
Abstract
Previous studies have shown that the AtAGIP promoter derived from the Arabidopsis AGAMOUS (AG) second intron/enhancer specifies a carpel- and stamen-specific expression in its native host species, but not in heterologous species such as tobacco, which restricts its application in the engineering of male and female sterility. These findings also imply that the AG regulatory mechanism that has evolved in Arabidopsis may, to some extent, have diverged from that of tobacco. To test whether a similar chimeric promoter created using the AG second intron/enhancer can overcome this barrier of evolutionary divergence in closely related species, we generated forward- and reverse-oriented chimeric promoters, fPtAGIP and rPtAGIP, from the petunia AG second intron/enhancer (PtAGI) fragment and tested them in tobacco, which, like petunia, belongs to the Solanaceae family. Our results demonstrate that both fPtAGIP and rPtAGIP confer similar carpel- and stamen-specific expression without any leaky activity in vegetative tissues in tobacco as revealed by tissue-specific gene expression and tissue ablation. This pattern resembles that driven by the AtAGIP in Arabidopsis and indicates that the AG regulatory mechanism is more conserved between tobacco and petunia than between tobacco and Arabidopsis. The petunia-derived promoters also exhibited petal-specific activity, and their activities in floral organs were substantially influenced by the orientation of the PtAGI enhancer, with reverse-oriented enhancers displaying approximately double the effectiveness of forward-oriented enhancers. These two properties are novel and have not been observed previously with AtAGIP promoters. Furthermore, we found that PtAGIP promoter-driven tissue ablation is effective for engineering complete sterility in plants, and the resulting sterile trait is stable for at least three mitotic generations at various temperature regimes, which is important for the complete containment of seed-, pollen-, and fruit-mediated gene flow in field conditions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The tissue-specific ablation of floral organs or tissues provides an effective means for generating complete sterility, which is useful for containing the flow of transgenes into wild or closely related species, potentially leading to the creation of super weeds or highly adaptable pests. Successful engineering of stable sterility in plants requires the identification and utilization of promoters that can drive the expression of chosen genes specifically in targeted tissues or organs. Earlier work has shown that male organ-specific promoters can, when fused to the Barnase gene coding for an extracellular ribonuclease, specifically ablate tapetal cells and generate plant sterility (Mariani et al. 1990, 1992). This approach has been widely used for engineering male sterility in numerous species, including Arabidopsis (Konagaya et al. 2008), Brassica (Block and Debrouwer 1993; Roque et al. 2007), cabbage (Lee et al. 2003), creeping bentgrass and rice (Luo et al. 2006), tobacco (Hofig et al. 2006; Twell 1995), tomato (Gomez et al. 2004; Roque et al. 2007), and wheat (Block et al. 1997). As promoters specific for floral meristems or reproductive tissues become available, it is more feasible to generate both male and female sterility in transgenic plants. For example, the SLG promoter from Brasicca, which drives expression primarily in the stigmatic tissues of gynoecia and to a lesser extent in tapetal cells of anthers, is one of the pioneer promoters that has been used for engineering complete sterility. SLG is able to, when fused to the DT-A gene, which encodes a ribosomal inactivation factor, arrest the development of targeted tissues in tobacco and Arabidopsis, making plants completely sterile (Thorsness et al. 1991, 1993). Similar sterility was also produced with the use of the Arabidopsis LFY promoter, which is specific for floral meristem cells, and when fused to DT-A completely ablated sepals, petals, stamens, and carpels (Nilsson et al. 1998). Meiosis-specific promoters, such as the AtDMC promoter, further improve the accuracy and specificity of tissue ablation. As such, when this promoter is fused to Barnase, it enables the precise ablation of both male and female gametes without sacrificing flower appearance and architecture (Kobayashi et al. 2006), which is of aesthetic value for economically important ornamental cultivars. Furthermore, with an array of flower-specific promoters that have now been characterized in woody species, engineering male and female sterility in tree species is also becoming feasible. PTLF and PTD promoters from poplar, as well as BpFULL and BpMADS1 from birch, are able to direct either Barnase or DT-A expression specifically in floral tissues, rendering transgenic trees completely sterile (Lannenpaa et al. 2005; Lemmetyinen et al. 2004; Skinner et al. 2003; Wei et al. 2007).
