
Abstract
Spartina species play an important ecological role on salt marshes. Spartina maritima is an Old-World species distributed along the European and North-African Atlantic coasts. This hexaploid species (2n = 6x = 60, 2C = 3,700 Mb) hybridized with different Spartina species introduced from the American coasts, which resulted in the formation of new invasive hybrids and allopolyploids. Thus, S. maritima raises evolutionary and ecological interests. However, genomic information is dramatically lacking in this genus. In an effort to develop genomic resources, we analysed 40,641 high-quality bacterial artificial chromosome-end sequences (BESs), representing 26.7 Mb of the S. maritima genome. BESs were searched for sequence homology against known databases. A fraction of 16.91 % of the BESs represents known repeats including a majority of long terminal repeat (LTR) retrotransposons (13.67 %). Non-LTR retrotransposons represent 0.75 %, DNA transposons 0.99 %, whereas small RNA, simple repeats and low-complexity sequences account for 1.38 % of the analysed BESs. In addition, 4,285 simple sequence repeats were detected. Using the coding sequence database of Sorghum bicolor, 6,809 BESs found homology accounting for 17.1 % of all BESs. Comparative genomics with related genera reveals that the microsynteny is better conserved with S. bicolor compared to other sequenced Poaceae, where 37.6 % of the paired matching BESs are correctly orientated on the chromosomes. We did not observe large macrosyntenic rearrangements using the mapping strategy employed. However, some regions appeared to have experienced rearrangements when comparing Spartina to Sorghum and to Oryza. This work represents the first overview of S. maritima genome regarding the respective coding and repetitive components. The syntenic relationships with other grass genomes examined here help clarifying evolution in Poaceae, S. maritima being a part of the poorly-known Chloridoideae sub-family.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The grass (Poaceae) genus Spartina is member of the Chloridoideae subfamily, an important group with more than 400 species in approximately 140 genera exhibiting a worldwide distribution (Peterson et al. 2010), but remarkably poorly-investigated with regard to genomic information. Chloridoideae belong to the PACMAD (Panicoideae, Arundinoideae, Chloridoideae, Micrairoideae, Aristidoideae and Danthonioideae) clade (Grass Phylogeny Working Group GPWG II 2012). So far, genomic efforts have concentrated on three economically important grass subfamilies, the Panicoideae (containing maize, sorghum, and sugarcane), the Ehrhartoideae (rice) and Pooideae (wheat, Brachypodium). Divergence times between Chloridoideae and Panicoideae were estimated about 34.6–38.5 million years ago and about 40–60 million years ago between Chloridoideae and the Erhartoideae–Pooideae respectively (Christin et al. 2008; Kim et al. 2009; Prasad et al. 2011). Phylogenetic relationships among Chloridoideae genera are not fully resolved and still under debate (Hilu and Alice 2001; Peterson et al. 2010). Most species exhibit C4-type metabolism, which confers higher productivity under warm, saline or arid conditions (Christin et al. 2009). Common base chromosome number is x = 10, sometimes 9 (Roodt and Spies 2003a), with widespread polyploidy and hybridization (Roodt and Spies 2003b). Genomic organization in Chloridoideae is particularly poorly known: Only a few studies have resulted in genetic maps for tropical crops such as finger millet Eleusinecoracana (Dida et al. 2006; Srinivasachary et al. 2007) or Eragrostis tef (Zhang et al. 2001; Yu et al. 2006). Recent but still limited transcriptome analyses have contributed to expressed sequence databases and gene annotation in the turfgrass Cynodon dactylon (Kim et al. 2008), or the salt-marsh species Spartina alterniflora (Baisakh et al. 2008; Ferreira de Carvalho et al. 2013), Spartina maritima (Ferreira de Carvalho et al. 2013) and the prairie cord grass Spartina pectinata (Gedye et al. 2010).
The Spartina genus is attracting a growing interest for various fundamental and economical perspectives. Spartina species play an important ecological role in the saltmarsh dynamics by protecting the coastline from erosion and modifying the physical structure of intertidal coastal zones where they are considered as “ecosystem engineers”. Some species (Larher et al. 1977; Otte et al. 2004) are able to produce DMSP (dimethylsulfoniopropionate). This putative osmoprotectant molecule plays an important ecological role as it is a precursor of DMS (dimethylsulfide) released in the atmosphere where it contributes to cloud formation. Moreover, some Spartina species have gained attention as suitable crop with high cellulosic biomass for producing biofuel (Gonzalez-Hernandez et al. 2009). They also proved to be useful for phytoremediation purposes: they are able to tolerate heavy metal pollution and hydrocarbon (Lee 2003; Cambrollé et al. 2008; Ramanarao et al. 2011). Also, electricity production using Spartina microbial fuel cells seems promising as a new sustainable technology (Timmers et al. 2010).
From a fundamental perspective, the Spartina genus offers many opportunities in evolutionary ecology, in studies on polyploid speciation (Ainouche et al. 2004a) and to understand biological invasion processes following interspecific hybridization (Ayres et al. 2004; Ainouche et al. 2009). This genus is composed of 13–15 perennial species, (Mobberley 1956) with ploidy levels ranging from tetraploid (2n = 40) to dodecaploid (2n = 120–24) levels (reviewed in Ainouche et al. 2012). In recent molecular phylogenies, Spartina appears closely related to Sporobolus and Calamovilfa representatives (Peterson et al. 2010). The genus evolved through two main lineages respectively tetraploid and hexaploid (Baumel et al. 2002a; Fortuné et al. 2007) that diverged sometimes between 7–11 MYA, as estimated from chloroplast sequences (Bellot et al. in prep). Recurrent events of hybridization and polyploidy have arisen within and between these two lineages, and include one of the best documented example of recent allopolyploid speciation (reviewed in Ainouche et al. 2004b, 2009). The unintentional introduction of the native American species Spartina alterniflora (hexaploid, 2n = 62) to Western Europe and its subsequent hybridization (as maternal genome donor, Ferris et al. 1997; Baumel et al. 2001, 2003) with the native European S. maritima (hexaploid 2n = 60), resulting in two independently formed hybrids. In England, hybridization resulted in Spartina x townsendii, a perennial sterile hybrid first recorded around 1870 (Groves and Groves 1880), and still forming a vigorous population (Renny-Byfield et al. 2010) that gave rise (by chromosome doubling) around 1890 to a fertile and highly invasive allo-dodecapolyploid species Spartina anglica, which is now introduced on several continents. In South-west France, hybridization between S. alterniflora and S. maritima resulted in another sterile hybrid, S. x neyrautii which is still surviving in spite of severe habitat destruction (Baumel et al. 2003). This system is now used to explore early evolutionary changes following interspecific hybridization and whole genome duplication, and the genomic determinants of biological invasion (Ainouche et al. 2004a, b, 2009, 2012 and references therein).
In the perspectives of exploring the genome of these species, we have first chosen the Euro-African native hexaploid species Spartina maritima, which is involved in the paternal parentage of the hybrids and newly formed invasive allopolyploid S. anglica. Spartina maritima is usually confined to open habitat of short and long-established salt marshes, but also soft mud of low-marsh flooded at every high tide (Marchant 1967). Therefore, S. maritima is able to tolerate a wide range of substrates including lower marshes and long period of flooding (Marchant 1967; Castillo et al. 2000). Studies on the role of S. maritima in phytostabilization show a high potential to retain heavy metals such as cobalt, chromium and nickel in the rhizosphere (in Spanish estuaries: Luque et al. 1999; Cambrollé et al. 2008; and Portuguese salt marshes: Caetano et al. 2008). Moreover, S. maritima is able to accumulate cobalt in roots as well as copper, zinc and iron in leaves (Cambrollé et al. 2008). Spartina species function as excluders (Alberts et al. 1990) through external or internal exclusion mechanisms to delay translocation of heavy metals in the leaves (Hansel et al. 2001). In Southern England and Brittany, native populations are currently regressing in its northern range limit. This is interpreted as a consequence of climate change and anthropogenic habitat disturbance (Raybould et al. 1991) but has also to be related with its biological and morphological traits. Spartina maritima is a non-rhizomatous, genetically depauperate species (Yannic et al. 2004) with very low seed production (Marchant and Goodman 1969; Castellanos et al. 1994; Castillo et al. 2010).
