
Abstract
The BcMF22 (Brassica campestris Male Fertility 22) gene was isolated from Chinese cabbage-pak-choi (B. campestris L. ssp. chinensis Makino, syn. B. rapa ssp. chinensis). The cDNA clone of BcMF22 was 1,870 bp in length and contained an open reading frame (OFF) of 1,260 bp, while the genomic sequence contained no introns. Sequence prediction indicated that BcMF22 might encode a methytransferase. Spatial and temporal expression analyses indicated BcMF22 was preferentially expressed in pollen. Transcripts of BcMF22 were first detected in pollen mother cells and continued to be present in mature pollen. In addition, transcripts were detected in pollinated pistils, approximately 4 h after pollination (4HAP). These results indicated that BcMF22 might be a pollen-preferential gene and was closely involved in pollination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Both pollination and fertilization are important processes in plant reproduction. Pollination involves the transfer of pollen from the anthers to stigmatic surfaces, and it is critical for subsequent fertilization and for seed set.
Pollen development is a process regulated by a wide variety of genes (Kim et al. 2010; Li et al. 2010, 2011; Singh and Grover 2010; Tsuwamoto et al. 2010). Transcriptional profiles of pollen tissues have revealed that several specific genes are involved during the development of mother cell meiosis into mature pollen (Becker et al. 2003; Honys and Twell 2003; Whittle et al. 2010). In tobacoo (Nicatiana tabocum), there were about 11,000 anther-specific mRNAs (Kamalay and Goldberg 1980), in maize (Zea mays), approximately 2,000 to 7,000 pollen-specific genes among 20,000 pollen expressed mRNAs (Wing et al. 1990), and in Arabidopsis, there were 992 pollen expressed mRNAs, nearly 40% of which were pollen-specific (Honys and Twell 2003). Moreover, there were almost 9.7% of the 13,997 male gametophyte-expressed mRNAs that were gametophyte-specific in Arabidopsis (Honys and Twell 2004). So, a large number of pollen-specific transcripts are being recognized in plants. In recent years, many pollen-related or pollen-specific genes were isolated, characterized and analyzed. These genes include “early genes” (Zhang et al. 2011a, b), “late genes” and genes expressed in both periods of male organ development (Chen et al. 2011; Fourgoux-Nicol et al. 1999; Honys and Twell 2004; Kato et al. 2010; Robert et al. 1993; Zhang et al. 2011). However, these studies are mainly associated with genes which were involved in pollen development, and the current knowledge on the constant process of pollen development and fertilization is scarce, which is greatly appealing to us.
Recently, many works focused on the microarray analysis in plants providing important information on the genomewide regulatory networks of pollination and fertilization (Boavida et al. 2011; Lan et al. 2004; Wang et al. 2008). However, such transcriptional profile cannot provide a complete outline of the molecular events during this process. So, isolation and identification pollen development and fertilization-related genes and illustration their functions are demanding.
In our previous work, we found that one homologous gene of At1g58120 was upregulated in flower buds and the pollinated pistils (unpublished data) by ATH1 microarray of ‘Aijiaohuang’ genic male sterile AB line Bajh97-01A/B in Chinese cabbage-pak-choi (B. campestris L. ssp. chinensis, Makino, syn. B. rapa ssp. chinensis). Thus, we speculate that this gene might be one that participated both in pollen development and fertilization, which is valuable for understanding this entire process in plants. In this paper, we analyzed the expression pattern of the homologous gene of At1g58120, BcMF22 in Chinese cabbage-pak-choi using quantitative real-time RT-PCR (qRT-PCR) and in situ hybridization and discussed its association with pollen development and fertilization.
Materials and Methods
Plant Material
A Chinese cabbage-pak-choi ‘Aijiaohuang’ genic male sterile system named Bajh97-01A/B was selected in our laboratory (Cao et al. 2006). It comprised a homozygous male sterile line Bajh97-01A and a heterozygous male fertile line Bajh97-01B. The segregation ratio of male fertile plant Bajh97-01B and the sterile plant Bajh97-01A is 1:1. When comparing it to the male fertile plant Bajh97-01B, the anther of a sterile plant could not produce pollen grains; however, the other floral tissues such as the sepal, petal and pistil remain normal and the female functions are not affected (Huang et al. 2010). All the plants were cultivated in the experimental farm of Zhejiang University.