Despite its great effectiveness, current tissue- or cell-specific ablation systems suffer several undesirable drawbacks. The most prevailing of these is the presence of leaky promoter activity in non-targeted tissues, which often causes adverse effects on vegetative growth (Kobayashi et al. 2006; Lannenpaa et al. 2005; Lemmetyinen et al. 2004; Nilsson et al. 1998; Skinner et al. 2003; Wei et al. 2007). Although co-expression of Barnase together with its inhibitor, Barstar, could lessen such vegetative damage, this attenuation appears to be restricted to certain developmental stages and/or growth conditions (Wei et al. 2007). Whether this approach is effective and practically applicable under field conditions remains unknown. Another drawback of these ablation systems is the reversibility of the engineered sterility. For example, transgenic Arabidopsis plants harboring an SLG::DT-A cassette display perfect self-sterility, but become partially fertile when out-crossed with either wt male or female gametes (Thorsness et al. 1993). Since engineering sterile traits for transgene containment will eventually need to be deployed in the field, the influence of environmental or genetic factors could significantly impact the stringency of containment. Hence, engineering stable sterility without impairing non-targeted tissue is highly desirable, but is still a challenge and requires a continuous effort in the search and characterization of highly specific and tightly regulated promoters.
Recently, we have taken advantage of the tissue specificity of the enhancer within the AGAMOUS (AG) second intron for engineering complete sterility. AG encodes a floral homeotic MADS-box factor and acts to specify stamen and carpel identity and floral meristem determinacy in the inner two whorls of flowers, which has been very well characterized in Arabidopsis (Bowman et al. 1989, 1991). AG mRNA expression is highly specific for carpel and stamen primordial cells and tissues, but is absent in any other tissues (Drews et al. 1991; Yanofsky et al. 1990). An enhancer located within the AG’s second intron was found to primarily regulate this tissue-specific expression (Busch et al. 1999; Deyholos and Sieburth 2000; Sieburth and Meyerowitz 1997). Intriguingly, the AG promoter alone has been found to drive GUS expression ubiquitously (Sieburth and Meyerowitz 1997), while the isolated enhancer can, when fused with the minimal 35S promoter, confer a carpel- and stamen-specific expression pattern in transgenic plants (Busch et al. 1999; Deyholos and Sieburth 2000), a pattern that is identical to that of endogenous AG transcript accumulation in plants (Yanofsky et al. 1990). When the same enhancer fusion promoter was used for directing tissue-specific expression of DT-A, over 90% of transgenic Arabidopsis lines displayed precise ablation of both carpels and stamens without compromising vegetative growth (Liu and Liu 2008). The ensuing sterility was stable and persisted for several mitotic generations (Liu and Liu 2008). This AG enhancer-mediated tissue-specific ablation appears to be more efficient and precise than any other system reported to date. However, a similar construct, when introduced into tobacco, failed completely to ablate carpels and stamens in flowers, and instead, a mildly retarded floral phenotype was observed in only two of 25 lines analyzed (Wang et al. 2008). The failure of the Arabidopsis AG enhancer-derived promoter to instigate floral organ-specific ablation in tobacco suggests that the AG regulatory apparatus that has evolved in tobacco might, to a certain degree, have diverged from that in Arabidopsis, and the AG second intron may only interact or function effectively in more closely related species (e.g., same family). Since the tobacco AG ortholog(s) and its gene organization, as well as its second intron/enhancer, has not been characterized as of yet, we set out to analyze and evaluate the tissue specificity of the second intron/enhancer from the AG ortholog of petunia, pMADS3, and assess its potential application for engineering complete sterility in tobacco plants to determine whether an AG enhancer can function in closely related species.
Materials and Methods
Isolation and Cloning of Petunia AG Second-Intron Fragments
Genomic DNA was extracted from the leaves of Petunia hybrida cultivar V26 grown in a greenhouse using the D-TAB method (Gustincich et al. 1991). Three hundred nanograms of genomic DNA was used as template for amplifying the approximately 4-kb second intron of pMADS3 (PtAGI), one of two AG paralogs in petunia, using primer pair PMADS3U3151 (CTGTGCTCTGTGATGCTGAAGTTGCTTTGATT) and PMADS3L7275 (CTGAACAAGCTTTCTTGTACCTCTCAATTGTTGCT). These primers were designed to immediately flank the splice junctions of the PtAGI in pMADS3 (Kapoor et al. 2002; GenBank accession no. AB076051). Amplified fragments were cloned into the pGEM-T easy vector (Promega, Madison, WI) and verified by DNA sequencing. All PCR amplifications were performed using the following conditions: 2 min at 95°C, followed by 30 cycles of 40 s at 95°C, 1 min at 60°C, and 5 min at 72°C with a final extension of 10 min at 72°C.
