0.2 U of R Taq DNA polymerase, and 1.5 μl 10 × PCR buffer (TaKaRa, Dalian, China). The conditions for the PCR were as follows: 94 °C for 5 min, followed by 29 cycles each comprising 94 °C for 30 s, annealing temperature (55–60 °C) for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 7 min. All the markers were separated and visualized on 12% polyacrylamide gel stained with ethidium bromide.
QTL Mapping Analysis
QTL IciMapping software (Meng et al. 2015) was used to construct the genetic map, and its ICIM (Inclusive Composite Interval Mapping) method was employed to perform QTL mapping of stem and leaf waxy phenotypes in the F2 generation population. In this method, the mapping step size was set to 1 cM, the LOD value was set to 2.5, and the PIN value was set to 0.001 following the research of Liu et al. (2022), and the phenotypic variation and additive effects of QTL for wax-related traits were calculated.
Candidate Gene Screening
Based on the durum wheat genome annotation information, the genes with high confidence in the target region were screened, and the “HomologFinder” tool of the WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/) (Ma et al. 2021) was also utilized to find out the homologs of the candidate genes in Chinese spring. The expression of Chinese spring homologs of durum wheat candidate genes in various tissues of wheat growth from IWGSC (2014) were analyzed and obtained by using the “GENE EXPRESSION” tool of the WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/) (Ma et al. 2021), and the heatmap of their expression was plotted using the Pretty Heatmap tool of ImageGP website (https://www.bic.ac.cn/ImageGP/index.php/Home/Index/PHeatmap.html). The expression and annotation information of these genes were preliminarily analyzed to obtain candidate genes that might be related to the waxy trait in durum wheat.
Analysis of Key Candidate Genes
Differentially expressed genes (DEGs) analysis was performed separately for the stem epicuticle RNA-Seq data of two parents using the DESeq2 package (v 1.40.0) (Love et al. 2014) of R with the Benjamini and Hochberg’s approach controlling the false discovery rate. Only the genes with an adjusted P value < 0.05 and an absolute value of log2 fold change (FC) > 1 were considered DEGs. When the candidate genes identified above were found to be DEGs between the two parents, they were considered as key candidate genes. The counts of each key candidate gene between two parents stem was visualized using ImageGP online tool (https://www.bic.ac.cn/ImageGP/index.php/Home/Index/Barplot.html), and the significant differences marked with stars according to the adjusted P values of the genes calculated by DESeq2 package (v 1.40.0) (Love et al. 2014).
Results
Phenotypic Observations and Chi-Square Analysis
Phenotypic observations were made on a field-grown population of 237 F2 plants and their parents (Fig. 1, Table 1). The results exhibited that 41 individual plants showed distinctly waxy traits and were consistent with parent HX027. There were 185 individual plants that showed non-waxy traits similar to parent HX128. Moreover, an intermediate waxy trait (waxy traits but not distinct) was shown by 11 individual plants in total, different from the biparental traits. The segregation analysis of waxy traits in single plants of F2 population revealed that the intermediate phenotypes in this F2 could be categorized as waxy, and the segregation ratio of nonwaxy phenotypes to waxy phenotypes was close to 3:1 (Table 1). Therefore, we hypothesized that the waxy traits might be controlled by a single completely recessive locus in our population.

The waxy traits in durum wheat. A The stem waxy trait of two parents (HX027, HX128). B The stem waxy trait of three lines from (HX027 × HX128) F2. C Histogram of stem waxy trait distribution in F2 generation
Analysis of BSR Sequencing Results
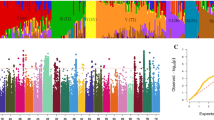
By comparing the two extreme phenotypic bulks, a total of 3887 SNPs were detected, and 461 of them were located on chromosome 2B (Fig. 2A). The wheat BSR analysis online website PLANT GMAP (http://183.221.124.252:4100/) (Zhang et al. 2021) was used to locate QTL. Distribution of the SNPs with differences between the waxy and non-waxy pool on the chromosome arm 2B (0.8–23.4 Mb) based on the Euclidean Distance (ED) and Gprime algorithm (Fig. 2 B and C).