At the flowering stage, the tissue samples were marked and frozen in liquid nitrogen immediately, and then stored at −80°C. Developmental stages of flower buds were selected as described in our previous work (Huang et al. 2008): four different organs (open flowers, germinal siliques, scapes and leaves) in fertile plants and five floral parts (petals, sepals, stamens, pistils and nectarys) of flower buds in sterile and fertile plants at Stage V. In addition, we pollinated the pistil of the Bajh97-01A lines (sterile plants) with the pollen grains from the Bajh97-01B lines (fertile plants), and then selected pistils at 1, 2, 4, 8, 12 and 24 h after pollination (HAP) and the corresponding controls in sterile plants.
Bioinformatics Analysis
Database searches were performed at the NCBI World Wide Web server using the Basic Local Alignment Search Tool (BLAST) network service (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Gene sequence was analyzed by using the DNAStar software (DNASTAR, Madison, WI, USA). The nucleotide sequence and the deduced amino acid sequence of gene were analyzed by GENETYX. The instability index of the gene was computed by the ProtParam (http://ca.expasy.org/tools/protparam.html), and its transmembrane regions were predicted by TMHMM (http://www.cbs.dtu.dk/servies).
DNA, RNA Extraction and cDNA Synthesis
DNA was extracted from the fresh leaves of the fertile line Bajh97-01B using cetyl-trimethylammonium bromide (CTAB) plant DNA extraction method. Total RNA was extracted from the flower buds at five developmental stages and five floral parts of the flower bud in both the fertile and sterile plants at Stage V, different organs in fertile plants, and pistils at 1, 2, 4, 8, 12, and 24 HAP and the corresponding controls using Trizol reagents (Invitrogen, USA) according to the manufacturer's instruction. The first-stranded cDNA was then synthesized using a SMART™ PCR cDNA synthesis kit (Clontech, USA) according to the manufacturer's instruction.
Isolation of cDNA and DNA Sequences
The full length cDNA was amplified by homologous cloning; P1 and P2 primers (Table 1) were designed according to At1g58120. The genomic DNA sequence was amplified using the same primer. A 3′-rapid amplification of its cDNA ends was performed to obtain its 3′end sequence. The gene-specific primer, P3 (Table 1), was designed according to its full length cDNA. The 3′end anchor primers, P4 (Table 1), was designed according to the cDNA Synthesis Kit. Because we failed to obtain the 5′end sequence by 5′RACE, we then performed BLAST (http://brassica.bbsrc.ac.uk/BrassicaDB/blast_form.html) to find the homology of the gene and found that GR4545551 fragment in Brassica napus had 83% similarities with it. The primers of P5 and P6 (Table 1) were designed according to the GR455551 fragment. Then, a 5′ end sequence of the gene was performed by the homologous fragment of GR4545551.
PCR was carried out in a total volume of 25 μl containing 0.5 μl template (0.5 μg), 2.5 μl 10 × PCR buffer, 0.5 μl 10 mmol/l dNTPs, 0.5 Taq polymerase (5 U/μl), 1 μl gene-specific primer (10 mM) and 19 μl ddH2O.The PCR program was as follows: 94°C for 3 min, followed by 34 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 2 min, and the final extension step at 72°C for 7 min.
The amplified PCR products were purified using AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA), cloned into a pGEM-T easy vector (Promega, USA), and then transformed into Escherichia coli competent cells. The recombinant clone was sequenced by Invitrogen Co. (Shanghai, CN).
Gene Expression Analysis by qRT-PCR
For gene expression analysis, first-stranded cDNAs from the flower buds at the five developmental stages and five floral parts of both the fertile and the sterile plants, four different organs of fertile plants, and pistils at different times after pollination of sterile plants were used as templates.
The qRT-PCR primers were selected from a specific region in the gene. Gene-specific primers (RT1 and RT2) and internal control gene Actin-1 (GenBank accession number: EU012495) primers (actin-1-F and actin-1-R) were shown in Table 1. Analysis was performed using an Mx3005P multiplex quantitative PCR system (Stratagene, La Jolla, CA, USA). Amplification was carried out with the following cycling parameters: heating for 3 min at 95°C, 40 cycles of denaturation at 95°C, annealing for 15 s at 55°C, and extension for 30 °s at 72°C. Triplicate quantitative PCR experiments were performed for each sample; the expression values obtained were normalized against Actin-1. Analysis of the relative gene expression data was done using the 2−Δ − ΔCT method (Livak and Schmittgen 2001).