Plasmid Construction and Plant Transformation
The PtAGI fragment was fused with the 60-bp minimal 35S promoter at its 5′ and 3′ ends to create functional fPtAGIP and rPtAGIP promoters, respectively. These chimeric promoters were isolated as Fse1-SbfI fragments and inserted 5′ of the GUS and DT-A coding regions in a pBIN19 background to generate fPtAGIP::GUS, rPtAGIP::GUS, fPtAGIP::DT-A, and rPtAGIP::DT-A fusions, respectively, as illustrated in Fig. 1a. Plasmid DNA was isolated and introduced into Agrobacterium tumeficiens strain GV3101, which was subsequently used for the transformation of tobacco SR1 cultivar leaves in vitro (Horsch et al. 1985). Transformed cells and shoots were selected on standard MS medium containing 1 mg/L BA, 500 mg/L carbenicillin, and 200 mg/L kanamycin and were subsequently rooted in the same media lacking BA.

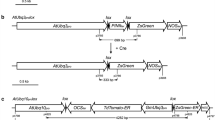
Plasmid construction and tissue-specific analysis of chimeric PtAGIP promoters. a Gene constructs. Petunia pMADS3’s second intron (PtAGI) was fused with the minimal 35S promoter (illustrated as filled bars) at its 5′ and 3′ ends, respectively, to create forward-oriented fPtAGIP and reverse-oriented rPtAGIP promoters. The orientation of PtAGI is indicated by an open arrowhead at one end. The resulting promoters were placed in front of GUS and DT-A coding regions, respectively. Transcriptional start sites and direction of transcription are indicated by angled arrows above the minimal 35S promoter (filled bars). All gene fragments were inserted between the right and left T-DNA transfer borders in the binary vector pBIN19. CaMV cauliflower mosaic virus, CsVMV cassava vein mosaic virus promoter, T nos terminator. b Summary of tissue-specific expression and ablation examined in T0 transgenic plants. Percentages (%) of the total evaluated lines (n) are indicated in each column. Floral organ tissues that express GUS are denoted by (+), while those that are completely ablated by DT-A expression are denoted by (X) and partially ablated or retarded by (X/−) symbols. The tissues that do not express detectable GUS or are not ablated by DT-A expression are denoted by (−)
Histochemical GUS Assays
Tobacco plantlets grown in vitro, as well as hand-sectioned flower buds from 2-month-old plants grown in a greenhouse, were histochemically analyzed according to a previously described protocol (Jefferson et al. 1987). Briefly, all tissues were incubated in X-gluc solution (10 mM EDTA, 100 mM NaH2PO4H2O, 0.5 mM K4Fe (CN)63H20, 0.1% triton X-100, and 1 mM X-gluc) at 37°C overnight and were subsequently depigmented with 95% ethanol three to five times before photographing.
RT-PCR Analysis of Gene Expression in Transgenic Plants
Flower buds at the 0.2- to 0.4-cm stages were harvested for isolation of total RNA using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA). Genomic DNA in the RNA samples was removed by DNase digestion with the DNA-free kit (Ambion, Austin, TX) before RT-PCR analysis with 20 ng of treated RNA as template and the Titan One Tube RT-PCR kit (Qiagen). Primer pairs DTAU363 (CTTCGTACCACGGGACTAAACTGGTTATGT) and DTAL800 (AAGTTCTACGCTTAACGCTTTCGCCTGT) were used for analysis of DT-A transcripts, while NTACT66U1 (GTTATCTGATTTGGCATAGCCT) and NTACT66L1 (TGGAATTGTAAGTTGTTTCGTG) were used to amplify the internal control, Actin 66. Amplification cycles included 45°C for 30 min for reverse transcription, 95°C for 15 min for the activation of Hotstart Taq DNA polymerase, followed by 27 cycles of 94°C for 1 min, 48–50°C for 1 min, and 72°C for 2 min. All amplified DNA fragments were resolved on a 1.2% agarose gel.
Evaluating the Stability of Engineered Sterility in Various Mitotic Generations
Three CsVMV::GUS, two fPtAGIP::DT-A, and three rPtAGIP::DT-A T0 lines were chosen for stability analysis and were visually and microscopically evaluated for floral phenotypes. After flower development had terminated in the T0 generation, the plants were cut back to approximately the fourth or fifth node from the base of the plant, and subsequent re-growth (termed T0C1) was subjected to similar phenotypic analyses. T0C1 plants were cut back one more time and T0C2 plants were reevaluated. The entire evaluation lasted approximately 8 to 9 months in a greenhouse. All data are summarized in Table 1.