SNP density distribution and Manhattan plot of Gprime and ED algorithms of BSR-seq analysis. A The SNP density distribution for BSR-seq data of two bulks. B Manhattan plot of Gprime algorithm (a. whole genome. b. Chromosome 2B). C Manhattan plot of ED algorithm (a. whole genome. b. Chromosome 2B)
Primer Polymorphism Screening
Indel information of the gene BSR-seq localization interval of 0.8–23.4 Mb was used for primer design of differential sequence information using the PrimerServer tool (Design&Check) on the WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/PrimerServer/) (Ma et al. 2021). Subsequently, these pairs of Indel primers were screened for polymorphism in the parental and selected F2 plants through PCR and polypropylene gel electrophoresis. A total of four polymorphic pairs of Indel primers were found: chr2B_6013821, chr2B_6847466, chr2B_10118300, and chr2B_18411436 (Table 2).
Linkage Analysis and QTL Mapping
The F2 population (237 plants) was screened using the above mentioned four pairs of polymorphic primers. Subsequently, a genetic map was constructed using the QTL IciMapping software (Meng et al. 2015). This map was generated by incorporating both the phenotypic data related to waxy traits (categorized as “1” for non-waxy material, “2” for intermediate material, and “3” for waxy material) and the genotypic data of the F2 population. The results showed that a major QTL (qWAX.2B1) was flanked by chr2B_6847466 (6,847,466 bp) and chr2B_10118300 (10,118,300 bp) on chromosome 2B with an LOD score of 129.53 and explained 44.65% of the phenotypic variance (Fig. 3).

Linkage map of chromosome 2B and the LOD graph of QTL mapping for wax trait analysis
The genotyping results of the two flanking markers, chr2B_6847466 and chr2B_10118300, within the localization interval, were used to correlate the genotypes with the wax phenotypic values of the F2 plants. The genotypes (AA, Aa, and aa) were classified as follows: AA represents the non-waxy parental HX128 genotype, Aa denotes the intermediate, and aa represents the waxy parental HX027 genotype. The statistical analyses were conducted using a one-way analysis of variance (ANOVA) followed by multiple comparisons using Fisher’s least significant difference (LSD) test with Bonferroni adjustment in the R package “agricolae” (de Mendiburu and de Mendiburu 2019). The results revealed significant differences in wax phenotypic assignment values between genotypes AA and Aa when compared to genotype aa (Fig. 4).

Statistical violin plot of the corresponding phenotypes of chr2B_6847466 and chr2B_10118300. An ANOVA analysis and LSD test with Bonferroni adjustment were performed, different capital letters show significantly differences at a p value of < 0.01
Candidate Gene Screening for Localization Intervals
Combining our findings, the waxy genes were localized to the 6.8 Mb to 10.1 Mb interval on chromosome 2B. According to the durum wheat gene annotation information, this interval contains a total of 149 annotated genes, among which 58 genes were found to be potential candidate genes. Based on the candidate genes screened, 58 candidate genes (Supplementary Table 1) were found to be homologous in Chinese spring using the HomologFinder tool on the WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/). The expression of Chinese spring homologs of 58 candidate genes in various tissues of common wheat was analyzed using the GENE EXPRESSION tool on the WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/).
The expression profiles of candidate genes in the target region were extracted from one public dataset of common wheat (IWGSC 2014) according to the corresponding homologous genes in common wheat using the “GENE EXPRESSION” tool of WheatOmics 1.0 website (http://wheatomics.sdau.edu.cn/) (Supplementary Table 2, Fig. 5A) and from our RNA expression data of the parental stems (Supplementary Table 3, Fig. 5B). After the initial expression analysis, 28 candidate genes exhibiting elevated expression levels were identified (Table 3).