In Situ Hybridization
The inflorescences of Bajh97-01B and pollinated pistils of Bajh97-01A were used for in situ hybridization. Samples were fixed in 4% paraformaldehyde PBS solution with 0.1% Triton X-100 and 0.1% Tween-20, serially dehydrated, cleared with dimethylbenzene, and embedded in paraffin. Then, 7- μm thick sections of flower buds and pistil were hybridized to specific digoxigenin (DIG)-labeled RNA probes (Roche, Branchburg, NJ, USA). Templates for the gene-specific probes were obtained from amplification with a primer pair SP1 and SP2 (Table 1). The sense and antisense probes were synthesized and DIG-labeled using a SP6/T7 transcription kit (Roche). Images were taken by a Leica DMLE camera (Leica, Wetzlar, Germany).
Results
The Structure Characterization Defined BcMF22 as a New Gene
The gene was isolated in Chinese cabbage-pak-choi by homology cloning according to Arabidopsis At1g58120. The full-length cDNA was 1,870 bp, with a 418-bp 5′-untranslated region and a 192-bp of 3′-untranslated region (Fig. 1). The 1,260-bp DNA sequence, amplified with the gene-specific primers, contained no introns. The NCBI BLAST showed that its nucleotide sequence had 86% similarities with At1g58120.

ORF with 1,260 bp and the deduced amino acid sequence of the BcMF22 gene (GenBank Accession No: JN398670). There is no intron in the BcMF22 sequence. The ORF of BcMF22 gene is composed of 419 amino acids. The deduced protein with a transmembrane (underlined). Initiation and stop code are shown in bold. Asterisk indicates the termination codon
The ORF consists of 1,260 bp, encoding a 419 amino acid polypeptide with molecular weight of 48.062 kDa and an isoelectric point of 6.69 (GenBank Accession No: JN398670) (Fig. 1). Among the 419 amino acids, 60 were strongly basic (+) (K and R), 62 were strongly acidic (−) (D and E), 136 were hydrophobic (A, I, L, F, W and V), and 68 were polar (N, C, Q, S, T and Y). Its instability index was computed as 45.68 by the ProtParam (http://ca.expasy.org/tools/protparam.html), which was classified as an unstable protein. Furthermore, the aliphatic index was 84.42, which defined it as aliphatic. The deduced amino acid sequence was predicted by TMHMM (http://www.cbs.dtu.dk/services) database, in which it contains one transmembrane region located in the first 60 amino acids. The gene only shared a significant sequence similarity to At1g58120 of Arabidopsis, which encodes a methyltransferases. Secondary structure composition prediction by PredictProtein server showed that there were 29.6% helix, 18.6% sheet, and 51.8% loop in the deduced protein.
BcMF22 is Differently Expressed in Reproductive Organs
The relative expression patterns of the gene in different stages of flower buds, different organs, floral parts, and pollinated pistils were investigated by qRT-PCR.
Our data showed that the gene was constantly expressed in flower buds at five developmental stages in fertile plants (Fig. 2a). However, the expression levels of the gene in the flower buds of fertile plants show an evident increase compared to those of the sterile plants, except Stage I (pollen mother stages), and the dramatic difference appeared at Stage V (mature pollen stage) (Fig. 2a). Therefore, the gene was named as B. campestris Male Fertility 22 (BcMF22).

Relative expression analysis of BcMF22 in various tissues by qRT-PCR. a Flower buds (bud1–bud5) of Bajh97-01A/B at Stages I–V, respectively. b Open flowers, siliques, scapes and leaves (F, Si, Sc and L) of the Bajh97-01B. c Petal, sepal, stamen, pistil and nectar (Pe, Se, St, Pi and Ne) of the flower buds at Stage V, respectively. d Black column indicates pistils at different hours after pollination (1, 2, 4, 8, 12 and 24 h), gray column indicates the corresponding control
The expression pattern of BcMF22 in the different organs was checked in fertile plants, and the results demonstrated that BcMF22 was expressed at higher level in open flowers than in other organs (siliques, scapes and leaves) (Fig. 2b). However, BcMF22 was expressed at a low expression level in germinal siliques, roots and leaves.
