
Abstract
Septo-optic dysplasia (SOD) is a highly heterogeneous condition comprising a variable phenotype of optic nerve hypoplasia, midline forebrain abnormalities and pituitary hypoplasia with consequent endocrine deficits. The majority of cases are sporadic and several aetiologies including drug and alcohol abuse have been suggested to account for the pathogenesis of the condition. However, a number of familial cases have been described and the identification of mutations in the key developmental gene HESX1 in patients with SOD and associated phenotypes suggests that a genetic causation is likely in the more common sporadic cases of the condition. More recently, we have implicated duplications of SOX3 and mutations of both SOX2 and SOX3 in the aetiology of variants of SOD. As with other developmental disorders such as holoprosencephaly, the precise aetiology is most likely multifactorial involving contributions from environmental factors in addition to an important role for crucial developmental genes. This potentially complex interaction between genetics and the environment is borne out by the variability of the penetrance and phenotypes in patients with genetic SOD, but at present, the understanding of these interactions is rudimentary. Further study of these critical factors may shed light on the aetiology of this complex disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Septo-optic dysplasia
Septo-optic dysplasia (SOD), also known as de Morsier Syndrome [1], is a rare condition initially described by Reeves [2] in a 7-month-old baby with absence of the septum pellucidum and optic nerve abnormalities. The condition is highly heterogeneous, currently defined by any combination of (1) optic nerve hypoplasia, (2) midline neuroradiological abnormalities, such as agenesis of the corpus callosum and absence of the septum pellucidum, (3) and pituitary hypoplasia with consequent panhypopituitarism [1–7].
The reported incidence of SOD is 1/10,000 live births [8] and it is thought to be equally prevalent in males and females. Although the condition is generally sporadic, familial cases have been described. Neurological deficit is common, often occurring in association with optic nerve hypoplasia (75–80% of patients), and ranges from global retardation to focal deficits such as epilepsy or hemiparesis [9, 10]. Associated anatomical features may include cavum septum pellucidum, cerebellar hypoplasia, schizencephaly and aplasia of the fornix.
Optic nerve hypoplasia (ONH) may be unilateral or bilateral although bilateral involvement is more common (88% as compared with 12% unilateral cases), and may be the first presenting feature, with later onset of endocrine dysfunction [4, 5, 11–15]. In rare cases, the eye abnormality may be more severe, resulting in microphthalmia or anophthalmia.
Pituitary hypoplasia may manifest as variable endocrine deficits ranging from isolated growth hormone (GH) deficiency to complete panhypopituitarism, and it has been suggested that abnormalities of the septum pellucidum and hypothalmo-pituitary axis on neuroimaging can predict the severity of endocrine dysfunction [16]. Decreased growth rate due to growth GH deficiency is the most common endocrinological feature followed by thyroid stimulating hormone (TSH) and adrenocorticotropic hormone (ACTH) deficiencies [17, 18], whereas gonadotrophin secretion may be retained in the face of other pituitary hormone deficiencies. However, the endocrinopathy may evolve with a progressive loss of endocrine function over time [7]. Commencement of growth hormone treatment in children with SOD involving GH deficiency may be associated with accelerated pubertal maturation [19] and either sexual precocity or failure of pubertal development may occur, with abnormal hypothalamic neuroanatomy or function [20–24]. Other features such as hypoglycemia, diabetes insipidus, and polyuria and polydipsia are less commonly associated [4, 25].
The phenotype is highly variable; each of the three components listed in the first paragraph can occur in isolation or in combination with at least one other, with a diagnosis of SOD usually made if two or more of the above features are present. According to Morishima and Aranoff, approximately 30% of SOD cases have complete manifestations, 62% have the complication of hypopituitarism and 60% have an absent septum pellucidum [26]. De Morsier [1] reported that optic nerve abnormalities were present in only nine of 36 cases with absent septum pellucidum; while Acers studied 45 individuals with optic nerve hypoplasia and found that 12 of these patients had associated midline brain defects, with six of these also demonstrating hypopituitarism. Additionally, there appears to be little correlation between the size of the optic nerve and its visual function.