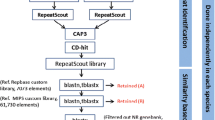
Complementing ongoing studies at the transcriptome level (Ferreira de Carvalho et al. 2013), we take here advantage of a BAC (Bacterial Artificial Chromosome) library constructed for S. maritima by analyzing 40,641 BES to provide a first glimpse on the Spartina genome composition. This study represents the first large genomic investigation performed for Spartina species. The analyses focused on the detection of repeated elements, microsatellite and protein coding regions content (Fig. 1). Additionally, comparisons with related plant lineages of the grass family (rice, Sorghum and Brachypodium) provide new insights into the evolution of a Chloridoideae subfamily representative, then contributing filling a gap regarding this poorly investigated lineage.

Analyses conducted on the BAC-end sequences
Materials and methods
BAC library construction
Spartina maritima individuals were sampled on the Etel river marshes (Presqu’île du Verdon, Morbihan, France) and transferred into pots in the greenhouse. As S. maritima populations are genetically depauperate in Western Europe with low inter-individual genetic variation and predominant vegetative propagation (Yannic et al. 2004), the sampled plants are expected to represent the same genetic background. About 40 g of etiolated young leaves were collected, kept in liquid nitrogen and stored at −80 °C until DNA extraction for the construction of the BAC library at the Centre National des Ressources Génomiques Végétales (CNRGV, Toulouse, France). High Molecular Weight (HMW) DNA was prepared from leaves of Spartina maritima. Approximately 20 g of frozen leaf tissue was ground to powder in liquid nitrogen with a mortar and pestle used to prepare megabase-size DNA embedded in agarose plugs. HMW DNA was prepared as described by Peterson et al. (2000) and modified as described in Gonthier et al. (2010). Embedded HMW DNA was partially digested with HindIII (New England Biolabs, Ipswich, Massachusetts), subjected to two size selection steps by pulsed-field electrophoresis, using a BioRad CHEF Mapper system (Bio-Rad Laboratories, Hercules, California), and ligated to pIndigoBAC-5 HindIII-Cloning Ready vector (Epicentre Biotecnologies, Madison, Wisconsin). Pulsed-field migration programs, electrophoresis buffer, and ligation desalting conditions were performed according to Chalhoub et al. (2004). To evaluate the average insert size of each library, BAC DNA was isolated from about 384 randomly selected clones in each library, restriction enzyme digested with the rare cutter NotI, and analyzed by Pulsed-Field Gel Electrophoresis (PFGE). All fragments generated by NotI digestion contained the 7.5 kb vector band and various insert fragments. In total, 44,544 clones with a mean insert size of 110 kb were retained, representing 4,900 Mb or 1.5X the genome of S. maritima (3,700 Mb, estimated from Fortuné et al. 2008). As this genome is hexaploid, the BAC library would represent 8X the basic genome (estimated as 616 Mb if we assume equivalent genome size of the 3 duplicated homoeologous genomes). More than 20,000 paired BAC-ends were sequenced by the Genoscope (Evry, France) using the BigDye Termination kit on Applied Biosystems 3730xl DNA Analysers.
Organellar DNA content
To identify organellar DNA sequences, BESs were first compared to the Oryza sativa indica and Sorghum bicolor chloroplast and mitochondrial genomes (NC_008155.1, NC_007886.1, NC_008602.1 and NC_008360.1 downloaded from the NCBI website) using BLASTn with a stringent threshold of 10−6 and a minimum hit length of 70 bp. BESs were also compared to the assembled chloroplast genome of S. maritima from 454 Roche pyrosequencing data (Bellot et al. in prep).
Identification of repetitive sequences
A survey of the composition in repeat sequences of Spartina maritima was performed using RepeatMasker version 3.2.9 (http://www.repeatmasker.org/) with Oryza sativa as the query species in Repbase (Jurka et al. 2005). BESs were annotated based on their best match to the repeat database and categorized according to the reference database used.
All BESs containing retro-elements were extracted and aligned (BLASTx with an e-value of 10−6) to the Repbase database (Jurka et al. 2005) including Reverse Transcriptase (RT) protein sequences from Copia-like and Gypsy-like elements. Spartina maritima RT sequences were then translated into proteins and aligned against Repbase RT sequences. The alignments were conducted using MUSCLE (Edgar 2004) and a maximum number of iterations of 8. Copia-like and Gypsy-like elements were analysed separately because of the high divergence between their RT domains. Phylogenetic analyses were performed using Geneious tree builder (Biomatters) with the Jukes-Cantor model and the Neighbour-joining method.
The BESs were also compared with Gramineae v3.3, O. sativa v3.3 and S. bicolor v3.0 databases downloaded from TIGR Plant repeat Databases (plantrepeats.plantbiology.msu.edu: Ouyang and Bell 2004). BLASTn analyses were conducted using an e-value cut off of 10−6 and a minimum hit length of 100 bp.
Simple sequence repeat (SSR) detection and primer design
Microsatellites were detected using the MISA perl script (MIcroSatellite research tool, Thiel et al. 2003). Parameters were set to find all SSRs with a motif length from one to six nucleotides (i.e. mono-, di-, tri-, tetra-, penta- and hexanucleotide repeats). SSR parameters were at least ten nucleotide long for mononucleotides, 12 for dinucleotides, 15 for trinucleotides, 20 for tetranucleotides, 25 for pentanucleotides and 30 for hexanucleotide motifs. The maximal number of bases interrupting two SSRs was set to 100 bp. The fasta file with BES containing SSR sequences was uploaded on the BatchPrimer3 web interface to design specific SSR primers (You et al. 2008). The criteria used for designing primer pairs included an optimum annealing temperature of 55ºC, amplicon size range of 100–300 bp with an average of 150 bp, primer length optimum of 21 ± 2 bp and GC % 50 ± 5 as suggested for SSR primer design (Bohra et al. 2011).
De novo identification of Spartina repeats
The masked file output from RepeatMasker (containing 39,910 sequences excised from repetitive elements and representing 26.2 Mb) was self-blasted with a highly-stringent e-value (10−50) to find potential novel uncharacterized repeat sequences from S. maritima genome. Sequences with at least six hits and a minimum of 90 % identity were then blasted against the NCBI GenBank non-redundant nucleic acid sequence database, the SwissProt database and a Poaceae EST database (including ESTs from Zea mays, Brachypodium distachyon, Sorghum bicolor and Oryza sativa) to find Spartina specific sequences. We also compared these sequences to different repeat databases namely TIGR Plant Repeat Databases including Gramineae v3.3, Zea mays v3.0, Oryza sativa v3.3 and Sorghum bicolor v3.0 repeat sequences, RepBase (Jurka et al. 2005) and TREP database (wheat.pw.usda.gov/ITMI/Repeats/) using BLASTn and an e-value cut off of 10−6 to assess their unique nature. BESs with no blast hits were then assembled using the Roche software (GS De Novo Assembler v. 2.5.3, Roche) with the following parameters: 90 % identity and a minimum overlap of 40 nucleotides.
