
Abstract
Key message
The main anthocyanin components were identified, and the transcriptional regulation pattern of anthocyanin related genes in leaves and stem bark was elucidated in a purple B. napus.
Abstract
Brassica napus is one of the most important oil crops planted worldwide, and developing varieties of dual-purpose for oil and vegetable is beneficial to improve economic benefits. Anthocyanins are a class of secondary metabolites that not only make plants present beautiful colors, but have a variety of important physiological functions and biological activities. Therefore, increasing the accumulation of anthocyanin in vegetative organs can improve vegetable value of rapeseed. However, anthocyanin enriched varieties in vegetative organs are rare, and there are few studies on category identification and accumulation mechanism of anthocyanin, which limits the utilization of anthocyanins in B. napus. In this study, 157 anthocyanin biosynthesis related genes (ABGs) were identified in B. napus genome by homology comparison and collinearity analysis of genes related to anthocyanin synthesis and regulation in Arabidopsis. Moreover, five anthocyanins were identified in the stem bark and leaves of the purple B. napus PR01 by high performance liquid chromatography-mass spectrometry (HPLC–MS), and the expression characteristics of ABGs in the leaves and stem bark of PR01 were analyzed and compared with the green cultivar ZS11 by RNA-Seq. Combining further weighted gene co-expression network analysis (WGCNA), the up-regulation of transcript factors BnaA07. PAP2 and BnaC06. PAP2 were identified as the key to the up-regulation of most of anthocyanin synthesis genes that promoted anthocyanin accumulation in PR01. This study is helpful to understand the transcriptional regulation of anthocyanin biosynthesis in B. napus and provides the theoretical basis for breeding novel varieties of dual-purpose for oil and vegetable.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brassica napus (AnAnCnCn, 2n = 38) is one of the most important oil crops, formed from a spontaneous hybridization of two diploid species, Brassica rapa (ArAr = 20) and Brassica oleracea (CoCo = 18) (Chalhoub et al. 2014). B. napus has a variety of commercial value, including the production of high-quality vegetable oil, feed and vegetable oil fuel (Aachary and Thiyam, 2012). B. napus stalk is also a popular and delicious vegetable in China, it has been used to eating since ancient times. Therefore, developing dual-purpose of oil and vegetable in B. napus can significantly increase the economic benefits of rapeseed production without affecting oil production.
Anthocyanins belong to the water-soluble flavonoid class of secondary metabolites, which exhibit a wide range of biological functions in plants (Tanaka and Ohmiya, 2008; Glover and Martin, 2012). Anthocyanins play an important role in plant defensive mechanisms against abiotic and biotic stresses (Qiu et al. 2016). Moreover, anthocyanins can endow plants with beautiful appearance, and attract insects for pollination and animals for spreading seeds by imparting attractive red, blue and purple colors to the flowers, fruit and vegetable tissues of plants (Cartea et al. 2011). As a result of their anti-inflammatory, antioxidant and chemoprotective properties, anthocyanins may reduce the risk of inflammation and protect certain types of cancer, cardiovascular, neurodegenerative, and various age-related diseases (Butelli et al. 2008).
There are purple varieties rich in anthocyanins in common cabbage and Chinese cabbages (Yan et al. 2019; He et al. 2020), but there are few reports of rapeseed. It may be because people only pay attention to their oil producing seed traits, or the diversity is lost in the process of domestication. Considering the various health-promoting benefits of anthocyanins, B. napus varieties with high anthocyanin content may be welcomed by consumers. Therefore, the study on anthocyanin accumulation in rapeseed is of great significance.
The biosynthesis pathways and regulatory of anthocyanins have been deeply studied in some plants, such as Arabidopsis thaliana (Shi and Xie, 2014), tomato (Sun et al. 2020), rice (Zhu et al. 2018), B. rape (Guo et al. 2014) and B. oleracea (Guo et al. 2019), and the majority of the structural and regulatory genes have been identified and functionally characterized (Zhang et al. 2014). Anthocyanin synthesis pathway is a branch of flavonoid synthesis pathway, which is synthesized under the catalysis of a series of enzymes (Tanaka et al. 2008). In the anthocyanin biosynthesis pathway, structural genes can be divided into two categories: early biosynthesis genes (EBGs) and late biosynthesis genes (LBGs). Early biosynthesis stages start with the catalysis of phenylalanine to produce cinnamic acid and coumaroyl-CoA. Then, chalcone isomerase (CHI) isomerized chalcone to flavanone. The enzyme flavanone 3-hydroxylase (F3H), flavonoid 3′ -hydroxylase (F3′H) and flavonoid 3′-5′-hydroxylase (F3′5′H) catalyzes flavanone to dihydroflavonols. In the late biosynthetic step, dihydroflavonol reductase (DFR) reduces dihydroflavonols to leucoanthocyanins, which is then converted by leucoanthocyanin dioxygenase/anthocyanin synthetase (LDOX/ANS) to anthocyanidins (Nishihara and Nakatsuka, 2011). Finally, unstable anthocyanins were catalyzed by flavone 3-o-glucosyltransferase (UFGT) to produce stable anthocyanins (Yamazaki et al. 2002). The steps of anthocyanin synthesis occur in the cytoplasm of plant cells. Under the action of transporters such as glutathione s-transfer (GST), multidrug and toxicity (MATE) and ATP binding cassette (ABC), stable anthocyanins are transported from the cytoplasm to vacuoles for storage (Alfenito et al. 1998).