Results
The PtAGIP Promoter Confers Distinct Floral Organ-Specific Expression
The petunia genome contains two AG homologs, FBP6 and pMADS3, both of which are specifically expressed in carpel and stamen primordia and assume similar functions as demonstrated by ectopic expression studies (Kater et al. 1998). Further analyses showed that the pMADS3 genomic region contains nine exons and eight introns with sizes and locations similar to those characterized in the Arabidopsis AG gene (Busch et al. 1999; Deyholos and Sieburth 2000; Kapoor et al. 2002). The second intron in pMADS3 is about 1 kb longer than the Arabidopsis AG’s second intron, but they both share similar transcription factor binding sites (Hong et al. 2003; Kapoor et al. 2002), which strongly suggests that the second intron of pMADS3 assumes a similar regulatory role as its Arabidopsis counterpart. Based on this information, we cloned and verified the second intron fragment from pMADS3. For convenience and consistency with earlier work, we termed this fragment PtAGI. To create functional promoters, we fused a minimal 35S promoter sequence to the PtAGI fragment at its 5′ and 3′ ends, respectively, generating forward- and reverse-oriented chimeric promoters, fPtAGIP and rPtAGIP, which were used to drive GUS expression in tobacco plants (Fig. 1a). At least 30 in vitro grown plantlets harboring each gene fusion, fPtAGIP::GUS, rPtAGIP::GUS, CsVMN::GUS, and empty vector, were initially screened with an X-gluc staining assay. Like empty vector plants (Fig. 2b), no visible GUS activity was detected in root, leaf, or stem tissues in either fPtAGIP::GUS or rPtAGIP::GUS lines (Fig. 2c, d), which is in contrast to CsVMV::GUS lines (Fig. 2a). Similarly, greenhouse-grown fPtAGIP::GUS and rPtAGIP::GUS plants also showed no detectable GUS activity in vegetative tissues (data not shown). However, strong GUS expression was detected in flower tissue specifically in the carpels and stamens, and these plants also exhibited weak but detectable GUS activity in petal tissues (Fig. 2k, l), which has never been observed for the Arabidopsis AtAGIP promoter (Liu and Liu 2008). GUS activity could be detected in flower buds as early as stages 4–5 and persisted through the remaining flower development stages (Fig. 2g, h). Interestingly, we noticed that fPtAGIP::GUS and rPtAGIP::GUS plants differed in GUS expression efficiency. While approximately 70% of 20 rPtAGIP::GUS lines exhibited GUS expression in flowers, only 36% of 14 fPtAGIP::GUS lines displayed a GUS expression phenotype (Fig. 1b). Evidently, the activity of the chimeric promoters is influenced by the orientation of the incorporated PtAGI fragment.

Floral organ-specific GUS expression. Four- to five-week-old in vitro grown plantlets (a, b, c, d), longitudinally hand-sectioned flower buds at stages 9 – 12 (i, j, k, l) and floral inflorescences with stage 4 – 10 flower buds (e, f, g, h), which contain CsVMV::GUS (a, e, i), control vector pBIN19 (b, f, j), fPtAGIP::GUS (c, g, k) or rPtAGIP::GUS (d, h, l) transgenes, respectively, were histochemically analyzed with X-gluc staining. Flower developmental stages were defined based on earlier descriptions (Mandel et al. 1992). Note that only tissues that were cut or subjected to section were accessible to X-gluc staining and displayed blue coloration. se – sepal; pe – petal; st – stamen; ca – carpel
Floral Organ-Specific Ablation by PtAGIP Fused to a Cytotxic Gene
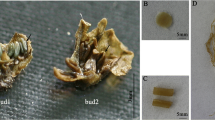
Arabidopsis AtAGIP promoters displaying carpel- and stamen-specific expression can direct a cytotoxic gene for specific ablation of both carpels and stamens, generating complete sterility (Liu and Liu 2008). To ascertain whether the PtAGIP promoters have a similar function, we fused them to the DT-A coding region, which encodes a ribosome inactivating protein (Palmiter et al. 1987), to create fPtAGIP::DT-A and rPtAGIP::DT-A fusions, respectively (Fig. 1a), and tested them in tobacco plants. As expected, both fPtAGIP::DT-A and rPtAGIP::DT-A lines showed normal vegetative growth, with similar numbers of leaves produced during development, and no growth damage was observed until the reproductive process began (Fig. 3b, c). Perturbed flower buds became apparent at very early stages of inflorescence development in fPtAGIP::DT-A and rPtAGIP::DT-A lines (Fig. 3b, c). The abnormal flower buds were small and lacked visible petals (Fig. 3e, f, g) compared to wt flowers with large, visible petals in CsVMV::GUS plants (Fig. 3d). Longitudinal tissue sections revealed that all carpel and stamen tissues were completely ablated and the residual tissues observed within perturbed flower buds in the early stages of floral development were primarily retarded petal tissue that eventually died (Fig. 3k, m), again confirming weak activity of the chimeric promoters in petal tissues. Consistent with the influence of the orientation of the PtAGI on GUS expression, the rPtAGIP::DT-A fusion was much more efficient than fPtAGIP::DT-A in directing the tissue-specific ablation of floral organs in transgenic plants. Figure 1b shows that as many as 94% of 16 rPtAGIP::DT-A plants exhibited floral organ ablation in comparison to only 17% of 21 fPtAGIP::DT-A plants with ablated floral organs.