The expression heatmap of candidate genes. A The expression data of the candidate genes from IWGSC.et al. (IWGSC 2014). B The expression of candidate genes in stem of two prenatal materials. The scales of (A) and (B) represent the transformed TPM (transcripts per million) values of candidate genes by log2 (TPM + 1), and the red and cyan colors represent the higher and lower relative abundance of the transcripts, respectively
It has been demonstrated that waxes on the surface of stems, leaves, and spikes are deposited from epidermal β-diketone waxes and that cytochrome P450 is related to fatty acid hydroxylases and belongs to a cluster of metabolic genes that mediate β-diketone biosynthesis (Hen-Avivi et al. 2016). Chalcone synthase is a key enzyme in the flavonoid/isoflavonoid biosynthesis pathway and is the best-known member of the plant polyketide synthase gene superfamily, which provides the starting material for a wide range of metabolites (flavonoids) (Dao et al. 2011), and the enzyme is also one of the best-known plant PKSs, which is an important gene located in hexaploid bread wheat capable of synthesizing β-diketones. The interaction between sterol 3-β-glucosyltransferases and lipids is highly specific (Pa̧zkowski et al. 2001). Sulfotransferases can catalyze the transfer of a sulfuryl group (SO3-) from the universal donor 3′-phosphoadenosine-5′-phosphonosulfate to the hydroxyl or amino groups of exogenous compounds, hormones, proteins, and other substrates and play an important role in the regulation of growth and development and adaptation to adversity (Jin et al. 2019). The alpha/beta-hydrolases superfamily protein is a widespread and plastic functioning protein fold that includes a variety of enzymes, including lipases that adsorb at the water/lipid interface of neutral water-insoluble ester substrates (Holmquist M 2000), which are importantly linked to lipid synthesis. O-acyltransferase WSD1 is involved in the biosynthesis of cuticular waxes, which reduces leaf water loss, especially during drought and plays a key role in stem wax ester synthesis (Li et al. 2008). In this study, of the 28 candidate genes screened, five were annotated as cytochrome P450 (family) proteins: TRITD2Bv1G003910, TRITD2Bv1G004510, TRITD2Bv1G004620, TRITD2Bv1G004670, and TRITD2Bv1G004740. Six genes were annotated as chalcone synthase: TRITD2Bv1G004520, TRITD2Bv1G004640, TRITD2Bv1G004680, TRITD2Bv1G004710, TRITD2Bv1G004720, and TRITD2Bv1G004770. Gene TRITD2Bv1G003590 annotated as sterol 3-beta-glucosyltransferase. Gene TRITD2Bv1G003900 annotated as sulfotransferase. Gene TRITD2Bv1G004660 was annotated as alpha/beta-hydrolases superfamily protein. And gene TRITD2Bv1G004790 was annotated as O-acyltransferase WSD1. So, we figured out that these 15 genes might be key candidate genes involved in the regulation of durum wheat waxes and played important roles in their synthesis.
Expression Analysis of Key Candidate Genes
Differentially expressed genes (DEGs) analysis in the biparental stems was conducted by DEseq2; the results indicated that the 15 candidate genes obtained from the screening were differentially expressed in the biparental stems (Fig. 6, Supplementary Table 4), and these candidate genes were expressed at greater than 0.97 TPM in the stems of the waxy parent HX027, whereas they were not expressed or were expressed at very low levels in the non-waxy parent HX128, namely TRITD2Bv1G003590, TRITD2Bv1G003900, TRITD2Bv1G003910, TRITD2Bv1G004510, TRITD2Bv1G004520, TRITD2Bv1G004620, TRITD2Bv1G004640, TRITD2Bv1G004660, TRITD2Bv1G004670, TRITD2Bv1G004680, TRITD2Bv1G004710, TRITD2Bv1G004720, TRITD2Bv1G004740, TRITD2Bv1G004770, and TRITD2Bv1G004790 (Fig. 6, and genes marked red in Supplementary Table 3). Among these 15 genes, 12 genes were expressed at more than 0.74 TPM, 0.83 TPM in stems and leaves of Chinese spring wheat, respectively (genes marked red in Supplementary Table 2).