The condition is thought to be more frequent in children born to younger mothers (mean maternal age 22 years) [4, 27], although this has been disputed [15] and in some studies there is a preponderance of primigravida mothers [5, 15]. Recently, cases of both isolated ONH as well as SOD have been shown to cluster in high population density, inner city areas with high rates of unemployment and teenage pregnancy [8].
Several aetiologies have been postulated to account for the sporadic occurrence of SOD, such as viral infections, environmental teratogens, and vascular or degenerative damage [24, 28]. However, the precise aetiology of the condition still remains unknown and is most likely to be multifactorial, with a combination of genetic and environmental factors.
The development of the forebrain is highly complex occurring at a very early stage of embryonic development, and has been extensively studied in the mouse [29]. Non-neural structures such as the anterior pituitary, the olfactory placode and the nasal cavity ectoderm and neural structures such as the hypothalamus, the posterior pituitary, the optic vesicles, the optic nerves and the forebrain develop from the same region of the embryo, the anterior neural plate. Any insult at this critical stage of embryonic development could account for the features of SOD. Such developmental insults would have to take place during the critical period of embryogenesis for these structures, which corresponds to 4–6 weeks of gestation in humans, during which two significant developmental events occur. First, the telencephalic optic vesicles and retinal ganglion cells differentiate, and second, the lamina terminalis thickens, and its subsequent differentiation results in formation of the corpus callosum, anterior commisure, and fornix. Any insult arising at this stage has the potential to produce failure of ganglion cell formation with subsequent hypoplasia of optic nerves and chiasm in the first instance and lack of commisural or septal formation in the second instance. It is important to stress that hypoplasia can result from an incomplete commitment of progenitor cells, or a failure to commit adequate numbers of precursors, which could then give rise to variable degrees of hypoplasia, as well as aplasia, of forebrain structures.
Familial cases of SOD are rare, but have the obvious advantage that they implicate a genetic defect pointing to the developmental mechanisms involved. They are more frequently associated with an autosomal recessive inheritance [30–32], although dominant inheritance has been described [33–35].
Development of the forebrain and anterior pituitary gland
Animal studies have recently shed a considerable amount of light on the normal development of the forebrain and pituitary gland. There is now good evidence that anterior patterning in the extraembryonic endoderm, or anterior visceral endoderm (AVE), of the early mammalian conceptus occurs before there is any sign of primitive streak formation, i.e., prior to gastrulation; the process resulting in the formation of the primitive streak, the notochord, and the three germ layers that lead to the development of all embryonic tissues and organs [36]. The AVE has been shown to be functional in providing the embryo with anterior characteristics and is vital for normal forebrain development.
Current knowledge of the expression of several genes, including Otx2, Lim1, goosecoid, cerberus-related 1, and Hex, shows they are restricted to a medial strip of the AVE underlying approximately the anterior third of the epiblast at least 12 h before the primitive streak has formed. As the streak forms, the most anterior extreme of the AVE, in the region where the heart will develop, starts to express the gene Mrg1. Slightly more posteriorly, where it overlies the portion of epiblast fated to give rise to oral ectoderm and forebrain, Hesx1 (also known as Rpx) is expressed. Hence, as gastrulation starts, the AVE already exhibits a restricted pattern of expression of transcription factors. Mutations in a number of these genes, including Hesx1, Lim1 and Otx2 [37–39], which are first expressed in the AVE and only subsequently in epiblast derivatives, affect anterior patterning in the mouse, with variable defects in forebrain development.
In humans, the first known event in the development of the nervous system is neural induction or neurulation, whereby the neural plate, the embryonic precursor of the brain, forms in the third week of gestation. The developing notochord induces the overlying ectoderm to thicken and form the neural plate, the primordium of the central nervous system. The ectoderm of the neural plate, the neurectoderm, gives rise to the brain and spinal cord. The neural plate forms as a result of the cellular interaction between the ectoderm and the mesoderm of the gastrula-stage embryo and is a planar sheet of pseudostratified neuroepithelium produced during gastrulation. The neural plate becomes regionally patterned along each of its axes and is converted into the neural tube which then differentiates into the central nervous system structures in the brain and spinal cord. Again, restricted patterns of differential gene expression are evident at the anterior neural plate stage in the human embryo, and these foreshadow the later development of morphological and functional subdivisions of the neural tube.