Tissue-specific ablation and generation of complete sterility. Two-month-old CsVMV::GUS plants grown in a greenhouse displayed normal flower buds (d, h, i), inflorescence development (a, h) and reproductive processes (a), but both fPtAGIP::DT-A (b, e, f, j, k) and rPtAGIP::DT-A (c, g, l, m) plants manifested a very similar abnormal flower phenotype and reproductive processes with severely perturbed inflorescences (j, l), flower buds (e, f, g), as well as completely ablated carpels and stamens (k, m). Wt flower buds from the CsVMV::GUS plants at stages of 0.7, 1.0 and 1.5 cm (i) and perturbed flower buds of from fPtAGIP::DT-A (k) and rPtAGIP::DT-A (m) plants at corresponding developmental stages were longitudinally hand-sectioned
The relationship between DT-A expression and floral organ-specific ablation was examined using RT-PCR analysis. Figure 4 shows that DT-A mRNA was detected in perturbed flowers from all six lines analyzed harboring each PtAGIP::DT-A fusion, but not in two CsVMV::GUS lines. Conversely, tobacco Actin 66 gene expression was detected in both CsVMV::GUS and PtAGIP::DT-A lines, indicating that the abnormal flower phenotype corresponded to DT-A expression.

Verification of DT-A expression in perturbed flowers by RT-PCR. Total RNA samples were isolated from young flower buds at the 0.2- to 0.4-cm stages and treated with DNase prior to RT-PCR amplification. Tobacco Actin 66 served as an internal control
Analysis of the Mitotic Stability of Engineered Sterility
The effective containment of transgene flow in ornamental, forestry, landscape, and bioenergy crops requires stable and irreversible sterility. Because of their sterile nature, it was impossible to analyze the meiotic stability of engineered sterility in either fPTAGIP::DT-A or rPtAGIP::DT-A plants. Instead, we analyzed their mitotic stability by continuously cutting back the plants into successive generations (Liu and Liu 2008). Two fPtAGIP::DT-A and three rPtAGIP::DT-A lines bearing the same flower phenotype, as well as three CsVMV::DT-A control lines, were chosen for this analysis. All T0 plants were subjected to two consecutive cutbacks with the mitotic generations designated T0C1 and T0C2, respectively. Table 1 shows that the more than 700 flowers examined in the CsVMV::GUS plants from T0, T0C1, and T0C2 generations exhibited the same flower phenotype with no altered flower or floral organ observed. Similarly, over 2,000 flowers evaluated in the fPtAGIP::DT-A and rPtAGIP::DT-A lines over three mitotic generations, respectively, displayed identical ablated flower phenotypes with no revertants observed. The entire evaluation process took more than 8 months in a greenhouse and experienced a wide range of temperature fluctuations ranging from as low as 10°C in the winter and as high as 40°C in the summer. The fact that no flower revertants were observed during our analyses strongly suggests that the perturbed phenotype induced by either rPtAGIP::DT-A or fPtAGIP::DT-A in tobacco is thermally stable.
Discussion
In this study, we analyzed chimeric promoters derived from the AG enhancer of petunia, which is taxonomically closely related to tobacco, and showed that this promoter can, unlike its Arabidopsis counterpart, confer floral organ-specific expression and tissue ablation in tobacco (Figs. 2 and 3). The distinct behaviors of the petunia and Arabidopsis AGIP promoters in the same heterologous tobacco host likely reflects the degree of evolutionary conservation and divergence of the AG regulatory mechanism in tobacco, Arabidopsis, and petunia.