Mean counts of 15 candidate genes on parental stems. The significant differences marked with stars according to the adjusted p values of the genes calculated by DESeq2, “**” is for p value < 0.01 and “***” for p value < 0.001
The gene TRITD2Bv1G003590 was expressed at 14.64 TPM on the stems of the waxy parent HX027, 1.75 TPM on the stems of the nonwaxy parent HX128, 13.78 TPM on wheatstem_Z30, and 7.27 TPM on wheatleaf_Z23. The gene was annotated as sterol 3-β-glucosyltransferase, which had a high specificity for interaction with lipids (Pa̧zkowski et al. 2001).
The gene TRITD2Bv1G003900 was expressed at 0.34–0.98 TPM on stems of waxy parent HX027, compared to 0–0.08 TPM on the non-waxy parent HX128, whose gene was annotated as sulfotransferase.
The five candidate genes TRITD2Bv1G003910, TRITD2Bv1G004510, TRITD2Bv1G004620, TRITD2Bv1G004670, and TRITD2Bv1G004740 were annotated as cytochrome P450, which had been shown to be associated with fatty acid hydroxylases and to belong to the cluster of metabolic genes mediating β-diketone biosynthesis (Hen-Avivi et al. 2016). These genes were expressed at 0.98–151.27 TPM on the stems of the waxy parent HX027, compared to 0–0.07 TPM on the stems of the non-waxy parent HX128, and at 0.74–9.42 TPM on wheat stem_Z30 or stem_Z65.
The gene TRITD2Bv1G004660 was expressed at 9.79 TPM on stems of the waxy parent HX027, but unexpressed on stems of the non-waxy parent HX128, and at 2.36 TPM on wheat stem_Z65, whose gene was annotated as an alpha/beta-hydrolase superfamily protein.
Six genes, TRITD2Bv1G004520, TRITD2Bv1G004640, TRITD2Bv1G004680, TRITD2Bv1G004710, TRITD2Bv1G004720, and TRITD2Bv1G004770, were annotated as chalcone synthases (CHS, EC 2.3.1.74). These genes were expressed at 42.09–235.85 TPM on the stems of the waxy parent HX027, whereas they were largely unexpressed on the stalks of the non-waxy parent HX128 and were expressed at 2.5–31.75 TPM on wheat stem_Z65 or leaf_Z71.
The gene TRITD2Bv1G004790 was expressed at 30.58 TPM on stems of the waxy parent HX027, but not on stems of the non-waxy parent HX128, and at 6.36 TPM on stem_Z65, which was annotated as O-acyltransferase WSD1.
According to previous reports, a more complex inheritance of waxy traits is already indicated (Wang et al. 2019). We also conclude that in our population, the synthesis of waxy traits was controlled by many genes, and there might be gene deletions in non-waxy populations that resulted in the inability to express the genes, and the 15 candidate genes identified above might be in clusters to regulate the synthesis of waxy traits.
Discussion
Waxes cover the stems, leaves, and other parts of the plant and play an important role for the plant, such as improving the stability of wheat leaf cell membranes, protecting the plant from mechanical damage, reducing the rate of water loss, and increasing the ratio of photosynthesis to transpiration thereby protecting plants from detrimental high temperatures and intense ultraviolet radiation (Reina-Pinto and Yephremov 2009; Yeats et al. 2012). Moreover, epicuticular wax provides plants with a natural physical protective layer that reduces mechanical damage. It allows the shedding of water droplets, dust particles, and spores and insects that may land on the plant surface, thereby reducing the chance of attack from insects and pathogens (Arif et al. 2022; Li et al. 2021). In summary, the wax provides effective protection for plants against other abiotic and biotic environmental stresses.