Gene content and functional annotation
BESs were masked for repeat sequences and low-complexity sequences with RepeatMasker v3.2.9 as described above. The masked BESs (39,910 sequences) were then compared to coding sequences of Oryza sativa and Sorghum bicolor (version 120 and 79 respectively, downloaded from www.phytozome.com). For all tBLASTx searches, an e-value cut off of 10−6 was used. The BESs showing homology with Sorghum bicolor transcripts were then analysed with the BLAST2GO software (Conesa et al. 2005; Götz et al. 2008) to assign GO terms. BLASTx alignments were conducted using the non-redundant database of NCBI and a 10−6 stringency. In parallel, the BESs were compared against the reference transcriptome of five Spartina species (Ferreira de Carvalho et al. 2013; Ferreira de Carvalho et al. unpublished). The reference transcriptome was built using 454 technology cDNA sequencing from 5 species of Spartina: S. maritima, S. alterniflora, S. x townsendii, S. x neyrautii and S. anglica. From the 420 Mb sequenced, 52,347 contigs were assembled using the Roche Software GS De Novo Assembler and annotated following the method described in Ferreira de Carvalho et al. (2013).
Comparative genome mapping
To explore areas of potential microsynteny between Spartina maritima and selected model plants, all 39,910 masked BESs were mapped to the sequenced genomes of Arabidopsis thaliana, Brachypodium distachyon, Oryza sativa and Sorghum bicolor (Athaliana_167.fa, Bdistachyon_192_hardmasked.fa, Sbicolor_79_RM.fa and Osativa_120_RM.fa downloaded from www.phytozome.com). The e-value cut off was set to 10−6 and best blast hits were retained if they had a minimum identity of 70 %. A given BAC was then considered collinear to the targeted genome if both ends were correctly orientated within 15–250 kb of each other on the same chromosome. Otherwise, the region was considered rearranged between the two species. The synteny between Spartina maritima and Sorghum bicolor, and between Spartina maritima and Oryza sativa was visualized using the CIRCOS program (V.0.55, Krzywinski et al. 2009). BESs showing a hit with the repeatmasked genome of Sorghum bicolor were mapped onto the 10 chromosomes using BLASTn (e-value of 10−6 and a minimum identity of 70 %). Similar comparisons were performed between Spartina maritima and the 12 chromosomes of Oryza sativa.
Results
After trimming BES for vector and low read quality sequences, 40,641 BAC ends were retained for further analyses. Among those, 37,354 sequences were paired-end (Table 1). The BESs ranged in size from 57 to 938 bp with an average of 656 bp corresponding to a total of 26,682,959 nucleotides that would represent about 4.3 % of the basic genome of Spartina maritima (x = 10,616 Mb assuming equivalent genome size of the 3 duplicated genomes in this hexaploid species). The GC content is of 45.6 %.
On the 40,641 BAC end sequences aligned against chloroplast databases, 699 found a match with the Spartina maritima chloroplast genome (representing 1.72 % of the BESs) (Table 1). Respectively, 683 (1.68 %) and 668 (1.64 %) BESs matched with the S. bicolor and the O. sativa chloroplast genomes. Regarding the mitochondrial genome, 175 (0.43 %) and 91 (0.22 %) BESs were found in comparison with the S. bicolor and O. sativa genomes, respectively (Table 1). When combining the two largest sets of blasted sequences (chloroplast sequences from S. maritima and mitochondrial sequences from O. sativa), 731 sequences are retrieved representing 1.80 % from the original BESs database. In total, 39,910 BESs were analysed in the following steps (Fig. 1).
Repetitive DNA content and composition
The 39,910 Spartina maritima BESs were compared to different databases of known repeat elements to identify repeat sequences from similarity searches. The first analysis was conducted with RepeatMasker. Class I (retrotransposons) elements are predominant among the Spartina repeat sequences and represent a significant portion (14.42 %) of the BESs analysed (Table 2). Class I elements can be subclassified into long terminal repeat (LTR elements) and non-LTR retrotransposons. LTR retrotransposons represent 13.67 % of the BESs analysed. Non-LTR retrotransposons represented by short interspersed elements (SINEs, 0.02 %) and long interspersed elements (LINEs, 0.73 %) are less abundant, accounting for 0.75 % of the BESs.
As LTR elements represent a large proportion of the repeat sequences present in the genome of Spartina maritima, we conducted a phylogenetic analysis of the different families of Copia and Gypsy-like elements. Respectively, 739 and 884 protein sequences were extracted from the Copia-like and Gypsy-like dataset of BESs. Sequences of at least 400 bp long were retained to build the trees. In the Copia analysis, 211 Spartina maritima sequences are aligned with 722 RT protein sequences from RepBase (Fig. 2). Spartina maritima RT sequences identified with red branches are present in the Ivana-Oryco, Maximus, and Hopscotch clades and at the base of the lineage including the Angela, Tar and Tork clades. The larger number of repeats is in the Hopscotch clade with the Hopscotch (previously found in Oryza sativa), Shacop20 (Medicago truncatula), Castor (Arabidopsis thaliana) and Retrofit (Oryza longistamina) elements. The Maximus clade is also well-represented with a specific branch of S. maritima RT sequences. In the Gypsy tree, 123 sequences are aligned with 163 RT protein sequences from Repbase. The tree is partitioned into three clades including Athila, Tat-Ogre and Chromovirus elements (Fig. 3). Spartina maritima RTs are predominantly present in the Tat lineage, with Grande1 and ACinful elements previously found in the genus Zea. The second most represented lineage is composed of Tekay chromoviruses including Sukkala (Hordeum vulgare) and RIRE3 (O. sativa) elements.

Phylogenetic tree (Neighbour Joining analysis) of Ty3-Gypsy elements based on Reverse Transcriptase sequence alignments of Spartina maritima repeats (red branches) and the Repbase (black branches)

Phylogenetic tree (Neighbour Joining analysis) of Ty1-Copia elements based on Reverse Transcriptase sequence alignments of Spartina maritima repeats (red branches) and the Repbase (black branches)
Among the Class II DNA transposons (0.99 %) the most abundant elements are from the sub-class En-Spm corresponding to 0.58 % of the BESs. The Superfamily Tc1-IS630-Pogo is represented by 198 sequences accounting for 0.13 % of the BESs. The hobo-activator superfamily is also represented, accounting for 0.10 % of the BESs, as well as the MuDR-IS905 superfamily (0.09 %). A total of 65 Miniature Inverted Repeat transposable elements (MITEs) from the Superfamily Tourist/Harbinger are identified in the dataset representing 0.06 % of the genomic sequences analysed. With other repetitive elements present in the Repbase database, such as small RNA (0.78 %), simple repeats (0.21 %) and low complexity sequences (0.39 %), the total of known repeat elements in the genomic sequences of Spartina maritima corresponds to 16.91 %.
In parallel, the 39,910 BESs were also aligned against the TIGR databases using tblastx and a cut-off e-value of 10−6. We found 12,481 hits against the Gramineae database, 10,479 against the O. sativa database and 6,440 against the S. bicolor database (data not shown). This is consistent with the number of repeat elements found using RepeatMasker.
To identify de novo repetitive sequences in the Spartina maritima genome a self-blast analysis was conducted on the sequences first filtered with RepeatMasker. Self-blastn analysis of repeatmasked BESs revealed 8,146 sequences (20.4 % of BESs) with at least six hits (Fig. 4). This dataset was then blasted against the non-redundant GenBank database and 1,915 BESs found a hit. Among those, 196 sequences found also a hit in the Uniprot protein database. Then, homologies were searched against known repeat elements databases. In total, 79 BESs correspond to known repeat sequences and 22 BESs show homology with the ESTs Poaceae database. At the end, 6,145 BESs (representing 14.97 % of nucleotides) remained with unknown annotation representing potential novel repeat sequences from the Spartina maritima genome. Among these, 4,324 (representing 2.7 Mb) BESs were assembled into 272 contigs (containing 1,826 BESs and representing 858,686 bp) and 2,498 BESs resulted as singletons.