AG plays a critical role in specifying both male and female floral organ identity in plants (Bowman et al. 1989, 1991; Yanofsky et al. 1990) and is specifically expressed in carpel and stamen primordial cells and tissue (Drews et al. 1991; Yanofsky et al. 1990), which is dictated by a enhancer element that resides within the second intron rather than its promoter (Busch et al. 1999; Deyholos and Sieburth 2000; Sieburth and Meyerowitz 1997). The AG enhancer bears a multitude of protein factor binding sites where both positive and negative regulators such as LEAFY, WUSCHEL, APETALA2, LEUNIG, SEUSS, BELLRINGER, and SEP3 bind to help define AG enhancer-governed tissue specificity in response to developmental cues (Bao et al. 2004; Bomblies et al. 1999; Busch et al. 1999; Deyholos and Sieburth 2000; Lohmann et al. 2001; Mayer et al. 1998; Parcy et al. 1998; Weigel et al. 1992). Such a complex interaction probably demands a high degree of host specificity of the AG’s enhancer function because any significant divergence in either enhancer or regulatory factors could impair their intimate interactions. Hence, the inability of the Arabidopsis AG second intron-derived AtAGIP promoters to properly function in tobacco (Wang et al. 2008) could be attributable to the physical and functional divergence of both the introduced Arabidopsis AG enhancer and native transcription factors in tobacco. If this notion holds true, the AG second intron/enhancer from closely related species (e.g., tomato or petunia) should faithfully function in tobacco, assuming that all AG enhancers in their native hosts play similar regulatory roles. In agreement with this prediction, the isolated petunia AG enhancer and its derived chimeric PtAGIP promoters were able to drive carpel- and stamen-specific GUS expression and DT-A-mediated tissue ablation in tobacco (Figs. 1b, 2, and 3), which resembles that observed with the AtAGIP promoter in Arabidopsis, indicating that the role of the AG enhancer in the regulation of their residential gene expression is broadly conserved in plants but its regulatory mechanism and apparatus is relatively conserved in evolutionarily related species (e.g., tobacco and petunia).
The observation of additional petal-specific activity manifested by GUS expression and a partially retarded petal phenotype in PtAGIP::DT-A lines suggests that the petunia AG enhancer has gained additional tissue specificity that has not been found in its counterpart in Arabidopsis. However, previous studies showed that pMADS3 is specifically expressed in stamens and carpels in petunia and its transcript is undetectable in petal tissue (Kapoor et al. 2002; Kater et al. 1998), but this research was based on RNA blot analysis, which may not be as sensitive as the transgenic expression and tissue ablation used in our study to detect weak activity. Hence, whether the observed petal-specific activity is a result of its inherent activity or stochastically gained in tobacco awaits further characterization of the function of the AG enhancers in their native petunia and tobacco hosts.
We previously demonstrated that Arabidopsis AtAGIP promoters with either forward- or reverse-oriented AG enhancer fragments yielded similar GUS expression activity and tissue ablation efficiencies (Liu and Liu 2008). However, the activity of the petunia PtAGIP promoters is substantially influenced by the orientation of the AG second intron/enhancer (Fig. 1b), which raises interesting questions about the underlying mechanisms. In Arabidopsis, the AG enhancer is located within the 3-kb second intron, but the 1.7-kb region located at its 3′ end is critical for its function (Busch et al. 1999; Deyholos and Sieburth 2000). This critical region has also been found to house eight of the nine binding sites for regulatory factors discussed above (Bao et al. 2004; Bomblies et al. 1999; Busch et al. 1999; Lohmann et al. 2001; Mayer et al. 1998; Parcy et al. 1998). These transcription factor binding sites were also found to be conserved in the enhancer of pMADS3 from petunia, but their locations are quite different (Hong et al. 2003). Instead, eight of the nine binding sites were found to reside in the first 2-kb region of the 4-kb enhancer fragment. Conceivably, the reverse orientation of the petunia AG enhancer positions the key regulatory region in closer proximity to the minimal 35S promoter, allowing more efficient or intimate physical interactions between the AG enhancer and minimal 35S promoter, which could contribute to the observed orientation-dependent promoter activity.
Our work revealed that the petunia PtAGIP promoters can effectively function in evolutionarily closely related tobacco species, while the Arabidopsis AtAGIP promoter fails in this same species. PtAGIP-conferred tissue specificity is analogous to that conferred by the AtAGIP promoter in general (Liu and Liu 2008), indicating that AG enhancer-mediated regulation of its residential gene is widely conserved among plants. Importantly, the chimeric promoters created here can direct the tissue-specific ablation of carpels and stamens without compromising vegetative growth, and the rPtAGIP promoter in particular can achieve up to 94% ablation efficiency in evolutionarily closely related tobacco plants (Fig. 1b), which is comparable to the efficiency obtained with the AtAGIP::DT-A fusion in its native host (Liu and Liu 2008). This signifies that AG enhancer-derived promoters are ideal for engineering male and female sterility in their native hosts as well as in closely related species. Given that AG orthologs and their tissue-specificity are widely conserved in many species (Kramer et al. 2004; Zahn et al. 2006), engineering complete sterility for containing pollen-, fruit-, and seed-mediated gene flow could be achieved across a wide range of species using a similar approach. The demonstrated stability of the engineered sterile trait in various mitotic generations in a greenhouse with significant temperature fluctuations further validates our earlier report on the mitotic stability of sterility with the AtAGIP promoters in Arabidopsis (Liu and Liu 2008) and is of significance for the application of this engineered sterility as a practical tool for gene containment in field conditions.