Anthocyanin biosynthesis is regulated by several transcription factors (TFs), such as MYB1, MYB10, PAP1, PAP2, TT2 and TT8 (Quattrocchio et al. 1999; Ramsay and Glover, 2005). These transcription factors recognize the specific binding region of the structural gene promoter and bind to the structural gene promoter to promote or inhibit anthocyanin biosynthesis (Yao et al. 2017; Jia et al. 2021). Studies have shown that MYB, basic helix-loop-helix (bHLH) and WD40 are the three most important transcription factors in the anthocyanin biosynthesis pathway, which regulate anthocyanin biosynthesis through the formation of ternary protein complex MYB-bHLH-WD40 (MBW) (Gonzalez et al. 2008; Xu et al. 2015). The complex has a stronger ability to activate target genes than MYB alone, and can effectively turn on the expression of anthocyanin synthesis, modification and transport related genes (Lloyd et al. 2017). MYB protein mainly regulates the upstream gene group of anthocyanin biosynthesis pathway, such as MYB111/MYB12, while MBW complex mainly regulates downstream gene group (Hichri et al. 2011). According to reports, TT8 is the key bHLH protein in the MBW complex that regulates anthocyanin accumulation in purple cabbage, and TT8 is the only bHLH transcription factor consistent with the expression pattern of LBGs and positively regulates anthocyanin biosynthesis in purple cabbage (Mushtaq et al. 2016). In purple mustard, the late biosynthesis genes involved in anthocyanin biosynthesis were found to be highly up-regulated in purple mustard leaves. The up-regulation of BjTT8 and BjMYC2 and anthocyanin biosynthesis genes (BjC4H, BjDFR and BjANS) may activate the formation of purple leaves of mustard (Heng et al. 2020). In addition to the above three transcription factor families regulating anthocyanin biosynthesis, Jasmonate ZIM-domain (JAZ) proteins in Arabidopsis thaliana can negatively regulate anthocyanin biosynthesis by destroying bHLH-MYB to form complexes (An et al. 2021).
Considering the various health-promoting benefits of anthocyanins, the bolts and leaves with high anthocyanin content (appears purple) may be welcomed to consumers. Anthocyanins have beneficial roles in human health, cultivating purple bolting rapeseed rich in anthocyanins is of great significance to improve rapeseed production efficiency. In this study, (1) the B. napus anthocyanin biosynthesis related genes (ABGs), involved in anthocyanin biosynthesis, transport and regulation, was identified by comparative genomics; (2) the type and content of anthocyanins in a purple B. napus strain was determined by high performance liquid chromatography-mass spectrometry (HPLC–MS); (3) the differential expression of ABGs between green cultivar ZS11 and purple inbred line PR01 was analyzed by transcriptome data, and revealed the anthocyanin biosynthesis and regulation by combining HPLC–MS. Our study will promote the understanding of the genetic and molecular mechanism of anthocyanin biosynthesis and accumulation in B. napus.
Materials and methods
ABGs identification in B. napus
Anthocyanin biosynthetic genes of B. napus were identified by syntenic and non-syntenic homolog analysis with A. thaliana. The reference genome of B. napus used for anthocyanin biosynthetic gene identification was ZS11.v0 (http://cbi.hzau.edu.cn/cgi-bin/rape/download_ext). We identified syntenic orthologs between A. thaliana and B. napus from the B. napus pan-genome information resource (BnPIR; http://cbi.hzau.edu.cn/bnapus/index.php) using both sequence similarities (cutoff: E ≤ 1E − 20) and the collinearity levels of flanking genes. The homologous relationships were analyzed by BLASTN and BLASTP algorithm-based searches using gene and protein sequences, respectively, with a cut off E-value ≤ 1E − 10 and coverage ≥ 0.75.
Plant materials and phenotypic characterization
The purple B. napus self-bred lines PR01 (produced by Department of Biotechnology, College of Life Science and Technology, Huazhong University of Science and Technology) were used for anthocyanin accumulation related research, and the green cultivar Zhongshuang 11 (ZS11) was used as a control in this study. Under the same cultivation conditions, PR01 and ZS11 were planted in greenhouse (16 h of light, 8 h of dark, 28℃), leaf edge and stem bark samples were collected at 60 days after sowing. Each sample was analyzed in triplicate, and three biological replicates were conducted. All samples were then frozen in liquid nitrogen and stored at—80° C for RNA extraction and other analysis. The phenotype of the leaf and stem from PR01 and ZS11 was compared. In addition, in order to examine the accumulation site of purple, some stems and leaves were cut horizontally and longitudinally by hand respectively, and observed by intelligent depth of field microscope Smartzoom 5 (Zeiss, Germany).
Anthocyanin extraction and HPLC–MS analysis
Anthocyanin extraction of B. napus was carried out as the same way as for Brassica juncea Coss Variety (Lin et al. 2011). The samples were then analyzed by an UHPLC-Q-Orbitrap-MS (Thermo, USA). The results were analyzed by Agilent 1200 HPLC ChemStation software. The chromatographic separation was performed on a ACQUITY UPLC BEH C18 Column (2.1 mm × 150 mm, 1.7 μm). The injection sling was 10 μL. Elution was performed using mobile phase A (aqueous 5% formic acid solution) and mobile phase B (methanol). The detection was at 520 nm, and the column oven temperature was set at 40 °C. The flow rate was 0.3 mL.min−1. The gradient program is described as follows: 0 − 4 min, 4% B; 4 − 8 min, 12 − 16% B; 8 − 9 min, 16 − 20% B; 9 − 26 min, 20 − 25% B; 26 − 27 min, 25 − 4% B; 27 − 32 min, 4% B (Shao et al. 2014).
RNA extraction, cDNA synthesis, and Illumina deep sequencing
Total RNA was extracted from the tissue using Plant RNA Purification Reagent for plant tissue according the manufacturer’s instructions (Invitrogen) and genomic DNA was removed using DNase I (TaKara). Then RNA quality was determined by 2100 Bioanalyser (Agilent) and quantified using the ND-2000 (NanoDrop Technologies). Only high-quality RNA sample (OD260/280 = 1.8 ~ 2.2, OD260/232.0, RIN ≥ 6.5, 28S:18S ≥ 1.0, > 1 μg) was used to construct sequencing library (Li and Dewey, 2011; Kim et al., 2015; Pertea et al., 2015).