By the fourth week of gestation, a series of ring-like constrictions mark the approximate boundaries between the primordia of the major brain regions: the forebrain or prosencephalon, midbrain or mesencephalon and hindbrain or rhombencephalon. As the rostral neuropore closes at about 25 days of gestation, the optic vesicles appear as two lateral outgrowths on each side of the forebrain, and are the primordia of the retinae and optic nerves. A second pair of diverticula arise more dorsally and rostrally; these are the cerebral or telencephalic vesicles which are the primordia of the cerebral hemispheres, their cavities forming the lateral ventricles. During the fifth week, the forebrain partly divides into two regions, the telencephalon and the diencephalon. The diencephalon will give rise to the thalamus and hypothalamus.
Fate map studies in a number of species such as the chick, Xenopus, Axolotl, and Zebrafish have shown that the prospective telencephalon occupies the rostral end of the neural plate, partly overlapping the anterior neural ridge. Distributed laterally along the anterior neural ridge are sites fated to form the olfactory bulbs and the lateral, dorsal, and medial regions of cerebral cortex. The neural plate medial to the telencephalon contains sites destined to form the preoptic, optic, and hypothalamic areas. Of note is the finding that the site destined to form the anterior pituitary or adenohypophysis also maps onto the anterior neural ridge. Additionally, in Xenopus, the mid-anterior ridge produces a triangular wedge of tissue extending at the midline from the chiasma ridge to the lamina terminalis and laterally into the optic stalks [40].
Several lines of molecular and genetic evidence now suggest that medial and ventral specification of the forebrain is regulated by the prechordal plate, an axial mesendodermal structure. Forebrain development is highly complex and it is clear that a number of genes are implicated in this process. These include Sonic Hedgehog, Fgf8, Hesx1, HNF3β, Nkx2.1, Nkx2.2, Lim1, Otx1, Otx2, Cerberus, goosecoid, Pax6, Six3, Emx1, and Emx2. The exact role of many of these genes remains to be defined; nevertheless, as the forebrain, eyes, optic chiasm, olfactory bulbs, adenohypophysis and hypothalamus originate from the same region in the embryo, defects of some of the genes expressed in the anterior neural ridge could potentially lead to developmental defects of these structures. However, it is important to stress that many of these genes have multiple spatial and temporal domains of expression throughout the embryo and are not solely involved with forebrain and pituitary development.
The pituitary gland is a midline structure consisting of the anterior, intermediate, and posterior lobes of varying size and complexity in different species. Developmentally, the pituitary has a dual embryonic origin; fate map studies in the chick and Xenopus have demonstrated that the derivatives of the oral roof ectoderm (which include the pituitary and nasal epithelium) are initially derived from the anterior neural ridge (ANR). As the embryonic head turns, the ANR is displaced ventrally and forms the stomodeum, the ectoderm which gives rise to the roof of the mouth and its derived structures [41, 42]. In the mouse, the onset of pituitary organogenesis coincides with a thickening of the initially uniform stomodeal ectoderm on embryonic day (E)8.5 which then invaginates on E9.0 to form a structure called Rathke’s pouch, from which the anterior and intermediate lobes of the pituitary are derived. The posterior lobe develops as a result of the evagination of the neural ectoderm (infundibulum) at the base of the developing diencephalon which comes into direct contact with Rathke’s pouch [43]. In humans, the diverticulum forming Rathke’s pouch projects dorsally from the roof of the stomodeum around the fourth week of gestation and grows towards the brain. By the fifth week, the pouch has elongated, has become constricted at its attachment to the oral epithelium and has come into contact with the infundibulum. The apposition of Rathke’s pouch and the diencephalon is maintained throughout the early stages of pituitary organogenesis, and this close relationship has long suggested that inductive tissue interactions are involved in the process.