It has previously been reported that the W1 locus is located in the region of 9.2–9.9 Mb on 2BS, and three candidate genes at the W1 loci for β-diketone synthesis in hexaploid bread wheat have been reported: diketone metabolism-PKS (DMP), hydrolase (DMH), and CYP450 (DMC) (Hen-Avivi et al. 2016). However, there were still several wax genes that had not been identified and validated. In this study, we utilized the F2 mapping population derived from a cross between HX027 (with obvious waxy traits) and HX128 (without waxy traits) and performed BSR-seq sequencing and QTL mapping.
As a result, a QTL (qWAX.2B1) associated with the waxiness trait was detected, and two InDel markers, 2B_6847466 and 2B_10118300 were screened, which were tightly linked to this QTL at the site of chromosome 2B. The interval obtained was similar to the results of previous studies, and four candidate genes TRITD2Bv1G004620, TRITD2Bv1G004660, TRITD2Bv1G004720, and TRITD2Bv1G004790 were screened within the interval corresponding to the reported genes DMC(P450)CYP709J4, DMH (Hydrolase), DMP (PKS-like), and WES (wax ester synthase). These four genes were annotated as cytochrome P450, alpha/beta-hydrolases (ABH) superfamily proteins, chalcone synthase, and O-acyltransferase WSD1, respectively. And these four genes were highly expressed in the stems of the waxy parental parent HX027, as well as in wheat stems and leaves, and it can be determined that these four genes may be important genes in the synthesis of wheat waxes based on the annotation information.
In addition, nine genes were significantly higher expressed in the stem of waxy parental HX027 rather than HX128. Among which, four genes TRITD2Bv1G003910, TRITD2Bv1G004510, TRITD2Bv1G004670, and TRITD2Bv1G004740 were annotated as cytochrome P450, and five genes TRITD2Bv1G004520, TRITD2Bv1G004640, TRITD2Bv1G004680, TRITD2Bv1G004710, and TRITD2Bv1G004770 were annotated as chalcone synthase. Therefore, it was more likely that these 9 genes were also the members of the cluster genes involved in the regulation of waxy traits like the four previously reported genes. The two genes TRITD2Bv1G003590 and TRITD2Bv1G003900, annotated as sterol 3-β-glucosyltransferase and sulfotransferase, respectively, were also highly expressed in the waxy parent HX027; they are likely the members of the cluster genes regulating the waxy trait.
Conclusion
BSR-seq sequencing and QTL localization of F2 populations created from parents with significant differences in durum wheat waxy traits detected a QTL located on 6.8 Mb to 10.1 Mb of chromosome 2B, explaining 44.65% of the phenotypic variation. There were 15 high confidence candidate genes in this located region, including 6 chalcone synthase genes and 5 cytochrome P450 (family) genes. Among which, four candidate genes TRITD2Bv1G004620, TRITD2Bv1G004660, TRITD2Bv1G004720, and TRITD2Bv1G004790 corresponding to the reported genes DMC(P450)CYP709J4, DMH (Hydrolase), DMP (PKS-like), and WES (wax ester synthase) were detected within the interval; moreover, eleven new genes were also detected with the four reported as a gene cluster to regulate wax synthesis. This study provides additional information for reveling the mechanism of gene cluster regulation of waxy traits.
Data Availability
The data is provided within the manuscript or supplementary information files.