Gene differential expression analysis and functional enrichment
To identify DEGs (differential expression genes) between two different samples, the expression level of each transcript was calculated according to the fragments per kilobases per million reads (FPKM) method. RSEM (http://deweylab.biostat.wisc.edu/rsem/) (Li and Dewey, 2011) was used to quantify gene abundances. Essentially, differential expression analysis was performed using the DESeq2 (Love et al. 2014) /DEGseq (Wang et al. 2010) /EdgeR (Robinson et al. 2010) with Q value ≤ 0.05, DEGs with |log2FC|> 1 and Q value < = 0.05 (DESeq2 or EdgeR) /Q value < = 0.001 (DEGseq) were considered to be significantly different expressed genes). In addition, functional-enrichment analysis including GO and KEGG were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways at Bonferroni-corrected P-value ≤ 0.05 compared with the whole-transcriptome background. GO functional enrichment and KEGG pathway analysis were carried out by Goatools (https://github.com/tanghaibao/Goatools) and KOBAS (http://kobas.cbi.pku.edu.cn/home.do).
qRT-PCR analysis
Five reported anthocyanin synthesis related structural genes and three MYB transcription genes were selected to analyze the expression levels of anthocyanin synthesis related genes in purple and green B. napus. Specific primers were designed for qRT-PCR (Table S1). Each reaction mixture (10 μL) contained 2 μL of ddH2O, 2 μL of cDNA templates, 0.5 μL of forward primers, 0.5 μL of reverse primers, and 5 μL of SYBR Green qPCR Mix. PCR amplification was carried out at 95 ℃ for 1 min, followed by 40 cycles at 95 ℃ for 20 s, 58 ~ 60 ℃ for 30 s and 72 ℃ for 30 s. The melting curve analysis of qRT-PCR samples showed that there was only one product for each gene primer reaction, which confirmed the specific amplification. The expression levels of the above genes were analyzed by the qRT-PCR method on the ‘LightCycler®480 II’ (Roche, Swizerland). PCR analysis was performed using SYBR Green PCR master mix, as previously described. Three independent experiments are realized, and three technical copies of each experiment are realized. Finally, 2−ΔΔCt method was applied to calculate the relative expression value.
Construction of gene co-expression network module
Weighted gene co-expression network analysis (WGCNA) was analyzed based on FPKM values using WGCNA R software package (v1.68) (Langfelder and Horvath, 2008). Topology overlap measure (TOM) is used for network construction and module identification. Select the calculation parameters "soft threshold power" = 14, "minModuleSize" = 30 and "mergeCutHeight" = 0.25 to analyze the transcriptome dataset. These modules were used to calculate the relationship between modules, anthocyanin content and representative genes in 12 samples. The key module network is visualized using Cytoscape with threshold = 0.25.
Results and discussion
Identification of ABGs in B. napus
Arabidopsis thaliana is the ancestral species of B. napus, which belongs to Cruciferae and has a very close genetic relationship. The biosynthetic pathway and regulation of anthocyanins in Arabidopsis have been deeply studied (Snowdon, 2007; Mason and Snowdon, 2016). According to the report, 41 ABGs, consisting of 24 structural genes encoding anthocyanin biosynthesis enzymes, 16 regulatory genes encoding TFs and one anthocyanin transport gene, involved in anthocyanin biosynthesis pathway have been well characterized in the Arabidopsis genome (Holton and Cornish, 1995; Heng et al. 2020).
Based on a combination method of syntenic and non-syntenic homology analysis, a total of 157 B. napus anthocyanin biosynthetic genes (BnaABGs) were identified, representing homologs for 33 of the 41 Arabidopsis thaliana anthocyanin biosynthesis related genes (AtABGs), and orthologs of the other eight AtABGs (AtFSL2, AtFLS4, AtFLS5, AtFLS6, AtMYB11, AtMYB113, AtMYB114 and AtLBD39) were not detected in B. napus (Table 1). A total of 56 transcription factors were identified, including 35 transcription factors that positively regulate anthocyanin synthesis and 21 transcription factors that negatively regulate anthocyanin synthesis. MYB113 and MYB114 were not identified, and PAP1 had multiple copies. It may be that the gene belongs to R2R3-MYB and the homology similarity is too high, resulting in MYB113 and MYB114 genes being divided into PAP1 after homology comparison, so they were not identified in B. napus database (Fig. S5).
Of the 157 BnaABGs, 156 were located on the 19 chromosomes of B. napus, with one, eight, 11, nine, nine, six, seven, one, 13, four BnaABGs located on chromosomes A01 to A10, and three, 12, nine, 18, seven, eight, eight, nine, 13 BnaABGs located on chromosomes C01 to C09, respectively (Fig. 1). The remaining gene, Bnascaffold0027G0063600ZS, an ortholog of F3H was anchored on Scaffold0027 which has not yet been mapped onto a chromosome. Based on B. napus subgenome information established by whole genome analysis (Song et al. 2021), all BnaABGs genes were divided into three subgenomes: LF, MF1 and MF2. According to statistics, 58 genes are located in LF, 22 genes are located in MF1 and 38 genes are located in MF2. It can be seen that there are more genes on LF, but the distribution of genes on MF1 and MF2 is almost equal (Table S2). These results show that the distribution of BnaABGs is consistent with the gene fractionation status at the whole genome level (Wang et al. 2011; Cheng et al. 2012). Through the identification of BnaABGs, the biosynthetic pathway of anthocyanins in B. napus was preliminarily established.

Distribution of 157 BnaABGs on the 19 chromosomes of B. napus. The bars indicate the chromosomes of B. napus and relative positions of BnaABGs were marked on the chromosomes. The scale ruler on the left side showed the physical distance of the chromosomes
Anthocyanin distribution in purple B. napus at seedling stage
B. napus lines ZS11 (green B. napus) and PR01 (purple B. napus) were planted in greenhouse and observed their growth and development. With the development of B. napus, there was a significant color difference in the stem bark between the two, but the change of leaves color was not obvious (Fig. 2a and 2b). In purple PR01 B. napus, the petiole and stem bark were purple, while the leaf edge was purple. However, in green ZS11 B. napus, petiole and stem bark were green, and purple could not be observed at the edge of leaves (Fig. 2b). The contents of different anthocyanins in leaves and stem bark of ZS11 and PR01 were determined (Table 2 and 3).