Furthermore, the expression pattern of BcMF22 in the floral parts of fertile and sterile open flowers were also detected, and the results showed that BcMF22 was expressed at higher transcript levels in stamens of fertile plants as compared to other floral parts (petals, sepals, pistils and nectarys) (Fig. 2c). However, transcript levels of BcMF22 were very low in each floral part of sterile plants (Fig. 2c).
When we address the expression pattern in the pollinated pistils of BcMF22 in sterile plants, qRT-PCR results illustrated that BcMF22 was expressed at higher levels in pistils at 1, 2 and 4 HAP than their corresponding controls (Fig. 2d). Moreover, BcMF22 mRNA was rather highly expressed in pistils at 4 HAP compared to pistils after other times of pollination (Fig. 2d). However, BcMF22 showed a detectable relative expression in nonpollinated pistils and pistils at 8, 12 and 24 HAP (Fig. 2d) but at a very low level.
BcMF22 is Highly Expressed in the Pollen
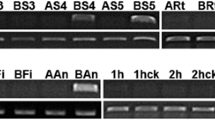
To further address the expression pattern and the cellular location of BcMF22 mRNA during pollen development and pollinated pistils, in situ hybridization was performed on transverse sections of flower buds at different developmental stages and pistil at 1, 4 and 24 HAP. Specific hybridization signals were first detected in the pollen mother cells and continued to the mature pollen grains (Fig. 3b) but not in other tissues, and the BcMF22 signal in the mature pollen grains reached its peak (Fig. 3e). There was also strong expression in the stigma and style of pistils at 4 HAP (Fig. 4m). However, no signal was observed at pollen mother stages (Fig. 3a), 1 and 24 HAP (Fig. 4k, l), as well as the sense control (Fig. 3f–j, 4n–p).

Detection of BcMF22 mRNA in different anther developmental stages in Brassica campestris ssp. chinensis by in situ hybridization. a–e Sections of anthers at pollen mother (Stage I), tetrads (Stage II), unicleate (Stage III), binucleate (Stage IV), and mature pollen (Stage V), respectively, hybridized with a BcMF22 antisense RNA probe. f–j Sections of the corresponding developmental stages hybridized with a BcMF22 sense RNA probe as a control. Probes were labeled with digoxigenin-UTP. The transcript-specific hybridization signal is visualized as blue black color. MC pollen mother cell, Tds tapetum, Up unicleate, Up binucleate, Bp binucleate, Mp mature pollen. Scale bars = 20 μm

Analysis of the expression pattern of BcMF22 in pistil at different times after pollination in Brassica campestris ssp. chinensis by in situ hybridization. k–l Sections of pistil at 1, 4 and 24 HAP, respectively, hybridized with a BcMF22 antisense RNA probe. n–p Sections of the corresponding stages hybridized with a BcMF22 sense RNA probe as a control. Probes were labeled with digoxigenin-UTP. The transcript-specific hybridization signal is visualized as blue black color. Sti stigma, Sty style. Scale bars = 50 μm
Discussion
Analysis based on bioinformatics tools indicated that BcMF22 encoded a methytransferase. The results of qRT-PCR showed that BcMF22 was significantly upregulated in flower buds of different stages compared to the sterile ones and reached its maximum expression level at Stage V (mature pollen stage). Moreover, BcMF22 was also specially expressed in the stamens of fertile plants. Since sterile flower buds differ from the fertile ones only in the failure of pollen formation and the pistils in sterile plants is normal (Huang et al. 2010), we, therefore, could speculate that BcMF22 is a pollen-preferential gene. Cellular location of BcMF22 mRNA demonstrated that it was specially expressed in pollen, and its maximal mRNA level appeared at the mature pollen stage. This result agrees well with the results of the qRT-PCR analysis. Additionally, researchers (Mascarenhas 1975) reported that the RNAs required for germination and early tube growths were already present in the mature pollen grain. Biochemical analysis showed that the mRNAs, required for germination, are synthesized during pollen maturation and persisted in the pollen grain until they are utilized for translation during the germination process (Mascarenhas 1988). So, BcMF22 showed the highest expression in the fertile flower buds at mature pollen stages that might be because of accumulated mRNA of BcMF22 was prepared for the next step of pollen germination and pollen tube growth.