References
Arif MAR, Attaria F, Shokat S, Akram S, Waheed MQ, Arif A, Börner A (2020) Mapping of QTLs associated with yield and yield related traits in durum wheat (Triticum durum Desf.) under irrigated and drought conditions. Int J Mol Sci 21(7):2372. https://doi.org/10.3390/ijms21072372
Arif MAR, Shokat S, Plieske J, Ganal M, Lohwasser U, Chesnokov YV, Kocherina NV, Kulwal P, Kumar N, McGuire PE, Sorrells ME, Qualset CO, Börner A (2021) A SNP-based genetic dissection of versatile traits in bread wheat (Triticum aestivum L.). Plant J 108(4):960–976. https://doi.org/10.1111/tpj.15407
Arif MAR, Waheed MQ, Lohwasser U, Shokat S, Alquddah AM, Volkmar C, Börner A (2022) Genetic insight into the insect resistance in bread wheat exploiting the untapped natural diversity. Front Genet 13:898905. https://doi.org/10.3389/fgene.2022.828905
Bassi FM, Sanchez-Garcia M (2017) Adaptation and stability analysis of ICARDA durum wheat elites across 18 countries. Crop Sci 57(5):2419–2430. https://doi.org/10.2135/cropsci2016.11.0916
Bernard A, Joubès J (2013) Arabidopsis cuticular waxes: advances in synthesis, export and regulation. Prog Lipid Res 52(1):110–129. https://doi.org/10.1016/j.plipres.2012.10.002
Chai GQ, Li CL, Xu F, Li Y, Shi X, Wang Y, Wang ZH (2018) Three endoplasmic reticulum-associated fatty acyl-coenzyme a reductases were involved in the production of primary alcohols in hexaploid wheat (Triticum aestivum L.). BMC Plant Biol 18:41. https://doi.org/10.1186/s12870-018-1256-y
Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17):i884–i890. https://doi.org/10.1093/bioinformatics/bty560
Dao TTH, Linthorst HJM, Verpoorte R (2011) Chalcone synthase and its functions in plant resistance. Phytochem Rev 10(3):397–412. https://doi.org/10.1007/s11101-011-9211-7
de Mendiburu F, de Mendiburu MF (2019) Package ‘agricolae.’ R Packag Vers 1(13):1143–1149
Hen-Avivi S, Savin O, Racovita RC, Lee WS, Adamski NM, Malitsky S, Almekias-Siegl E, Levy M, Vautrin S, Bergès H, Friedlander G, Kartvelishvily E, Ben-Zvi G, Alkan N, Uauy C, Kanyuka K, Jetter R, Distelfeld A, Aharoni A (2016) A metabolic gene cluster in the wheat W1 and the barley Cer-cqu loci determines β-diketone biosynthesis and glaucousness. Plant Cell 28(6):1440–1460. https://doi.org/10.1105/tpc.16.00197
Hill JT, Demarest BL, Bisgrove BW, Gorsi B, Su YC, Yost HJ (2013) MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res 23(4):687–697. https://doi.org/10.1101/gr.146936.112
Holmquist M (2000) Alpha beta-hydrolase fold enzymes structures, functions and mechanisms. Curr Protein Pept Sci 1(2):209–235. https://doi.org/10.2174/1389203003381405
Huggins TD (2014) Understanding the genetic interactions that regulate heat and drought tolerance in relation to wax deposition and yield stability in wheat (Tricticum aestivum L.). Texas A&M University
IWGSC (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345(6194):1251788. https://doi.org/10.1126/science.1251788
Jin L, Ouyang N, Huang Y, Liu CL, Ruan Y (2019) Genome-wide analysis of sulfotransferase genes and their responses to abiotic stresses in Chinese cabbage (Brassica rapa L.). PLoS ONE 14(8):e0221422. https://doi.org/10.1371/journal.pone.0221422
Kidane YG, Gesesse CA, Hailemariam BN, Desta EA, Mengistu DK, Fadda C, Pè ME, Dell’Acqua M (2019) A large nested association mapping population for breeding and quantitative trait locus mapping in Ethiopian durum wheat. Plant Biotechnol J 17(7):1380–1393. https://doi.org/10.1111/pbi.13062
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37(8):907–915. https://doi.org/10.1038/s41587-019-0201-4