Anthocyanin accumulation in different tissues of purple B. napus PR01 and green B. napus ZS11 at Seedling Stage. a Whole plant, b Blade, petiole and stem (Green ZS11 is above purple PR01), c The content of total anthocyanins in different tissues of the two materials (Green ZS11 is above purple PR01)
As shown in Fig. 2c, the anatomical structures of leaves and stems between two B. napus strains were observed. The distribution of anthocyanins in the leaves of the purple line “PR01” is completely concentrated in the leaf edge epidermis, but no anthocyanins are accumulated in the mesophyll. The accumulation of anthocyanins in PR01 stems and petiole accumulates in the epidermal layer, but does not accumulate in the flesh (Fig. 2c). In contrast, the photomicrograph of the green line “ZS11” showed that the accumulation of anthocyanins in the leaves and stems of ZS11 was not obvious compared with that of PR01. The results showed that the accumulation of anthocyanins was mainly concentrated in the epidermis of leaves and stems.
Anthocyanins categories and content analysis
There are six kinds of common anthocyanins in plants. Due to the different amount of hydroxyl groups attached to the skeleton, each kind of anthocyanin has a different color (Yanlin et al. 2014). Five anthocyanins (Cy-3-digly(FA)-5-gly(MA), Cy-3-digly(SA-FA)-5-gly, Cy-3-digly(di-SA)-5-gly, Cy-3-digly(SA-pCoA)-5-gly(MA)/Cy-3-digly(di-FA)-5-gly(MA) and Cy-3-digly(SA-FA)-5-gly(MA)/Cy-3-digly(SA)-5-digly(SuA)) were identified in purple B. napus PR01 and green B. napus ZS11 by LC–MS/MS (Table 2 and Fig. S6). The results showed that cyanidin was a major anthocyanin in B. napus, and the anthocyanin content of PR01 in different tissues was much higher than that of ZS11. Furthermore, four anthocyanins (Cy-3-digly(SA-FA)-5-gly, Cy-3-digly(di-SA)-5-gly, Cy-3-digly(SA-pCoA)-5-gly(MA)/Cy-3-digly(di-FA)-5-gly(MA) and Cy-3-digly(SA-FA)-5-gly(MA)/Cy-3-digly(SA)-5-digly(SuA)) in ZS11 leaves were not identified (Table 3). The main anthocyanins identified in ZS11 and PR01 were cyanidin, and these five anthocyanins are formed by different acylation modifications of cyanidin. Anthocyanins mainly exist in the form of anthocyanin glycosides, and glucose is the most common glycosylated sugar in anthocyanins (Jeon et al. 2018). Subsequently, the gylcogroups in anthocyanins are modified by aromatics and fatty acid acylation to form stable anthocyanins (Li et al. 2012). The identification of anthocyanins in B. napus was determined by analyzing and comparing with the anthocyanins in Brassica species identified in previous references (Long-Ze et al. 2011; Matera et al. 2012; He et al. 2016).
DEGs analysis between green and purple B. napus
To obtain a genome-wide gene expression profile for purple color formation, four sequencing libraries were constructed from ZS11 (green B. napus) and PR01 (purple B. napus) with three replicates each, using Illumina sequencing technology. A total of 595.79 million raw reads, low-quality sequences, adapator-polluted reads, and ambiguous reads were removed and 590.18 million clean reads were obtained. The Q20 (sequencing error rate < 1%) and Q30 (sequencing error rate < 0.1%) values were ≥ 97.69% and 93.42%, respectively. The G + C percentage was 47.99% (Table S3). These results show that the quality of the data is high enough for the following analysis.
According to the reference transcriptome, the data obtained by RNA-seq technology were used for correlation analysis among samples: PCA and cluster analysis, and the results showed that the data were reliable (Fig. S3a and S3b). The 18,663 and 12,490 DEGs in leaves of two varieties (ZS11-L vs. PR01-L) and stems of two varieties (ZS11-S vs. PR01-S) were identified, respectively. In ZS11-L vs. PR01-L, 8280 genes up-regulated and 10,383 genes down-regulated. The 5358 genes were up-regulated and 7132 genes were down-regulated in ZS11-S vs. PR01-S (Fig. S3c and Fig. S1b). Venn diagram analysis revealed differences between different tissues of the two varieties (Fig. S1a). Cluster heat map shows the gene expression patterns of purple PR01 and green ZS11 (Fig. S2).
Gene Ontology (GO) terms and divided gene products into three categories: molecular functions, biological processes and cellular component (Fig. S2 and S4). Among the 949 GO terms in ZS11-S vs. PR01-S, the top 20 in significantly enriched, and among the 1532 GO terms in ZS11-L vs. PR01-L, the top 20 in significantly enriched. Significant enrichment of flavonoid biosynthetic process and phenylpropanoid biosynthetic process in ZS11-S vs. PR01-S, but not in ZS11-L vs. PR01-L (Fig. 3a and 3b). In order to understand their biological functions and gene interactions, DEGs were annotated in KEGG database. When using KEGG pathway and enrichment analysis to classify DEGs and highlight the biological relevance, the flavonoid biosynthesis pathway, phenylpropanoid biosynthesis and anthocyanin biosynthesis pathway of DEGs were significantly enriched in the stem bark, while the photosynthesis pathway and carbon metabolism pathway of DEGs were significantly enriched in the leaves (Fig. 3c and 3d). This result is consistent with the phenotype of ZS11 and PR01, which provides a basis for screening candidate genes related to anthocyanin biosynthesis.