Between E10.5 and E12 in the mouse, the pouch epithelium continues to proliferate as it closes and separates from the underlying oral ectoderm. Subsequent to these initial patterning events, the progenitors of the hormone-secreting cell types proliferate ventrally from the pouch between E12.5 and E15.5 to populate what will form the anterior lobe [44]. The remnants of the dorsal portion of the pouch form the intermediate lobe destined to form melanotropes. The size of the intermediate lobe varies markedly between species, and in humans, its exact role remains unclear.
The development of the anterior pituitary gland is now known to be dependent on various intrinsic and extrinsic transcription factors and signalling molecules. Extrinsic factors include Ttf1, BMP4, and Fgf8 [45–47], which are expressed in the diencephalon and not Rathke’s pouch, but are nevertheless implicated in normal pituitary development. It appears that Rathke’s pouch develops in a two-step process that requires at least two sequential inductive signals from the diencephalon, which are present only in the infundibulum and developing hypothalamus and not in Rathke’s pouch itself. First, the induction and formation of the pouch rudiment is dependent upon Bone Morphogenetic Protein 4 (BMP4), followed by a second signal, Fibroblast Growth Factor 8 (FGF8), which activates the key regulatory genes Lhx3 and Lhx4 (see below) essential for subsequent development of the pouch rudiment into a definitive pouch structure [47].
A number of transcription factors are expressed within the pituitary primordium itself. These include Six3 [48], Pax6 [49], and Hesx1/Rpx [50, 51], all of which continue to be expressed in Rathke’s pouch. Concurrent with organ commitment, two LIM-homeodomain factors, Lhx3 or P-Lim [52, 53], and Lhx4 [54], are also expressed in the pouch. Deletion of the Lhx3/P-Lim gene in mice results in the failure of the pituitary gland to grow and differentiate, despite initial formation of Rathke’s pouch [55], whereas mutation of both Lhx3 and Lhx4 results in complete failure of pouch formation [54]. The OTX-related factor Ptx1/P-OTX is also expressed throughout early pituitary development [56, 57].
The mature anterior pituitary gland is populated by five endocrine cell types, at least as defined by the hormones they produce: somatotropes (producing GH), corticotropes (ACTH), thyrotropes (TSH), gonadotropes (luteinizing hormone, LH; follicle stimulating hormone, FSH) and lactotropes (prolactin, PRL). These endocrine cell types progressively differentiate in a temporally and spatially regulated manner [58, 59], and this process is dependent upon an increasing list of transcription factors. One of the first such factors to be discovered, was the tissue-specific POU domain factor POU1F1 (OMIM 173110; previously referred to as PIT1) which is required for terminal differentiation, growth and survival of somatotropes, lactotropes, and thyrotropes [60]. Additionally, a paired-like homeodomain factor Prop1 is also required for the determination of the POU1F1-dependent lineages [61], however the phenotype of patients with mutations in PROP1 (OMIM 601538) suggests that the gene is also involved in the determination of gonadotropes [62].
Midline defects and pituitary dysfunction
Congenital midline defects encompass a large array of clinical phenotypes, ranging from those that are incompatible with life to severe palato-facial cleft associated with neuroanatomical defects. The conditions include various forms of holoprosencephaly, septo-optic dysplasia and agenesis of the corpus callosum. At the less severe end of the spectrum are isolated cleft lip or palate. As the pituitary is a midline structure, the association between hypopituitarism and extensive congenital midline brain defects has long been recognized, of which septo-optic dysplasia phenotypes are the most common [63–66].
HESX1 and SOD
Hesx1 is a member of the paired-like class of homeobox genes [50] and is first expressed during mouse embryogenesis in a small patch of cells in the anterior midline visceral endoderm as gastrulation commences [29, 51, 67]. Following a critical signal from the visceral endoderm, expression is then induced in the adjacent ectoderm in an area destined to give rise to the ventral prosencephalon, which will in turn form the forebrain. Subsequently, Hesx1 is detected at the anterior extreme of the rostral neural folds; expression is then restricted to the ventral diencephalon by E9.0 in the mouse, and also in the thickened layer of oral ectoderm that will give rise to Rathke’s pouch. Hesx1 continues to be expressed in the developing anterior pituitary until E12 when expression is attenuated in a spatiotemporal sequence corresponding to progressive pituitary cell differentiation, finally becoming undetectable by E13.5.