Kong L, Zhi P, Liu J, Li H, Zhang X, Xu J, Zhou J, Wang X, Chang C (2020) Epigenetic activation of enoyl-coa reductase by an acetyltransferase complex triggers wheat wax biosynthesis. Plant Physiol 183(3):1250–1267. https://doi.org/10.1104/pp.20.00603
Li F, Wu X, Lam P, Bird D, Zheng H, Samuels L, Jetter R, Kunst L (2008) Identification of the wax ester synthase/acyl-coenzyme a:diacylglycerol acyltransferase WSD1 Required for stem wax ester biosynthesis in Arabidopsis. Plant Physiol 148(1):97–107. https://doi.org/10.1104/pp.108.123471
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Li TT, Sun YL, Liu TX, Wu HQ, An PP, Shui ZJ, Wang JH, Zhu YD, Li CL, Wang Y, Jetter R, Wang ZH (2019) is involved in cuticular wax alkane biosynthesis in hexaploid wheat and responds to plant abiotic stresses. Plant Cell Environ 42(11):3077–3091. https://doi.org/10.1111/pce.13614
Li L, Chai L, Xu H, Zhai H, Wang T, Zhang M, You M, Peng H, Yao Y, Hu Z, Xin M, Guo W, Sun Q, Chen X, Ni Z (2021) Phenotypic characterization of the glossy1 mutant and fine mapping of GLOSSY1 in common wheat (Triticum aestivum L.). Theor Appl Gen 134(3):835–847. https://doi.org/10.1007/s00122-020-03734-6
Li LH, Zhang Z, Song WJ, Su ZQ, Zhang YF, You MS, Ni ZF, Xing JW (2022) The essential role of W5 in wax metabolism in wheat (Triticum aestivum L.). J Plant Biol 65(4):311–317. https://doi.org/10.1007/s12374-021-09325-2
Liao Y, Smyth GK, Shi W (2013) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930. https://doi.org/10.1093/bioinformatics/btt656
Liu D, Megerssa SH, Ammar K, Acevedo M, Bergstrom GC, Dreisigacker S, Randhawa M, Brown-Guedira G, Ward B, Sorrells ME (2022) QTL mapping of seedling and field resistance to stem rust in DAKIYE/Reichenbachii durum wheat population. PLoS ONE 17(10):e0273993. https://doi.org/10.1371/journal.pone.0273993
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. https://doi.org/10.1186/s13059-014-0550-8
Ma S, Wang M, Wu J, Guo W, Chen Y, Li G, Wang Y, Shi W, Xia G, Fu D, Kang Z, Ni F (2021) WheatOmics: a platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol Plant 14(12):1965–1968. https://doi.org/10.1016/j.molp.2021.10.006
Maccaferri M, Harris NS, Twardziok SO, Pasam RK, Gundlach H, Spannagl M, Ormanbekova D, Lux T, Prade VM, Milner SG, Himmelbach A, Mascher M, Bagnaresi P, Faccioli P, Cozzi P, Lauria M, Lazzari B, Stella A, Manconi A, Gnocchi M, Moscatelli M, Avni R, Deek J, Biyiklioglu S, Frascaroli E, Corneti S, Salvi S, Sonnante G, Desiderio F, Marè C, Crosatti C, Mica E, Özkan H, Kilian B, De Vita P, Marone D, Joukhadar R, Mazzucotelli E, Nigro D, Gadaleta A, Chao S, Faris JD, Melo ATO, Pumphrey M, Pecchioni N, Milanesi L, Wiebe K, Ens J, MacLachlan RP, Clarke JM, Sharpe AG, Koh CS, Liang KYH, Taylor GJ, Knox R, Budak H, Mastrangelo AM, Xu SS, Stein N, Hale I, Distelfeld A, Hayden MJ, Tuberosa R, Walkowiak S, Mayer KFX, Ceriotti A, Pozniak CJ, Cattivelli L (2019) Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 51(5):885–895. https://doi.org/10.1038/s41588-019-0381-3
Magwene PM, Willis JH, Kelly JK (2011) The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput Biol 7(11):e1002255. https://doi.org/10.1371/journal.pcbi.1002255
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283. https://doi.org/10.1016/j.cj.2015.01.001
Pa̧zkowski C, Kalinowska M, Wojciechowski ZA (2001) Phospholipids modulate the substrate specificity of soluble UDP-glucose:steroid glucosyltransferase from eggplant leaves. Phytochemistry 58(5):663-669. https://doi.org/10.1016/s0031-9422(01)00292-8