Functional annotation of differentially expressed genes (DEGs) in leaves and stems of purple and green Brassica napus L. a-b GO annotation. c-d KEGG pathway enrichment
Expression analysis of ABGs in green and purple B. napus
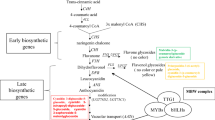
The 157 homologous genes were involved in anthocyanin biosynthesis pathway in B. napus, 97 structural genes, 56 TFs and 4 transport genes were identified. Phenylpropanoid pathway genes (PAL, 4CL and C4H) and EBGs (CHS, CHI, F3H, F3′H and FLS) were not differentially expressed in different leaves and stem bark, indicating that these genes are not the key to anthocyanin accumulation. However, the LBGs (BnaC09. DFR, BnaC01. ANS, BnaUGT79B1s, BnaA02. UGT78D2a and BnaUGT75C1s) showed differential expression levels in the leaf and stem bark of PR01 and ZS11. These genes were highly expressed in the leaf and stem bark of PR01, and the expression level in ZS11 was lower than that in PR01 (Fig. 4). Meanwhile, it has been reported that the expression of late structural genes in anthocyanin biosynthesis pathway is mainly regulated by R2R3-MYB, bHLH and WD40 transcription factors and their interactions (Yang et al. 2021).

B. napus unigenes that might be involved in the anthocyanin biosynthetic pathway and their expression levels. PAL, phenylalanine ammonia lyase; C4H, cinnamic acid 4-hydroxylase; 4CL, 4-coumarate-CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone-3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; DFR, dihydroflavonol reductase; ANS, anthocyanidin synthase; UFGT, UDP-glucose: flavonoid-3-O-glucosyltransferase; bHLH, basic helix–loop–helix; WD40, WD-repeat protein
In the study, a total of 157 genes related to the anthocyanin pathway were identified, of which 44 were differentially expressed, and only some functional genes play a role in anthocyanin biosynthesis. For example, BnaC01. ANS is significantly expressed in the leaves and stems of PR01, while BnaC07. ANS is not different from ZS11 in each tissue of PR01 (Fig. 4). The expression levels of BnaC09. DFR, BnaC01. ANS and BnaC09. UGT75C1 LBGs in leaves and stem barks of PR01 and ZS11 were different. The expression level of these genes was the highest in PR01-L, followed by PR01-S, and the lowest in leaves and stem bark of ZS11, which was green and almost free of anthocyanins. The expression of structural genes in anthocyanin biosynthesis is mainly regulated by the interaction of R2R3-MYB, bHLH and WD40 transcription factors (Petroni and Tonelli, 2011). The expression levels of R2R3-MYB proteins, such as MYB11, MYB12 and MYB111, which can independently regulate EBGs, are relatively low in PR01 and ZS11, and there is no regular difference (Fig. 4). These results show that the large expression of late structural genes is the key to anthocyanin accumulation in the biological process of regulating anthocyanin synthesis.
The transcriptional expression of LBGs is mainly activated by MBW ternary complex (Zheng et al. 2019). We found nine R2R3-MYB genes (BnaC06. PAP1a, BnaC02. PAP1, BnaA07. PAP1, BnaA02. PAP1, BnaC03. PAP1, BnaC06. PAP1b, BnaA03. PAP1, BnaA07. PAP2 and BnaC06. PAP2), BnaC06. PAP1a, BnaA07. PAP1 and BnaA02. PAP1 were almost not expressed in ZS11-S and ZS11-L; BnaC02. PAP1 was significantly higher in PR01-L than PR01-S, and the overall relative expression was higher; BnaA07. PAP2 and BnaC06. PAP2 was significantly higher in all tissues of PR01 than ZS11; BnaC06. PAP1b was significantly higher in PR01 than ZS11, but the overall expression level was lower (Fig. 4). In addition, the expression level of bHLH gene (BnaA09. TT8) was higher in PR01 than in ZS11. TT8 expression was found to be directly regulated by TT8 itself through a positive feedback regulatory loop involving redundant MBW complexes in Arabidopsis (Xu et al. 2013).Therefore, the level of TT8 expression is closely related to anthocyanin biosynthesis. The WD40 protein coding genes BnaC07. TTG1 and BnaA06. TTG1 were expressed in PR01 and ZS11, and the overall expression level in PR01 was higher than that in ZS11, while the expression level in stem bark was higher than that in leaves in ZS11 (Fig. 4). The expression levels of four TT19 genes BnaC02. TT19, BnaC09. TT19, BnaA10. TT19 and BnaA02. TT19, BnaC02. TT19 were the highest in PR01-S, followed by PR01-L, and the lowest in ZS11; the overall expression levels of BnaC02. TT19, BnaC09. TT19 and BnaA10. TT19 in PR01 were higher than those in ZS11.
Based on these results, the expression of anthocyanin biosynthesis pathway genes in different tissues of two varieties of B. napus can be observed directly. The change of anthocyanin content in plants is related to the differential expression of key genes encoding structural enzymes in anthocyanin biosynthesis pathway (Naing and Kim, 2018). The expression of structural genes in flavonoid anthocyanin biosynthesis pathway is mainly regulated by the ternary complex MBW formed by MYB, bHLH and WD40 (Zhou et al. 2012; Xu et al. 2013). Therefore, it is speculated that the activation of anthocyanin biosynthesis of the late structural genes is caused by the up regulation of the expression of PAP1/PAP2 and TT8, which is the main mechanism controlling PR01 anthocyanin biosynthesis in purple B. napus.