References
Bao X, Franks RG, Levin JZ, Liu Z (2004) Repression of AGAMOUS by BELLRINGER in floral and inflorescence meristems. Plant Cell 16:1478–1489
Block M, Debrouwer D (1993) Engineered fertility control in transgenic Brassica napus L: histochemical analysis of anther development. Planta 189:218–225
Block M, Debrouwer D, Moens T (1997) The development of a nuclear male sterility system in wheat: expression of the barnase gene under the control of tapetum specific promoters. Theor Appl Genet 95:125–131
Bomblies K, Dagenais N, Weigel D (1999) Redundant enhancers mediate transcriptional repression of AGAMOUS by APETALA 2. Dev Biol 216:260–264
Bowman JL, Smyth D, Meyerowitz EM (1989) Genes directing flower development in Arabidopsis. Plant Cell 1:37–52
Bowman JL, Smyth DR, Meyerowitz EM (1991) Genetic interactions among floral homeotic genes of Arabidopsis. Development 112:1–20
Busch MA, Bomblies K, Weigel D (1999) Activation of a floral homeotic gene in Arabidopsis. Science 285:585–587
Deyholos MK, Sieburth LE (2000) Separable whorl-specific expression and negative regulation by enhancer elements within the AGAMOUS second intron. Plant Cell 12:1799–1810
Drews GN, Bowman JL, Meyerowitz EM (1991) Negative regulation of the Arabidopsis homeotic gene AGAMOUS by the APETALA2 product. Cell 65:991–1002
Gomez MD, Beltran JP, Canas LA (2004) The pea END1 promoter drives anther-specific gene expresión in different plant species. Planta 219:967–981
Gustincich S, Manfioletti G, Del Sal GC, Schneider C, Carnici PC (1991) A fast method for high-quality genomic DNA extraction from whole human blood. Biotechniques 11:298–302
Hofig KP, Moller R, Donaldson L, Putterill J, Walter C (2006) Towards male sterility in Pinus radiata—a stilbene synthase approach to genetically engineer nuclear male sterility. Plant Biotechnol J 4:333–343
Hong RL, Hamaguchi L, Busch MA, Weigel D (2003) Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell 15:1296–1309
Horsch RB, Fry JE, Hoffmann NL, Wallroth M, Eichholtz D, Rogers SG, Fraley RT (1985) A simple and general method for transferring genes into plants. Science 227:1229–1231
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Kapoor M, Tsuda S, Tanaka Y, Mayama T, Okuyama Y, Tsuchimoto S, Takatsuji H (2002) Role of petunia pMADS3 in determination of floral organ and meristem identity, as revealed by its loss of function. Plant J 32:115–127
Kater MM, Colombo L, Franken J, Busscher M, Masiero S, Campagne MML, Angenent GC (1998) Multiple AGAMOUS homologs from cucumber and petunia differ in their ability to induce reproductive organ fate. Plant Cell 10:171–182
Kobayashi K, Munemura I, Hinata K, Yamamura S (2006) Bisexual sterility conferred by the differential expression of Barnase and Barstar: a simple and efficient method of transgene containment. Plant Cell Rep 25:1347–1354
Konagaya K, Ando S, Kamachi S, Tsuda M, Tabei Y (2008) Efficient production of genetically engineered, male-sterile Arabidopsis thaliana using anther-specific promoters and genes derived from Brassica oleracea and B. rapa. Plant Cell Rep 27:1741–1754
Kramer EM, Alejandra J, Stilio VSD (2004) Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS box genes in angiosperms. Genetics 166:1011–1023
Lannenpaa M, Hassinen M, Ranki A, Holtta-Vuori M, Lemmetyinen J, Keinonen K, Sopanen T (2005) Prevention of flower development in birth and other plants using a BpFULL1::BARNASE construct. Plant Cell Rep 24:69–78
Lee YH, Chung KH, Kim HU, Jin YM, Kim HI, Park BS (2003) Induction of male sterile cabbage using a tapetum-specific promoter from Brassica campestris L. ssp. pekinensis. Plant Cell Rep 22:268–273
Lemmetyinen J, Keinonen K, Sopanen T (2004) Prevention of the flowering of a tree, silver birch. Mol Breed 13:243–249
Liu Z, Liu Z (2008) The second intron of AGAMOUS drives carpel- and stamen-specific expression sufficient to induce complete sterility in Arabidopsis. Plant Cell Rep 27:855–863
Lohmann JU, Hong RL, Hobe M, Busch MA, Parcy F, Simson R, Weigel D (2001) A molecular link between stem cell regulation and floral patterning in Arabidopsis. Cell 105:793–803