Frequency of BESs showing similarity to other sequences in the same dataset for de novo identification of repeated regions
A total of 4,285 simple sequence repeats (SSRs) were detected in the 26.18 Mb of Spartina maritima BESs, representing 64,643 bp or 0.25 % of the BESs sequenced (Supplementary Table 1) which is equivalent to one microsatellite every 6.1 kb (Table 3). Mononucleotides (60.9 %) are the most abundant motifs, followed by dinucleotides (21.6 %), trinucleotides (16.1 %), tetra, penta and hexanucleotides (1.42 %) (Table 3). A list of 200 SSR designed primer pairs is provided in Supplemental Table 2.
Gene content and functional annotation
The 39,910 masked for repeats BESs were first compared against the CDS databases of O. sativa and S. bicolor downloaded from the phytozome.net website using tBLASTx and a cut-off e-value of 10−6. Among the BESs analyzed, 7,305 sequences were found matching at least one coding sequence of the Oryza sativa CDS database, representing 18.3 % of the analysed BESs. A total of 6,809 BESs were homologous to at least one coding sequence of the CDS database of Sorghum bicolor, representing 17.1 % of the total BESs. Using CDSs from O. sativa and S. bicolor, 4,070 and 4,098 different coding sequences were annotated. When comparing the BESs against the Spartina reference transcriptome (Ferreira de Carvalho et al. 2013), we found 8,968 best blast hits (e-value of 10−6 and a minimum identity of 90 %) representing 22.4 % of the BESs.
Among the 6,809 BESs of S. maritima showing significant homology with the coding sequence database of S. bicolor, 4,108 were associated with at least one GO term. Among the sequences assigned to a biological process category, most terms are associated with metabolic process (5,072 sequences: including primary, cellular macromolecules and nitrogen compound metabolic process), biosynthetic process (618 sequences) and regulation of biological process (337 sequences) (Fig. 5a). Among the BESs in the molecular function category, 2,362 sequences correspond to binding activities (including nucleic acid, nucleotide, ion and protein binding). Finally, 796 sequences are associated with transferase and 580 to hydrolase activities (Fig. 5b).

Classification of GO annotations, a for biological process and b molecular function
A summary of the Spartina maritima BES composition is presented in Fig. 6. Annotation of 52.31 % of the BESs is performed and provides a first overview of the composition of Spartina maritima genome. Cytoplasmic sequences account for 2.15 % of the sequences. Low complexity regions, small RNA and Simple sequence repeats occurred in 1.41 % of the BESs. Overall, interspersed repeats represent 15.48 % of the genome including LTR-Copia elements (5.45 %), LTR-Gypsy elements (8.16 %), LINEs and SINEs (0.75 %), unclassified repeats (0.13 %) and DNA transposons (0.99 %). Potential uncharacterized highly repeated sequences in the genome represent 14.97 %. Coding regions account for 22.40 % of the genome based on homology with ESTs data from close-related Spartina species. Nevertheless, unknown genomic regions still represent 43.59 % of the dataset.

Summary of Spartina maritima BES functional annotations by homology searches
Comparative genome mapping
The synteny between Spartina BES and other plants was characterized by searching for paired BES (1) on the same chromosome, (2) within a 15–250 kb region and (3) orientated correctly with respect to each other and the homologous region. To assess the right distance between paired BESs, a histogram showing the distribution of the distance between paired BESs (using the Sorghum bicolor genome as a reference) was built (Supplementary Figure 1). Most BESs are comprised in a distance range from 15 to 250 kb; BESs out of this range are thought to be rearranged. Syntenic relationships between S. maritima repeat-masked BESs and other plant species were identified using BLASTn searches against the full-sequenced genomes of Arabidopsis thaliana, Oryza sativa, Brachypodium distachyon and Sorghum bicolor. As shown in Table 4, 3.2 % of the Spartina maritima BESs only matched the non-Poaceae genome (A. thaliana) with the retained parameters (70 % identity, e-value 10−6), revealing the high divergence between the two taxa. The other Poaceae genomes matched Spartina BESs on levels ranging from 13.6 to 16.2 % (Table 4).
According to these parameters, Arabidopsis thaliana does not show syntenic relationships with S. maritima whereas about half of paired BESs are collinear with other Poaceae species (Table 4). The higher number of homologous BESs and synteny is found between Spartina maritima and Sorghum bicolor as expected from their phylogenetic relationships in the grass family. Among the 1,394 paired BESs, 826 are localized on the same S. bicolor chromosome, with 270 BESs situated outside the 15–250 kb distance “micro-synteny” range (i.e. rearranged) and 556 BESs within the distance window of 15–250 kb. Most of these (524) are collinear with Sorghum, whereas 32 exhibit a shift in the orientation of one of the BESs (Table 4). A substantial proportion of the paired BESs (568 representing 40.75 %) match to different Sorghum chromosomes (Table 4).
The Spartina BESs mapped on the ten Sorghum bicolor chromosomes and on the twelve Oryza sativa chromosomes are represented in Fig. 7a, b, respectively. We chose to represent rearranged paired BESs for collinear regions including at least two pairs of rearranged BES (Fig. 7a, b). These putative orthologous regions involve both rearrangements on the same chromosomes or paired BESs matching different chromosomes. Collinear paired BESs show a high concentration on Sorghum chromosomes 1, 3, 4 and 6. Eight intrachromosomic and 6 interchromosomic rearrangements were detected (Fig. 7a). More rearrangements occurred between Spartina and Oryza than between Spartina and Sorghum, as 8 interchromosomic and 11 intrachromosomic could be detected (Fig. 7b).

BES sequences mapped to the a Sorghum and b Oryza genomes. The 10 (Sorghum) and 12 (Oryza) individual chromosomes are shown in the outer circle. From outer to inner circles, all homologous BESs are mapped: single BESs (black tiles), collinear paired BESs (blue tiles) and finally rearranged paired BESs (orange tiles). Paired BESs are linked to each other with grey links
Discussion
This study provides a first overview of the composition and structure of the Spartina genome. A set of 39,910 high quality genomic sequences of Spartina maritima (2n = 6x = 60, c.a. 3,700 Mb) was analysed to improve our knowledge on the repetitive and coding components of its genome.
Repetitive DNA in Spartina
The analyses of BAC-end sequences provided estimations of the repetitive sequence component, representing a proportion of 30.45 % of the sequences analysed, with 15.48 % showing homology to known repeat elements and 14.97 % potential highly repeated sequences specific to Spartina maritima. Repetitive DNA content in Spartina is intermediate between rice (35 %, 2n = 2x = 24, 1C = 420 Mb; IRGSP 2005) and Brachypodium distachyon (28.1 %, 2n = 2x = 10, 1C = 270 Mb; IBI 2010). However, regarding the Spartina maritima basic genome size (x = 10, c.a. 616 Mb), a larger number of repeat sequences would be expected: Sorghum bicolor has a genome size of 740 Mb (2n = 2x = 20) and a repeat element fraction of 62 % (Paterson et al. 2009). The proportion of repeats in S. maritima is most likely underestimated regarding the dataset analysed.