Different expression verification of DEGs by qRT-PCR
To confirm the reliability of the high-throughput sequencing results, the key genes of anthocyanin synthesis related enzymes were further analyzed by Real-Time Quantitative RT-PCR (qRT-PCR) to determine the transcriptional expression pattern of DEGs in anthocyanin synthesis. Eight DEGs (BnaC09. DFR, BnaA10. F3′H, BnaC04. PAL2, BnaA04. C4H, BnaC07. ANS, BnaC06. PAP2 and BnaA07. PAP2, BnaC06. PAP1b) related to anthocyanin biosynthesis pathway were selected for qRT-PCR analysis (Fig. 5). The results showed that the expression of PAL, C4H, DFR, ANS and F3′H in purple material PR01 was significantly higher than that in green material ZS11. In addition, BnaC06. PAP2 and BnaA07. PAP2 belong to R2R3-MYB, which are significantly expressed in PR01 (Fig. 5). The expression pattern was consistent with that of high-throughput sequencing, which confirms the validity of our transcriptome data. Previous studies have shown that anthocyanin content is related to the expression of anthocyanin biosynthesis genes, such as potato (Liu et al. 2015), pear (Yang et al. 2015).

qRT-PCR analysis the expression pattern of genes involved in anthocyanin biosynthesis B. napus. PR01-L and PR01-S represent the leaves and stems of purple material PR01, respectively. ZS11-L and ZS11-S represent the leaves and stems of purple material ZS11, respectively
Co-expression network analysis and identification of key regulatory genes
In order to identify the genes related to anthocyanin biosynthesis in purple B. napus PR01, all differentially expressed genes were analyzed by WGCNA. According to the gene expression trend, all genes were divided into six WGCNA modules, including an unclustered gray module (Fig. 6a). These DEGs are grouped into six main branches, each representing a module (marked with different colors). The largest module (‘turquoise’) contained 3772 genes, while the smallest module (‘grey’) contained 312 genes. Analysis of the module-trait relationships revealed that the ‘blue’ (r = 0.764, p = 0.00382) and ‘brown’ (r = 0.781, p = 0.00271) modules were highly positively correlated with antho-cyanin accumulation, the ‘turquoise’ (r = -0.746, p = 0.00534) was highly negatively correlated with anthocyanin accumulation (Fig. 6b).

Weighted gene co-expression network analysis. a Hierarchical clustering tree (cluster dendrogram) showing six modules of co-expressed genes by WGCNA. Each leaf of tree corresponds to one gene. The major tree branches constitute six modules, labeled with different colors. b Module-trait relationships. Each row represents a module, and the number of genes in each module is displayed on the left. In the right column, the values in each cell at the row column intersection represent the correlation coefficient between the module and the phenotype and are displayed according to the color code on the right. The values in parentheses in each cell represent the P value. c Cytoscape representation of co-expressed anthocyanin metabolism-related genes with edge weights ≥ 0.25 in the “MEblue” module
In order to further determine the relationship between genes in the module and screen hub genes (highly connected genes), the blue module was constructed the relevant network. In the blue module, 28 genes related to anthocyanin biosynthesis showed high expressional correlation, including two MYB transcription genes, 24 structural genes and two transporter protein genes. In the interaction network diagram, including phenylpropanoid-biosynthesis genes: PAL, 4CL and C4H; early biosynthesis genes: CHS, F3H, and F3′H; Late biosynthesis genes: DFR, ANS and UFGT: R2R3-MYB: PAP2 (Fig. 6c). These highly connected genes are called central genes in the gene co-expression network, which play an important role in understanding the biological mechanism of anthocyanin synthesis regulation. In the middle of the network, two MYB genes and two transporter genes (BnaC06. PAP2, BnaA07. PAP2, BnaA10. TT19 and BnaA02. TT19) showed the highest node connectivity with other structural genes and two transporter genes (Fig. 6c).
In previous studies, heterologous expression of PAP2 in Arabidopsis induced anthocyanin accumulation in tomato (Li et al. 2018), PAP2 is a key gene controlling anthocyanin accumulation in B. napus (Chen et al. 2020). In addition, PAL, 4CL, C4H, CHS, F3H, F3′H, DFR, ANS, UFGT and TT19 have been identified to be related to anthocyanin biosynthesis (Petroni and Tonelli, 2011; Sun et al. 2012). Through the above analysis, it was found that BnaC06. PAP2 and BnaA07. PAP2 were located in the center of the co-expression network and were highly correlated with other genes. According to reports of other species, PAP2 is a key regulator of anthocyanin biosynthesis (Gonzalez et al. 2008). Therefore, we speculate that the differential expression of PAP2 is the key to anthocyanin accumulation in PR01.
Conclusions
The accumulation of anthocyanins in leaves and stems is conducive to the development of vegetable value for B. napus. Total 157 ABGs were identified in the B. napus genome based on homology analysis with Arabidopsis ABGs in this study, and 5 anthocyanins were detected in in purple B. napus PR01 by HPLC–MS. The expression patterns of these genes indicated that late biosynthesis genes (BnaC09. DFR, BnaA04. C4H, and BnaC07. ANS), positive regulatory genes (BnaC06. PAP2 and BnaA07. PAP2), negative regulatory genes (BnaC09. LBD37 and BnaA09. LBD38) and transport protein genes (BnaA10. TT19 and BnaA02. TT19) were differentially expressed in leaves and stem barks between PR01 and ZS11, and the study found that BnaC06. PAP2 and BnaA07. PAP2 were highly co-expressed with BnaA10. F3′H, BnaC09. F3′H, BnaC08. F3Hb, BnaA09. F3Hb, BnaC09. DFR, BnaC07. ANS, BnaC01. ANS BnaA02. UGT78D2a, BnaA08. UGT75C1, BnaC08. UGT75C1, BnaA10. TT19 and BnaA02. TT19, which showed that these genes play key roles in anthocyanin accumulation in B. napus. These results provided the insight of the genetic mechanism and regulatory network of anthocyanin accumulation in purple B. napus, and served as the theoretical basis for breeding vegetable varieties with high anthocyanin content.