Reina-Pinto JJ, Yephremov A (2009) Surface lipids and plant defenses. Plant Physiol Biochem 47(6):540–549. https://doi.org/10.1016/j.plaphy.2009.01.004
Rowland O, Zheng H, Hepworth SR, Lam P, Jetter R, Kunst L (2006) CER4 encodes an alcohol-forming fatty acyl-coenzyme a reductase involved in cuticular wax production in Arabidopsis. Plant Physiol 142(3):866–877. https://doi.org/10.1104/pp.106.086785
Sanzhen Liu CTY, Tang HM, Nettleton D, Schnable PS (2012) Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 7(5):e36406. https://doi.org/10.1371/journal.pone.0036406
Shaheenuzzamn M, Liu TX, Shi SD, Wu HQ, Wang ZH (2019) Research advances on cuticular waxes biosynthesis in crops: a review. Int J Agric Biol 21(5):911–921. https://doi.org/10.17957/Ijab/15.0975
Sharp PJ, Fau CS, Desai S, Fau DS, Gale MD, Gale MD (1989) The isolation, characterization and application in the Titmice of a set of wheat RFLP probes identifying each homoeologous chromosome arm. Theor Appl Gen 78(3):342–348. https://doi.org/10.1007/BF00265294
Sun Y, Ruiz Orduna A, Zhang Z, Feakins SJ, Jetter R (2023) Biosynthesis of barley wax β-diketones: a type-III polyketide synthase condensing two fatty acyl units. Nat Commun 14(1):7284. https://doi.org/10.1038/s41467-023-42917-9
Wang Y, Wang M, Sun Y, Hegebarth D, Li T, Jetter R, Wang Z (2015) Molecular characterization oftafar1involved in primary alcohol biosynthesis of cuticular wax in hexaploid wheat. Plant Cell Physiol 56(10):1944–1961. https://doi.org/10.1093/pcp/pcv112
Wang S, Xu SS, Chao S, Sun Q, Liu S, Xia G (2019) A genome-wide association study of highly heritable agronomic traits in durum wheat. Front Plant Sci 10:919. https://doi.org/10.3389/fpls.2019.00919
Yeats TH, Buda GJ, Wang ZH, Chehanovsky N, Moyle LC, Jetter R, Schaffer AA, Rose JKC (2012) The fruit cuticles of wild tomato species exhibit architectural and chemical diversity, providing a new model for studying the evolution of cuticle function. Plant J 69(4):655–666. https://doi.org/10.1111/j.1365-313X.2011.04820.x
Zhang L, Dong C, Chen Z, Gui L, Chen C, Li D, Xie Z, Zhang Q, Zhang X, Xia C, Liu X, Kong X, Wang J (2021) WheatGmap: a comprehensive platform for wheat gene mapping and genomic studies. Mol Plant 14(2):187–190. https://doi.org/10.1016/j.molp.2020.11.018
Funding
This work was supported by the Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (2021C02064-3–4), the National Natural Science Foundation of China (32001537), the Natural Science Foundation of Zhejiang Province, China (LQ20C130001), and the Program for Research and Development of the Zhejiang A&F University (2019FR0327).
Author information
Authors and Affiliations
Contributions
Original draft, visualization, validation, software, methodology was performed by Yijing Cai. Investigation, formal analysis, data curation was performed by Yijing Cai, Yasir Muhammad, Yujie Zhou, Yuefen Cao, and Xin Hu. Manuscript revisions and editing were handled by Yasir Muhammad, Xin Hu, and Junkang Rong. Supervision, resources, funding acquisition, conceptualization were provided by Xin Hu. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
All authors had given their consent to participate in this study.
Consent for Publication
All authors had gone over the manuscript and gave their consent.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cai, Y., Muhammad, Y., Zhou, Y. et al. QTL Mapping and Candidate Gene Analysis for Wax Trait of Stem and Leaf in Durum Wheat (Triticum turgidum L. ssp. durum (Desf.)). Plant Mol Biol Rep (2024). https://doi.org/10.1007/s11105-024-01497-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11105-024-01497-8