Transposable elements (TEs) are known to have important consequences on genome structure and functions (reviewed in Kejnovsky et al. 2012). Therefore, it is important to identify and evaluate the importance of the different families of repetitive elements in the genome. Identification of transposable elements in Spartina maritima is also essential to explore the effects of hybridization and genome duplication in S. anglica since S. maritima was the paternal genome donor to that species. Previous studies have shown no transposition burst in the allododecaploid Spartina anglica (Baumel et al. 2002b) most likely as a result of important methylation changes in regions flanking transposable elements (Parisod et al. 2009). In this study, analysis of TE distribution revealed that Class I TEs are significantly predominant in the genome of Spartina maritima compared to Class II TEs, with 14.42 % (10,582 elements) and 0.99 % (1,019 elements) of BESs, respectively. This contrasts from Oryza sativa for which Class II outnumbered Class I TEs with 61,900 and 163,800 TEs respectively. However, the nucleotide contribution of Class I elements in rice is larger than Class II due to the largest size of LTR retrotransposons compared to DNA transposons (IRGSP 2005). Nonetheless, our results are consistent with the contents observed in Brachypodium distachyon (Pooideae) and Sorghum bicolor (Panicoideae) where Class I elements outnumber and cover a larger fraction of the genome than Class II TEs. Indeed, in Brachypodium, Class I and Class II elements occupy 23.33 and 4.77 % of the genome respectively (IBI 2010). In Sorghum bicolor, transposable elements account for 62 % of the genome including 54.52 % of Class I TEs (Paterson et al. 2009). The comparison of TE composition in a broad range of species suggests no phylogenetic explanations but radical changes associated with TE proportions (Kejnovsky et al. 2012).
In Class I elements, LTR retrotransposons are the most abundant with a larger percentage of Ty3-Gypsy elements compared to Ty1-Copia elements, 8.16 and 5.45 %, respectively. A similar pattern is observed in other Grass genomes such as Sorghum bicolor (Ty3-Gypsy 19.00 % and Ty1-Copia 5.18 %; Paterson et al. 2009), Brachypodium distachyon (Ty3-Gypsy 16.05 % and Ty1-Copia 4.86 %; IBI 2010) and Oryza sativa (Ty3-Gypsy 10.90 % and Ty1-Copia 3.85 %; IRGSP 2005). To identify and annotate the detected elements, we performed a phylogenetic analysis including annotated elements from various databases. In the Gypsy-like element tree, all clades are represented with a larger number of sequences corresponding to the TAT clade (Spartina sequences are related to RIRE2 elements from Oryza sativa) and the Tekay clade (Spartina sequences are related to RIRE3 elements from O. sativa). In the Copia-like element tree, a larger number of Spartina repeats are present in clade 8 (corresponding to Hopscotch and Retrofit elements in Oryza) and repeats are phylogenetically close to elements of clade 3 including BARE1 and RIRE1 elements previously found in Hordeum (Manninen and Schulman 1993) and Oryza. These abundant retrotransposons have most likely undergone amplification events in Spartina maritima and now represent the largest component of repetitive DNA. Indeed, large-scale amplification rounds can lead to TE high copy number in plant genomes over short evolutionary timescales (Bennetzen 2005). Particularly, LTR retrotransposons contribute in genome size expansion (Vitte and Bennetzen 2006). One example of LTR retrotransposon family proliferation in Oryza australiensis shows a two-fold increase in genome size compared to O. sativa in less than 3 million years (Piegu et al. 2006).
Few genomic resources are available for the Spartina genus and more generally in the Chloridoideae sub-family. As a consequence, the identification of repetitive DNA using closely related species databases is challenging. In this study, we used an approach to identify Spartina maritima lineage-specific highly repeated sequences, which proved to be useful and efficient in other studies (Huo et al. 2007; Cavagnaro et al. 2008; Ragupathy et al. 2011). Such lineage-specific repetitive DNA comprised 14.97 % of the DNA analysed. In other studies, the same analysis provided also a large proportion of novel repetitive elements. As a comparison, Ragupathy et al. (2011) found 7.4 % of unique Linum usitatissimum repeats; Cavagnaro et al. (2008) found 8.45 % of carrot-specific repeat sequences and Huo et al. (2007) discovered 7.4 % of unique Brachypodium repeat sequences. These estimations are due to the high nucleotide divergence between species specific TEs and annotated TEs in databases. Indeed, most LTR-retrotransposons older than 5 million years are severely fragmented or deleted in rice (Ma et al. 2004). Nevertheless, these proportions can be underestimated as we only analyzed a small sample of the genome of Spartina maritima and some repeats located in centromeres and telomeres are frequently under-represented in BAC libraries (Zhong et al. 2002; Osoegawa et al. 2007).
SSR markers are widely used for polymorphism analyses within species. In our study, a total of 4,285 SSR regions (representing 64,643 bp) have been identified from the 26.7 Mb of genomic DNA analyzed. Also, a list of SSR marker primer pairs was designed and can be used for further genetic diversity analyses in Spartina. The density found is of one SSR every 6.1 kb in Spartina maritima, mononucleotides being the most abundant with 60.9 % of all SSRs and A/T motif the most frequent. This pattern is also most frequent in Arabidopsis thaliana (Hsu et al. 2011). The SSR frequency is consistent with the observations in Musa acuminata (1 SSR every 6.2 kb; Cheung and Town 2007) and A. thaliana (1 SSR every 6.4 kb); but lowest than O. sativa (1 SSR every 9.0 kb) and Z. mays (1 SSR every 16.1 kb) (Hsu et al. 2011). These findings are in agreement with Morgante et al. (2002), who found relationships between SSRs and low-copy DNA fraction. Indeed, SSR frequency is inversely correlated to the proportion of repetitive DNA and especially LTR retrotransposons in plants.
A previous study was performed by Gedye et al. (2010) in Spartina pectinata, where they found 841 SSRs in ESTs longer than 500 bp representing 3.2 % of their dataset. GC-rich trinucleotide repeats were the most abundant in the dataset and accounted for 18.5 % of all SSRs. Although SSR discovery by genome sequencing is easier, the development of microsatellite resources through transcriptome has many advantages as it gives the possibility to find associations with functional genes and phenotypes (Li et al. 2002). Moreover, the mutation rate in coding sequencing being lower, the numbers of SSRs and polymorphisms are expected to be lower (Blanca et al. 2011) which increases transferability of SSR markers across species (Zalapa et al. 2012).
Spartina coding sequences
Comparison of BES sequences with the non-redundant protein database of S. bicolor suggested that 6,809 are transcribed sequences representing 17.1 % of the dataset. Proportion of coding sequences identified using the Spartina reference transcriptome based on 5 Spartina species (Ferreira de Carvalho et al. 2013; Ferreira de carvalho et al. unpublished) suggests that 22.4 % of the BES sequences are coding sequences. In order to find homology despite presence of introns, the stringency must be lowered, thus increasing the possibility to find false positives. Difference of 5.3 % of putative genes between the Spartina and the Sorghum databases suggests unique transcripts and probably Spartina-specific genes (or genes that are lost in Sorghum bicolor). In the flax genome, Ragupathy et al. (2011) observed a proportion of 5.6 % unique flax transcripts, with 21.1 % of BESs showing homology to NCBI-ESTs and 26.8 % showing similarity to flax transcripts. The proportion of BESs with potential coding regions (22.4 %) is comparatively higher than the assessment of coding regions in most BES-based studies: carrot, 10 % (Cavagnaro et al. 2008); apple, 8.6 % (Han and Korban 2008); Musa, 11 % (Cheung and Town 2007) and comparable or lower than the coding fractions reported in walnut (24.9 %; Wu et al. 2011), Brachypodium (25.3 %; Huo et al. 2007) and Citrus clementina (36.0 %; Terol et al. 2008).