Data availability
The datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
References
Aachary AA, Thiyam U (2012) A pursuit of the functional nutritional and bioactive properties of canola proteins and peptides. Crit Rev Food Sci Nutr 52:965–979
Alfenito MR, Souer E, Goodman CD, Buell R, Mol J, Koes R et al (1998) Functional complementation of anthocyanin sequestration in the vacuole by widely divergent glutathione S-transferases. Plant Cell 10:1135–1149
An JP, Xu RR, Liu X, Zhang JC, Wang XF, You CX et al (2021) Jasmonate induces biosynthesis of anthocyanin and proanthocyanidin in apple by mediating the JAZ1-TRB1-MYB9 complex. Plant J 106:1414–1430
Butelli E, Titta L, Giorgio M, Mock HP, Matros A, Peterek S et al (2008) Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat Biotechnol 26:1301–1308
Cartea ME, Francisco M, Soengas P, Velasco P (2011) Phenolic compounds in Brassica vegetables. Molecules 16:251–280
Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X et al (2014) Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953
Chen D, Liu Y, Yin S, Qiu J, Jin Q, King GJ et al (2020) Alternatively spliced BnaPAP2.A7 isoforms play opposing roles in anthocyanin biosynthesis of Brassica napus L. Front Plant Sci 11:983
Cheng F, Wu J, Fang L, Sun SL, Liu B, Lin K et al (2012) Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS ONE 7:e36442
Glover BJ, Martin C (2012) Anthocyanins. Curr Biol 22:R147–R150
Gonzalez A, Zhao M, Leavitt JM, Lloyd AM (2008) Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J 53:814–827
Guo N, Cheng F, Wu J, Liu B, Zheng S, Liang J et al (2014) Anthocyanin biosynthetic genes in Brassica rapa. BMC Genom 15:426
Guo N, Han S, Zong M, Wang GX, Zheng SN, Liu F (2019) Identification and differential expression analysis of anthocyanin biosynthetic genes in leaf color variants of ornamental kale. BMC Genom 20:564
He Q, Ren Y, Zhao W, Li R, Zhang L (2020) Low Temperature promotes anthocyanin biosynthesis and related gene expression in the seedlings of purple head Chinese cabbage (Brassica rapa L.). Genes (basel) 11:81
He Q, Zhang ZF, Zhang LG (2016) Anthocyanin accumulation, antioxidant ability and stability, and a transcriptional analysis of anthocyanin biosynthesis in purple heading chinese cabbage (Brassica rapa L. ssp pekinensis). J Agr Food Chem 64:132–145
Heng SP, Wang L, Yang X, Huang H, Chen G, Cui MD et al (2020) Genetic and comparative transcriptome analysis revealed DEGs involved in the purple leaf formation in Brassica juncea. Front Genet 11:322
Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V (2011) Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Expl Bot 62:2465–2483
Holton TA, Cornish EC (1995) Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 7:1071–1083
Jeon J, Kim JK, Kim H, Kim YJ, Park YJ, Kim SJ et al (2018) Transcriptome analysis and metabolic profiling of green and red kale (Brassica oleracea var. acephala) seedlings. Food Chem 241:7–13
Jia L, Wang J, Wang R, Duan M, Qiao C, Chen X et al (2021) Comparative transcriptomic and metabolomic analyses of carotenoid biosynthesis reveal the basis of white petal color in Brassica napus. Planta 253:8
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinf 9:559
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf 12:323
Li HY, Deng ZY, Zhu HH, Hu CL, Liu RH, Young JC et al (2012) Highly pigmented vegetables: Anthocyanin compositions and their role in antioxidant activities. Food Res Int 46:250–259
Li N, Wu H, Ding Q, Li H, Li Z, Ding J et al (2018) The heterologous expression of Arabidopsis PAP2 induces anthocyanin accumulation and inhibits plant growth in tomato. Funct Integr Genomics 18:341–353
Lin LZ, Sun J, Chen P, Harnly J (2011) UHPLC-PDA-ESI/HRMS/MS(n) analysis of anthocyanins, flavonol glycosides, and hydroxycinnamic acid derivatives in red mustard greens (Brassica juncea Coss variety). J Agric Food Chem 59:12059–12072
Liu Y, Lin-Wang K, Deng C, Warran B, Wang L, Yu B et al (2015) Comparative transcriptome analysis of white and purple potato to identify genes involved in anthocyanin biosynthesis. PLoS ONE 10:e0129148
Lloyd A, Brockman A, Aguirre L, Campbell A, Bean A, Cantero A et al (2017) Advances in the MYB-bHLH-WD Repeat (MBW) pigment regulatory model: addition of a WRKY factor and co-option of an anthocyanin myb for betalain regulation. Plant Cell Physiol 58:1431–1441
Long-Ze L, Jianghao S, Pei C, James H (2011) UHPLC-PDA-ESI/HRMS/MS(n) analysis of anthocyanins, flavonol glycosides, and hydroxycinnamic acid derivatives in red mustard greens (Brassica juncea Coss variety). J Agr Food Chem 59:12059–12072
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Mason AS, Snowdon RJ (2016) Oilseed rape: learning about ancient and recent polyploid evolution from a recent crop species. Plant Bio 18:883–892
Matera R, Gabbanini S, Nicola GRD, Iori R, Petrillo G, Valgimigli L (2012) Identification and analysis of isothiocyanates and new acylated anthocyanins in the juice of Raphanus sativus cv. Sango Sprouts Food Chem 133:563–572
Mushtaq MA, Pan Q, Chen DZ, Zhang QH, Ge XH, Li ZY (2016) Comparative leaves transcriptome analysis emphasizing on accumulation of anthocyanins in Brassica: molecular regulation and potential interaction with photosynthesis. Front Plant Sci 7:311
Naing AH, Kim CK (2018) Roles of R2R3-MYB transcription factors in transcriptional regulation of anthocyanin biosynthesis in horticultural plants. Plant Mol Biol 98:1–18
Nishihara M, Nakatsuka T (2011) Genetic engineering of flavonoid pigments to modify flower color in floricultural plants. Biotechnol Lett 33:433–441