Luo H, Lee J-Y, Hu Q, Nelson-Vasilchik K, Eitas TK, Lickwar C, Kausch AP, Chandlee JM, Hodges TK (2006) RTS, a rice anther-specific gene is required for male fertility and its promoter sequence directs tissue-specific gene expression in different plant species. Plant Mol Biol 62:397–408
Mandel MA, Bowman JL, Kempin SA, Ma H, Meyerowitz EM, Yanofsky MF (1992) Manipulation of flower structure in transgenic tobacco. Cell 71:133–143
Mariani C, DeBeuckeleer M, Trueltner J, Leemans J, Goldberg RB (1990) Induction of male sterility in plants by a chimeric ribonuclease gene. Nature 347:737–741
Mariani C, Gossele V, Beuckeleer MD, Block MD, Goldburg RB, Greef WD, Leemans J (1992) A chimaeric ribonuclease-inhibitor gene restores fertility to male sterile plants. Nature 357:384–387
Mayer KF, Schoof H, Haecker A, Lenhard M, Jurgens G, Laux T (1998) Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95:805–815
Nilsson O, Wu E, Wolfe DS, Weigel D (1998) Genetic ablation of flowers in transgenic Arabidopsis. Plant J 15:799–804
Palmiter RD, Behringer RR, Quaife CJ, Maxwell FM, Maxwell IH, Brinster RL (1987) Cell lineage ablation in transgenic mice by cell-specific expression of a toxin gene. Cell 50:435–443
Parcy F, Nilsson O, Busch MA, Lee I, Weigel D (1998) A genetic framework for floral patterning. Nature 395:561–566
Roque E, Gomez MD, Ellul P, Wallbraun M, Madueno F, Beltran JP, Canas LA (2007) The PsEND1 promoter: a novel tool to produce genetically engineered male-sterile plants by early anther ablation. Plant Cell Rep 26:313–325
Sieburth LE, Meyerowitz EM (1997) Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell 9:355–365
Skinner JS, Meilan R, Ma C, Strauss SH (2003) The Populus PTD promoter imparts floral-predominant expression and enables high levels of floral-organ ablation in Populus, Nicotiana and Arabidopsis. Mol Breed 12:119–132
Thorsness MK, Kandasamy MK, Nasrallah ME, Nasrallah JB (1991) A Brassica S-Locus gene promoter targets toxic gene expression and cell death to the pistil and pollen of transgenic Nicotiana. Dev Biol 143:173–184
Thorsness MK, Kandasamy MK, Nasrallah ME, Nasrallah JB (1993) Genetic ablation of floral cells in Arabidopsis. Plant Cell 5:253–261
Twell D (1995) Diphtheria toxin-mediated cell ablation in developing pollen: vegetative cell ablation blocks generative cell migration. Protoplasma 187:144–154
Wang H-Z, Hu B, Chen G-P, Shi N-N, Zhao Y, Yin Q-C, Liu J-J (2008) Application of Arabidopsis AGAMOUS second intron for the engineered ablation of flower development in transgenic tobacco. Plant Cell Rep 27:251–259
Wei H, Meilan R, Brunner AM, Skinner JS, Ma K, Gandhi HT, Strauss SH (2007) Field trial detects incomplete barstar attenuation of vegetative cytotoxicity in Populus trees containing a poplar LEAFY promoter::barnase sterility transgene. Mol Breed 19:69–85
Weigel D, Alvarez J, Smyth DR, Yanofsky MF, Meyerowitz EM (1992) LEAFY controls floral meristem identity in Arabidopsis. Cell 69:843–859
Yanofsky MF, Ma H, Bowman JL, Drews GN, Feldmann KA, Meyerowitz EM (1990) The protein encoded by the Arabidopsis homeotic gene AGAMOUS resembles transcription factors. Nature 346:35–39
Zahn LM, Leebens-Mack JH, Arrington JM, Hu Y, Landherr L, dePamphilis CW, Becker A, Theissen G, Ma H (2006) Conservation and divergence in the AGAMOUS subfamily of MADS-box genes: evidence of independent sub- and neofunctionalization events. Evol & Dev 8:30–45
Acknowledgments
We thank Mr. Dennis Bennett for his excellent technical assistance. This study was funded by the United States Department of Agriculture (USDA)—Agricultural Research Service Headquarter 2007 classes of postdoctoral grants and a USDA Cooperative State Research, Education, and Extension Service Biotechnology Risk Assessment Research grant (2006-03701).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, Y., Singer, S.D. & Liu, Z. Petunia AGAMOUS Enhancer-Derived Chimeric Promoters Specify a Carpel-, Stamen-, and Petal-Specific Expression Pattern Sufficient for Engineering Male and Female Sterility in Tobacco. Plant Mol Biol Rep 29, 162–170 (2011). https://doi.org/10.1007/s11105-010-0215-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-010-0215-z