Based on the number of BESs matching at least one coding sequence of Sorghum bicolor in the CDS database (6,809), the mean sequence size of BESs (656 bp) and the total size of BESs sequenced (26.7 Mb), we estimated a percentage of 16.7 % of BESs containing potentially coding genes. Considering the basic genome size of S. maritima (616 Mb) and the mean size of an Oryza sativa gene (2.7 kb; IRGSP 2005), we estimated the transcriptome size of S. maritima to be around 103.21 Mb, representing 38,229 genes. This estimation is consistent with the gene number found in fully sequenced Poaceae such as Sorghum bicolor (34,008 genes; Paterson et al. 2009) and Oryza sativa (41,046 genes; Yu et al. 2005). Gene density predicts that a gene occurs every 16.1 kb based on the fact that we might expect 38,229 genes in the basic genome of Spartina maritima (estimated as 616 Mb). By comparison, S. bicolor has a gene density of one gene every 24.0 kb (Paterson et al. 2009) and O. sativa of one gene every 9.9 kb (IRGSP 2005). Musa acuminata is predicted to have one gene every 14.3 kb (D’Hont et al. 2012) and A. thaliana has one gene every 4.5 kb (AGI 2000).
Comparative genomics
Genus Spartina is part of the Chloridoideae subfamily, a poorly studied taxon of the Poaceae. The syntenic relationships remain unclear between Spartina and related grass species. Therefore, the comparative analysis of homologous regions facilitates the investigation of genome evolution and dynamics. In comparison with S. maritima, Arabidospis thaliana shows no syntenic paired BESs as they diverged 140–150 MYA (Chaw et al. 2004). Moreover, A. thaliana has undergone a recent duplication followed by the loss of 70 % of the duplicated genes (Bowers et al. 2003). The majority of the microsyntenic regions in grasses that existed before the duplication event have disappeared due to the contraction and diploidization of the genomes. Sorghum bicolor is the most comparable fully sequenced genome with an equivalent basic chromosome number (x = 10, 730 Mb for S. bicolor and 616 Mb for S. maritima) and similar gene density.
Grass genomes largely benefited from the high-throughput technologies. The sequencing of the Sorghum genome provided new insights into the synteny of cereal lineages (Paterson et al. 2009). Despite their divergence time (around 50 MYA; Christin et al. 2008), sorghum and rice are largely collinear with 57.8 % of Sorghum gene models assigned to blocks collinear with rice (Paterson et al. 2009). Kim et al. (2009) have compared Cynodon dactylon (Chlorioideae) ESTs to other grass subfamily representatives and have estimated that Chloridoideae and Panicoideae diverged about 34.6–38.5 million years ago. To our knowledge, the only physical comparative study involving a Chloridoideae member was performed by Srinivasachary et al. (2007) who compared a finger millet (Eleusine coracana, 2n = 4x = 36) genetic map with rice (2n = 2x = 24) and found that 30 % of millet BES end sequenced genomic clones and 73 % of millets ESTs identify putative rice orthologs. The recombination rate is increased in the distal chromosome regions (such as in wheat and rice, Akhunov et al. 2003; See et al. 2006) and can be caused by translocation and retention of duplicated gene copies in highly-recombinant regions. Moreover, six of the nine millet chromosomes correspond to six single rice chromosomes and the remaining three millet chromosomes are orthologous to rice chromosomes, each with one rice chromosome inserted in the centromeric region of a second rice chromosome to form a millet chromosomal conformation. Interestingly, homologous regions were identified between chromosome 2 of millet and chromosomes 2 and 10 of rice; chromosome 5 of millet and chromosomes 5 and 12 of rice; and chromosome 6 of millet and chromosomes 6 and 9 of rice. According to the known chromosome structures of rice and sorghum (Salse et al. 2008) chromosomes 1, 4, 8 and 9 of Eleusine are similar to chromosomes 3, 6, 7 and 5 of Sorghum, respectively and the synteny is potentially conserved as no major rearrangements are observed between Eleusine and rice regarding these four chromosomes. The other chromosomes seem to have undergone rearrangements since the divergence between Panicoideae and Chloridoideae 45–50 MYA. Therefore, those four conserved chromosomes should be less rearranged than the others in the Chloridoideae subfamily including Spartina species. We did not observe large macrosyntenic rearrangements using the mapping strategy employed in the present manuscript but some regions (respectively 8 among chromosomes and 6 between chromosomes) appeared to have experienced rearrangements between genera Spartina and Sorghum. We detected more rearrangements between Spartina and Oryza which is consistent with divergence times between Chloridoideae and these two respective lineages. Using BAC-End Sequence survey in sugarcane, Kim et al. (2013) also detected rearrangements between Saccharum and Sorghum that diverged about 7.8 MYA. These rearrangements were interpreted as a result of genome duplication rounds that occurred independently in the Saccharum lineage.
Among the Chloridoideae, Eleusine (x = 9) and Spartina (x = 10) have evolved separately into two sister clades: The Cynodonteae and the Zoysieae (Peterson et al. 2010). Furthermore, even though base chromosome number in the sub-family is x = 10, aneuploidy is frequent and lower base chromosome numbers (x = 7, 8, 9) are reported (Peterson et al. 2010). Duplication events are also frequent with ploidy levels ranging from diploid to 20-ploid (in Pleuraphismutica Buckley) with many of them allopolyploids as a consequence of extensive hybridization which complicates comparative analyses among genera (Roodt and Spies 2003a). Chloridoideae genome history needs definitely further investigation; The BAC library constructed and analysed in this study may provide more physical information on the putative rearrangements that occurred during Chloridoideae evolution.
In conclusion, this study represents the first overview of the Spartina maritima genome regarding the respective coding and repetitive components. This information will be particularly useful to explore genome evolution in hybrids and allopolyploid species deriving from S. maritima. The syntenic relationships with other grass genomes examined here help clarifying evolution in Poaceae, Spartina maritima being a part of the poorly-known Chloridoideae sub-family.