Petroni K, Tonelli C (2011) Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci 181:219–229
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295
Qiu Z, Wang X, Gao J, Guo Y, Huang Z, Du Y (2016) The tomato Hoffman’s anthocyaninless gene encodes a bHLH transcription factor involved in anthocyanin biosynthesis that is developmentally regulated and induced by low temperatures. PLoS ONE 11:e0151067
Quattrocchio F, Wing J, van der Woude K, Souer E, de Vetten N, Mol J et al (1999) Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell 11:1433–1444
Ramsay NA, Glover BJ (2005) MYB-bHLH-WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci 10:63–70
Robinson MD, McCarthy DJ, Smyth GK (2010) EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Shao Y, Jiang J, Ran L, Lu C, Wei C, Wang Y (2014) Analysis of flavonoids and hydroxycinnamic acid derivatives in rapeseeds (Brassica napus L. var. napus) by HPLC-PDA–ESI(–)-MS(n)/HRMS. J Agric Food Chem 62:2935–2945
Shi MZ, Xie DY (2014) Biosynthesis and metabolic engineering of anthocyanins in Arabidopsis thaliana. Recent Pat Biotechnol 8:47–60
Snowdon RJ (2007) Cytogenetics and genome analysis in Brassica crops. Chromosome Res 15:85–95
Song JM, Liu DX, Xie WZ, Yang Z, Guo L, Liu K et al (2021) BnPIR: Brassica napus pan-genome information resource for 1689 accessions. Plant Biotechnol J 19:412–414
Sun C, Deng L, Du M, Zhao J, Chen Q, Huang T et al (2020) A transcriptional network promotes anthocyanin biosynthesis in tomato flesh. Mol Plant 13:42–58
Sun Y, Li H, Huang J-R (2012) Arabidopsis TT19 functions as a carrier to transport anthocyanin from the cytosol to tonoplasts. Mol Plant 5:387–400
Tanaka Y, Ohmiya A (2008) Seeing is believing: engineering anthocyanin and carotenoid biosynthetic pathways. Curr Opin Biotechnol 19:190–197
Tanaka Y, Sasaki N, Ohmiya A (2008) Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J 54:733–749
Wang LK, Feng ZX, Wang X, Wang XW, Zhang XG (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Xu W, Grain D, Le Gourrierec J, Harscoet E, Berger A, Jauvion V et al (2013) Regulation of flavonoid biosynthesis involves an unexpected complex transcriptional regulation of TT8 expression, in Arabidopsis. New Phytol 198:59–70
Xu WJ, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci 20:176–185
Yamazaki M, Yamagishi E, Gong Z, Fukuchi-Mizutani M, Fukui Y, Tanaka Y et al (2002) Two flavonoid glucosyltransferases from Petunia hybrida: molecular cloning, biochemical properties and developmentally regulated expression. Plant Mol Biol 48:401–411
Yan C, An G, Zhu T, Zhang W, Zhang L, Peng L et al (2019) Independent activation of the BoMYB2 gene leading to purple traits in Brassica oleracea. Theor Appl Genet 132:895–906
Yang J, Song HD, Chen Y, Chen B, Kim M, Kim P et al (2021) A single amino acid substitution in the R2R3 conserved domain of the BrPAP1a transcription factor impairs anthocyanin production in turnip (Brassica rapa subsp. rapa). Plant Physiol Biochem 162:124–136
Yang YN, Yao GF, Zheng DM, Zhang SL, Wang C, Zhang MY et al (2015) Expression differences of anthocyanin biosynthesis genes reveal regulation patterns for red pear coloration. Plant Cell Rep 34:189–198
Yanlin S, Jinjin J, Liping R, Chunliang L, Cunxu W, Youping W (2014) Analysis of flavonoids and hydroxycinnamic acid derivatives in rapeseeds (Brassica napus L. var. napus) by HPLC-PDA–ESI(–)-MS(n)/HRMS. J Agr Food Chem 62:2935–2945
Yao G, Ming M, Allan AC, Gu C, Li L, Wu X et al (2017) Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J 92:437–451
Zhang Y, Chen G, Dong T, Pan Y, Zhao Z, Tian S et al (2014) Anthocyanin accumulation and transcriptional regulation of anthocyanin biosynthesis in purple bok choy (Brassica rapa var. chinensis). J Agric Food Chem 62:12366–12376
Zheng T, Tan W, Yang H, Zhang L, Li T, Liu B et al (2019) Regulation of anthocyanin accumulation via MYB75/HAT1/TPL-mediated transcriptional repression. PLoS Genet 15:e1007993
Zhou LL, Shi MZ, Xie DY (2012) Regulation of anthocyanin biosynthesis by nitrogen in TTG1-GL3/TT8-PAP1-programmed red cells of Arabidopsis thaliana. Planta 236:825–837
Zhu Y, Sun H, He S, Lou Q, Yu M, Tang M et al (2018) Metabolism and prebiotics activity of anthocyanins from black rice (Oryza sativa L.) in vitro. PLoS ONE 13:e0195754
Acknowledgements
Not applicable
Funding
The work was supported by the National Natural Science Foundation of China (U2004149), National Natural Science Foundation of China (32001583), Major science and technology projects in Henan Province (201400210600) and Chinese Postdoctoral Science Foundation (2019M662539).
Author information
Authors and Affiliations
Contributions
HF and HC participated in the experiments designing, data analysis, and drafting the manuscript. XZ and HW participated in gene and protein sequence retrieval and partial data analysis. HL and WZ collects and provides some transcriptome data and gives some proposals. TS did plant culturing and gene expression analysis. ML and JH took part in designing experiments and supervising the study. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the work was performed without any commercial or financial relationship that could be understood as a potential conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fu, H., Chao, H., Zhao, X. et al. Anthocyanins identification and transcriptional regulation of anthocyanin biosynthesis in purple Brassica napus. Plant Mol Biol 110, 53–68 (2022). https://doi.org/10.1007/s11103-022-01285-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-022-01285-6