In this paper, we pollinated the pistils of the Bajh97-01A (sterile plants) with pollen grains from the Bajh97-01B (fertile plants) for fixing the expression mode of BcMF22 in pollinated pistils. The qRT-PCR results showed that the expression levels of BcMF22 gradually increased during the three points of time after pollination (1, 2 and 4 HAP), and then declined from 4 HAP. Cellular localization of BcMF22 showed that the signal of hybridization was detected in the stigma and style of pistils at 4 HAP. There were studies which have already demonstrated that the pollen tubes passed through the stigma surface at 1HAP, and corresponded to the phase in which the pollen tubes had just penetrated the style at 4 HAP (Jiang et al. 2005; Tansengco et al. 2004; Wu et al. 2008). In Arabidopsis, the researchers found that during the first 0.5 HAP, most pollen grains have hydrated, germinated and invaded the stigmatic papilla cells; at 3.5 HAP, pollen tubes are growing through the style transmitting tissue cells (Boavida et al. 2011). As is known to all, after the pollen tube grows through the carpel's style, the sex cell nuclei from the pollen grains migrate into the ovule to fertilize the egg cell and endosperm nuclei within the female gametophyte in a process termed double fertilization. Thus, the expression features of BcMF22 and the cellular localization in pistil are strong evidence for BcMF22 that is closely related to fertilization in Chinese cabbage-pak-choi.
So, our work indicates that BcMF22 is a pollen-preferential gene and closely related to fertilization. However, more experiments should be performed to elucidate the function of BcMF22 in pollen formation and fertilization.
Abbreviations
- BcMF22 :
-
Brassica campestris Male Fertility 22
- ORF:
-
Open reading frame
- RACE:
-
Rapid amplification of cDNA ends
- HAP:
-
Time after pollination
- qRT-PCR:
-
Quantitative real-time RT-PCR
References
Becker JD, Boavida LC, Carneiro J, Haury M, Feijo JA (2003) Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiol 133:713–725
Boavida LC, Borges F, Becker JD, Feijo JA (2011) Whole genome analysis of gene expression reveals coordinated activation of signaling and metabolic pathways during pollen-pistil interactions in Arabidopsis. Plant Physiol 155:2066–2080
Cao J, Yu X, Ye W, Lu G, Xiang X (2006) Functional analysis of a novel male fertility CYP86MF gene in Chinese cabbage (Brassica campestris L. ssp. chinensis makino). Plant Cell Rep 24:715–723
Chen C, Liu S, Hao X, Chen G, Cao B, Chen Q, Lei J (2011) Characterization of a pectin methylesterase gene homolog, CaPME1, expressed in anther tissues of Capsicum annuum L. Plant Mol Bio Rep doi 10.1007/s11105-011-0358-6, in press
Fourgoux-Nicol A, Drouaud J, Haouazine N, Pelletier G, Guerche P (1999) Isolation of rapeseed genes expressed early and specifically during development of the male gametophyte. Plant Mol Biol 40:857–872
Honys D, Twell D (2003) Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol 132:640–652
Honys D, Twell D (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol 5:R85
Huang L, Cao J, Ye W, Liu T, Jiang L, Ye Y (2008) Transcriptional differences between the male-sterile mutant bcms and wild-type Brassica campestris ssp. chinensis reveal genes related to pollen development. Plant Biol (Stuttg) 10:342–355
Huang L, Zhao X, Liu T, Dong H, Cao J (2010) Developmental characteristics of floral organs and pollen of Chinese cabbage (Brassica campestris L. ssp. chinensis). Plant Systemat Evol 286:103–115
Jiang L, Yang S, Xie L, Puah C, Zhang X, Yang W, Sundaresan V, Ye D (2005) VANGUARD1 encodes a pectin methylesterase that enhances pollen tube growth in the Arabidopsis style and transmitting tract. Plant Cell 17:584–596
Kamalay JC, Goldberg RB (1980) Regulation of structural gene expression in tobacco. Cell 19:935–946
Kato H, Xie G, Sato Y, Imai R (2010) Isolation of anther-specific gene promoters suitable for transgene expression in rice. Plant Mol Bio Rep 28:381–387