References
Ainouche ML, Baumel A, Salmon A (2004a) Spartina anglica C. E. Hubbard: a natural model system for analysing early evolutionary changes that affect allopolyploid genomes. Biol J Lin Soc Lond 82:475–484
Ainouche ML, Baumel A, Salmon A, Yannic G (2004b) Hybridization, polyploidy and speciation in Spartina (Poaceae). New Phytol 161:165–172
Ainouche ML, Fortuné PM, Salmon A, Parisod C, Grandbastien M-A, Fukunaga K et al (2009) Hybridization, polyploidy and invasion: lessons from Spartina (Poaceae). Biol Invasions 11:1159–1173
Ainouche M, Chelaifa H, Ferreira J, Bellot S, Ainouche A, Salmon A (2012) Polyploid evolution in Spartina: dealing with highly redundant hybrid genomes. In: Soltis DE, Soltis PS (eds) Polyploidy and genome evolution. Springer, Berlin, pp 225–243
Akhunov ED, Goodyear AW, Geng S, Qi L–L, Echalier B, Gill BS et al (2003) The organization and rate of evolution of wheat genomes are correlated with recombination rates along chromosome arms. Genome Res 13:753–763
Alberts J, Price M, Kania M (1990) Metal concentrations in tissues of Spartina alterniflora (Loisel) and sediments of georgia salt marshes. Estuar Coast Shelf Sci 30:47–58
Ayres DR, Smith DL, Zaremba K, Klohr S, Strong DR (2004) Spread of exotic cordgrasses and hybrids (Spartina sp.) in the tidal marshes of San Francisco Bay, California, USA. Biol Invasions 6:221–231
Baisakh N, Subudhi PK, Varadwaj P (2008) Primary responses to salt stress in a halophyte, smooth cordgrass (Spartina alterniflora Loisel.). Funct Integr Genomics 8:287–300
Baumel A, Ainouche ML, Levasseur JE (2001) Molecular investigations in populations of Spartina anglica C.E. Hubbard (Poaceae) invading coastal Brittany (France). Mol Ecol 10:1689–1701
Baumel A, Ainouche ML, Bayer RJ, Ainouche AK, Misset MT (2002a) Molecular phylogeny of hybridizing species from the genus Spartina Schreb. (Poaceae). Mol Phylogenet Evol 22:303–314
Baumel A, Ainouche M, Kalendar R, Schulman AH (2002b) Retrotransposons and genomic stability in populations of the young allopolyploid species Spartina anglica CE Hubbard (Poaceae). Mol Biol Evol 19:1218–1227
Baumel A, Ainouche ML, Misset MT, Gourret JP, Bayer RJ (2003) Genetic evidence for hybridization between the native Spartina maritima and the introduced Spartina alterniflora (Poaceae) in South-West France: Spartina × neyrautii re-examined. Plant Syst Evol 237:87–97
Bennetzen JL (2005) Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev 15:621–627
Blanca J, Cañizares J, Roig C, Ziarsolo P, Nuez F, Picó B (2011) Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics 12:104
Bohra A, Dubey A, Saxena R, Penmetsa RV, Poornima KN, Kumar N et al (2011) Analysis of BAC-end sequences (BESs) and development of BES-SSR markers for genetic mapping and hybrid purity assessment in pigeonpea (Cajanus spp.). BMC Plant Biol 11:56
Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422:433–438
Caetano M, Vale C, Cesário R, Fonseca N (2008) Evidence for preferential depths of metal retention in roots of salt marsh plants. Sci Total Environ 390:466–474
Cambrollé J, Redondo-Gómez S, Mateos-Naranjo E, Figueroa ME (2008) Comparison of the role of two Spartina species in terms of phytostabilization and bioaccumulation of metals in the estuarine sediment. Mar Pollut Bull 56:2037–2042
Castellanos E, Figueroa M, Davy A (1994) Nucleation and facilitation in salt-marsh succession—interactions between Spartina maritima and Arthrocnemum perenne. J Ecol 82:239–248
Castillo JM, Fernández-Baco L, Castellanos EM, Luque CJ, Figueroa ME, Davy AJ (2000) Lower limits of Spartina densiflora and S. maritima in a Mediterranean salt marsh determined by different ecophysiological tolerances. J Ecol 88:801–812
Castillo JM, Ayres DR, Leira-Doce P, Bailey J, Blum M, Strong DR et al (2010) The production of hybrids with high ecological amplitude between exotic Spartina densiflora and native S. maritima in the Iberian Peninsula. Divers Distrib 16:547–558
Cavagnaro PF, Chung S-M, Szklarczyk M, Grzebelus D, Senalik D, Atkins AE et al (2008) Characterization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol Genet Genomics 281:273–288
Chalhoub B, Belcram H, Caboche M (2004) Efficient cloning of plant genomes into bacterial artificial chromosome (BAC) libraries with larger and more uniform insert size. Plant Biotechnol J 2:181–188
Chaw S-M, Chang C–C, Chen H-L, Li W-H (2004) Dating the monocot? Dicot divergence and the origin of core eudicots using whole chloroplast genomes. J Mol Evol 58:424–441
Cheung F, Town C (2007) A BAC end view of the Musa acuminata genome. BMC Plant Biol 7:29
Christin PA, Besnard G, Samaritani E, Duvall MR, Hodkinson TR, Savolainen V et al (2008) Oligocene CO2 decline promoted C4 photosynthesis in grasses. Curr Biol 18:37–43
Christin PA, Petitpierre B, Salamin N, Büchi L, Besnard G (2009) Evolution of C4 phosphoenolpyruvate carboxykinase in grasses, from genotype to phenotype. Mol Biol Evol 26:357–365
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
D’Hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O et al (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488:213–217
Dida MM, Srinivasachary Ramakrishnan S, Bennetzen JL, Gale MD, Devos KM (2006) The genetic map of finger millet, Eleusine coracana. Theor Appl Genet 114:321–332
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Ferreira de Carvalho J, Poulain J, Da Silva C, Wincker P, Michon-Coudouel S, Dheilly A et al (2013) Transcriptome de novo assembly from next-generation sequencing and comparative analyses in the hexaploid salt marsh species Spartina maritima and Spartina alterniflora (Poaceae). Heredity 110:181–193
Ferris C, King RA, Gray AJ (1997) Molecular evidence for the maternal parentage in the hybrid origin of Spartina anglica C.E. Hubbard. Mol Ecol 6:185–187
Fortuné PM, Schierenbeck KA, Ainouche AK, Jacquemin J, Wendel JF, Ainouche ML (2007) Evolutionary dynamics of Waxy and the origin of hexaploid Spartina species (Poaceae). Mol Phylogenet Evol 43:1040–1055
Fortuné PM, Schierenbeck K, Ayres D, Bortolus A, Catrice O, Brown S et al (2008) The enigmatic invasive Spartina densiflora: a history of hybridizations in a polyploidy context. Mol Ecol 17:4304–4316
Gedye K, Gonzalez-Hernandez J, Ban Y, Ge X, Thimmapuram J, Sun F, Wright C, Ali S, Boe A, Owens V (2010) Investigation of the transcriptome of prairie cord grass, a new cellulosic biomass crop. Int J Plant Genomics 3:69
Gonthier L, Bellec A, Blassiau C, Prat E, Helmstetter N et al (2010) Construction and characterization of two BAC libraries representing a deep-coverage of the genome of chicory (Cichorium intybus L., Asteraceae). BMC Res Notes 3:225
Gonzalez-Hernandez JL, Sarath G, Stein JM, Owens V, Gedye K, Boe A (2009) A multiple species approach to biomass production from native herbaceous perennial feedstocks. In Vitro Cell Dev Biol Plant 45:267–281
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ et al (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Grass Phylogeny Working Group II (2012) New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol 193:304–312
Groves H, Groves J (1880) Spartina × townsendii Nobis. Report of the Botanical Society and exchange club of the British Isles 1:37
Han Y, Korban SS (2008) An overview of the apple genome through BAC end sequence analysis. Plant Mol Biol 67:581–588
Hansel CM, Fendorf S, Sutton S, Newville M (2001) Characterization of Fe plaque and associated metals on the roots of mine-waste impacted aquatic plants. Environ Sci Technol 35:3863–3868
Hilu KW, Alice LA (2001) A phylogeny of Chloridoideae (Poaceae) based on matK sequences. Syst Bot 26:386–405
Hsu C–C, Chung Y-L, Chen T-C, Lee Y-L, Kuo Y-T, Tsai W-C et al (2011) An overview of the Phalaenopsis orchid genome through BAC end sequence analysis. BMC Plant Biol 11:3
Huo N, Lazo GR, Vogel JP, You FM, Ma Y, Hayden DM et al (2007) The nuclear genome of Brachypodium distachyon: analysis of BAC end sequences. Funct Integr Genomics 8:135–147
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Kejnovsky E, Hawkins JS, Feschotte C (2012) Plant transposable elements: biology and evolution. In: Wendel J, Greilhuber J, Dolezel J, Leitch I (eds) Plant genome diversity volume 1: plant genomes, their residents and their evolutionary dynamics. Vienna, pp 17–34
Kim C, Jang CS, Kamps TL, Robertson JS, Feltus FA, Paterson AH (2008) Transcriptome analysis of leaf tissue from Bermudagrass (Cynodon dactylon) using a normalised cDNA library. Funct Plant Biol 35:585–594
Kim C, Tang H, Paterson AH (2009) Duplication and divergence of grass genomes: integrating the Chloridoids. Trop Plant Biol 2:51–62
Kim C, Lee T-H, Compton RO, Robertson JS, Pierce GJ, Paterson AH (2013) A genome-wide BAC end-sequence survey of sugarcane elucidates genome composition, and identifies BACs covering much of the euchromatin. Plant Mol Biol 81:139–147
Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Larher F, Hamelin J, Stewart GR (1977) L’acide dimethylsulfonium propanoïque de Spartina anglica. Phytochemistry 16:2019–2020
Lee RW (2003) Physiological adaptations of the invasive cordgrass Spartina anglica to reducing sediments: rhizome metabolic gas fluxes and enhanced O2 and H2S transport. Mar Biol 143:9–15
Li Y-C, Korol AB, Fahima T, Beiles A, Nevo E (2002) Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol 11:2453–2465
Luque CJ, Castellanos EM, Castillo JM, Gonzalez M, Gonzalez-Vilches MC, Figueroa ME (1999) Metals in halophytes of a contaminated estuary (Odiel Saltmarshes, SW Spain). Mar Pollut Bull 38:49–51
Ma J, Devos KM, Bennetzen JL (2004) Analyses of LTR-Retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res 14:860–869
Manninen I, Schulman AH (1993) BARE-1, a Copia-like retroelement in barley (Hordeum vulgare L.). Plant Mol Biol 22:829–846
Marchant CJ (1967) Evolution in Spartina (Gramineae). I. The history and morphology of the genus in Britain. Bot J Linn Soc 60:1–24
Marchant C, Goodman P (1969) Spartina maritima (Curtis) Fernald. J Ecol 57:287–302
Mobberley DG (1956) Taxonomy and distribution of the genus Spartina. Iowa State Coll J Sci 30:471–574
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30:194–200
Osoegawa K, Vessere GM, Shu CL, Hoskins RA, Abad JP, de Pablos B et al (2007) BAC clones generated from sheared DNA. Genomics 89:291–299
Otte ML, Wilson G, Morris JT, Moran BM (2004) Dimethylsulphopropionate (DMSP) and related compounds in higher plants. J Exp Bot 55(404):1919–1925
Ouyang S, Bell R (2004) The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res 32:360–363
Parisod C, Salmon A, Zerjal T, Tenaillon M, Grandbastien M, Ainouche M (2009) Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol 184:1003–1015
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H et al (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Peterson DG, Tomkins JP, Frisch DA, Wing RA, Paterson AH (2000) Construction of plant bacterial artificial chromosome (BAC) libraries: an illustrated guide. J Agric Genomics 5. www.ncgr.org/research/jag.
Peterson PM, Romaschenko K, Johnson G (2010) A classification of the Chloridoideae (Poaceae) based on multi-gene phylogenetic trees. Mol Phylogenet Evol 55:580–598
Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H et al (2006) Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res 16:1262–1269
Prasad V, Stromberg CAE, Leache AD, Samant B, Patnaik R, Tang L et al (2011) Late Cretaceous origin of the rice tribe provides evidence for early diversification in Poaceae. Nat Commun 2:480
Ragupathy R, Rathinavelu R, Cloutier S (2011) Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L.) genome. BMC Genomics 12:217
Ramanarao MV, Weindorf D, Breitenbeck G, Baisakh N (2011) Differential expression of the transcripts of Spartina alterniflora Loisel (Smooth Cordgrass) induced in response to petroleum hydrocarbon. Mol Biotechnol 51:18–26
Raybould AF, Gray AJ, Lawrence MJ, Marshall DF (1991) The evolution of Spartina anglica CE Hubbard (Gramineae)—origin and genetic variability. Biol J Linn Soc Lond 43:111–126
Renny-Byfield S, Ainouche M, Leitch IJ, Lim KY, Le Comber SC, Leitch AR (2010) Flow cytometry and GISH reveal mixed ploidy populations and Spartina nonaploids with genomes of S. alterniflora and S. maritima origin. Ann Bot 105:527–533
Roodt R, Spies JJ (2003a) Chromosome studies in the grass subfamily Chloridoideae. I. Basic chromosome numbers. Taxon 52:557–566
Roodt R, Spies JJ (2003b) Chromosome studies in the grass subfamily Chloridoideae. II. An analysis of polyploidy. Taxon 52:736–746
Salse J, Bolot S, Throude M, Jouffe V, Piegu B, Quraishi UM et al (2008) Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell Online 20:11
See DR, Brooks S, Nelson JC, Brown-Guedira G, Friebe B, Gill BS (2006) Gene evolution at the ends of wheat chromosomes. Proc Natl Acad Sci USA 103:4162–4167
Srinivasachary, Dida MM, Gale MD, Devos KM (2007) Comparative analyses reveal high levels of conserved colinearity between the finger millet and rice genomes. Theor Appl Genet 115:489–499
Terol J, Naranjo MA, Ollitrault P, Talon M (2008) Development of genomic resources for Citrus clementina: characterization of three deep-coverage BAC libraries and analysis of 46,000 BAC end sequences. BMC Genomics 9:423
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
The International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Timmers RA, Strik DPBTB, Hamelers HVM, Buisman CJN (2010) Long-term performance of a plant microbial fuel cell with Spartina anglica. Appl Microbiol Biotechnol 86:973–981
Vitte C, Bennetzen JL (2006) Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci USA 103:17638–17643
Wu J, Gu YQ, Hu Y, You FM, Dandekar AM, Leslie CA et al (2011) Characterizing the walnut genome through analyses of BAC end sequences. Plant Mol Biol 78:95–107
Yannic G, Baumel A, Ainouche M (2004) Uniformity of the nuclear and chloroplast genomes of Spartina maritima (Poaceae), a salt-marsh species in decline along the Western European Coast. Heredity 93:182–188
You F, Huo N, Gu Y, Luo M, Ma Y, Hane D et al (2008) BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9:253
Yu J, Wang J, Lin W, Li S, Li H, Zhou J et al (2005) The genomes of Oryza sativa: a history of duplications. PLoS Biol 3:e38
Yu J-K, Sun Q, Rota ML, Edwards H, Tefera H, Sorrells ME (2006) Expressed sequence tag analysis in tef Eragrostis tef (Zucc) Trotter. Genome 49:365–372
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E et al (2012) Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot 99:193–208
Zhang D, Ayele M, Tefera H, Nguyen HT (2001) RFLP linkage map of the Ethiopian cereal tef: Eragrostis tef (Zucc) Trotter. Theor Appl Genet 102:957–964
Zhong CX, Marshall JB, Topp C, Mroczek R, Kato A, Nagaki K et al (2002) Centromeric retroelements and satellites interact with maize kinetochore protein CENH3. Plant Cell 14:2825–2836
Acknowledgements
This work was supported by CNRS INEE and University of Rennes 1, the Partner University Funds and by the Genoscope (GENOSPART Project). The analyses benefited from the Genouest (Bioinformatics) Plateform facilities. J. Ferreira de Carvalho benefited from a Ph.D. Grant (ARED EVOSPART) from the Regional Council of Brittany.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ferreira de Carvalho, J., Chelaifa, H., Boutte, J. et al. Exploring the genome of the salt-marsh Spartina maritima (Poaceae, Chloridoideae) through BAC end sequence analysis. Plant Mol Biol 83, 591–606 (2013). https://doi.org/10.1007/s11103-013-0111-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0111-7