Kim SG, Lee S, Kim YS, Yun DJ, Woo JC, Park CM (2010) Activation tagging of an Arabidopsis SHI-RELATED SEQUENCE gene produces abnormal anther dehiscence and floral development. Plant Mol Biol 74:337–351
Lan L, Chen W, Lai Y, Suo J, Kong Z, Li C, Lu Y, Zhang Y, Zhao X, Zhang X, Zhang Y, Han B, Cheng J, Xue Y (2004) Monitoring of gene expression profiles and isolation of candidate genes involved in pollination and fertilization in rice (Oryza sativa L.) with a 10 K cDNA microarray. Plant Mol Biol 54:471–487
Li T, Gong C, Wang T (2010) RA68 is required for postmeiotic pollen development in Oryza sativa. Plant Mol Biol 72:265–277
Li Y, Qiu L, Huang L, Cao J (2011) BcJMJ30, the gene encoding jmjC domain-containing histone demethylase is associated with pollen development and fertilization in Brassica campestris ssp. chinensis. Plant Mol Bio Rep doi 10.1007/s11105-011-0356-8, in press
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−Δ − ΔCT method. Methods 25:402–408
Mascarenhas JP (1975) The biochemistry of angiosperm pollen development. Bot Rev 41:259–314
Mascarenhas JP (1988) Anther- and pollen-expressed genes. In: D.P.S. Verma and Goldberg (ed) In temporal and spatial regulation of plant genes. Springler, Vienna, pp 97–115
Robert LS, Allard S, Gerster JL, Cass L, Simmonds J (1993) Isolation and characterization of a polygalacturonase gene highly expressed in Brassica napus pollen. Plant Mol Biol 23:1273–1278
Singh A, Grover A (2010) Plant Hsp100/ClpB-like proteins: poorly-analyzed cousins of yeast ClpB machine. Plant Mol Biol 74:395–404
Tansengco ML, Imaizumi-Anraku H, Yoshikawa M, Takagi S, Kawaguchi M, Hayashi M, Murooka Y (2004) Pollen development and tube growth are affected in the symbiotic mutant of Lotus japonicus, crinkle. Plant Cell Physiol 45:511–520
Tsuwamoto R, Yokoi S, Takahata Y (2010) Arabidopsis EMBRYOMAKER encoding an AP2 domain transcription factor plays a key role in developmental change from vegetative to embryonic phase. Plant Mol Biol 73:481–492
Wang Y, Zhang W, Song L, Zou J, Su Z, Wu W (2008) Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol 148:1201–1211
Whittle CA, Malik MR, Li R, Krochko JE (2010) Comparative transcript analyses of the ovule, microspore, and mature pollen in Brassica napus. Plant Mol Biol 72:279–299
Wing RA, Yamaguchi J, Larabell SK, Ursin VM, McCormick S (1990) Molecular and genetic characterization of two pollen-expressed genes that have sequence similarity to pectate lyases of the plant pathogen Erwinia. Plant Mol Biol 14:17–28
Wu J, Qin Y, Zhao J (2008) Pollen tube growth is affected by exogenous hormones and correlated with hormone changes in styles in Torenia fournieri L. Plant Growth Regul 55:137–148
Zhang A, Chen Q, Huang L, Qiu L, Cao J (2011a) Cloning and expression of an anther-abundant polygalacturonase gene BcMF17 from Brassica Campestris ssp. Chinensis. Plant Mol Bio Rep doi 10.1007/s11105-011-0298-1, in press
Zhang A, Qiu L, Huang Li, Yu X, Lu G, Cao J (2011b) Isolation and characterization of an anther-specific polygalacturonase gene, BcMF16, in Brassica campestris ssp. chinensis. Plant Mol Bio Rep doi: 10.1007/s11105-011-0341-2, in press
Zhang F, Liu X, Zuo K, Zhang J, Sun X, Tang K (2011) Molecular cloning and characterization of a novel gossypium barbadense L. RAD-Like gene. Plant Mol Bio Rep 29:324–333
Acknowledgement
This work was supported by the Natural Science Foundation of China (No. 30871715), the Natural Science Foundation of Zhejiang Province (2010 C12004) and the Key Sci-Technology Project of Zhejiang Province (No. Y3100300).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Y., Qiu, L., Huang, L. et al. Molecular Cloning and Characterization of BcMF22, a Novel Gene Related to Pollen Development and Fertilization in Brassica campestris ssp. chinensis . Plant Mol Biol Rep 30, 860–866 (2012). https://doi.org/10.1007/s11105-011-0393-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-011-0393-3