Homozygous disruption of Hesx1 in mice is associated with a phenotype closely resembling that of SOD, with all Hesx1 null mice being abnormal at birth [37]. Features include a reduction in prospective forebrain tissue, absence of developing optic vesicles, markedly decreased head size, craniofacial dysplasia with a short nose, and severe microphthalmia. Defects were often noted to be asymmetrical, with one side more severely affected than the other, and the severity of the mutant phenotype was also often variable, with mutants divided into either a severe Class I phenotype or a milder Class II phenotype. Histological analysis of the mutant mice revealed that severely affected Class I embryos had substantially reduced forebrains with no sign of telencephalic vesicle or infundibulum development, in addition to absence of the optic cups, the olfactory placodes, and Rathke’s pouch. Class II mutant embryos demonstrated a thin forebrain epithelium with retardation in prosencephalic development. Other abnormalities included reduced telencephalic vesicles with hypothalamic abnormalities and aberrant morphogenesis of Rathke’s pouch [37]. The olfactory pits and optic vesicles were also hypoplastic and often asymmetric in appearance.
Further analysis of neonatal and adult mutants revealed hypoplastic nasal cavities, hypoplastic olfactory bulbs, microphthalmia and anophthalmia, with abnormalities of the septum pellucidum and corpus callosum. A more detailed analysis of the Hesx1-null mutants revealed that, in the mice with the more severe phenotype (5%), no anterior pituitary gland was formed, although thickening of the oral ectoderm and minimal expression of Lhx3 were detected [68]. However, in the majority of the mice, pituitary development proceeded beyond formation of Rathke’s pouch; abnormalities were observed with multiple oral ectodermal invaginations leading to the formation of multiple pituitary glands, followed by dramatic cellular over-proliferation. It is notable that the late stages of pituitary development (E16.5) show normal terminal differentiation of the hormone-producing cell types with detectable expression of Pou1f1, GH, and TSHβ, in addition to α-GSU, the common subunit of FSH, LH and TSH, and the precursor of melanocorticotrophin, POMC. The expression domains of Lhx3 and Prop1 were noted to be expanded and extended into the more anterior region of oral ectoderm. Additionally, the expression domains of Fgf8 and Fgf10 in the infundibulum were expanded more rostrally, and these data therefore suggest that Hesx1 is required for the maintenance of proper FGF expression domains. Fgf8 in turn appears to regulate Hesx1 expression as expression of Fgf8 under the control of the early pituitary specific Pitx1 promoter leads to complete loss of Hesx1 expression [69]. These in vivo data are suggestive of a repressive function of Hesx1, which has been confirmed in vitro using transfection studies showing that HESX1 is a promoter-specific transcriptional repressor [68, 70]. This repressor action of HESX1 is mediated by 37 amino-acids at the N-terminus of the HESX1 protein (the engrailed homology domain; eh-1), which is also implicated in homo- and hetero-dimerization of HESX1 with partner proteins [70]. The exact mechanism of transcriptional repression remains unknown, although Dasen et al. [68] have shown that the mammalian homologue of the Drosophila co-repressor protein Groucho, TLE1 (Transducin-like enhancer of Split), is recruited by the eh-1 domain, whilst the homeodomain interacts with the nuclear receptor co-repressor N-CoR1, which in turn leads to the binding of histone deacetylases 1 and 2. TLE1 and Hesx1 lead to abrogation of the binding of Prop1, also a paired-like homeobox transcription factor, suggesting that Hesx1 and Prop1 bind to overlapping sites, and that the temporal control of expression of the two directs organ commitment and proliferation mediated by Hesx1 during early development and subsequently the specification of hormone producing cell lineages by Prop1. In the Ames dwarf mouse, the phenotype of which arises as a result of a mutation in the Prop1 gene, Hesx1 expression persists later than normal (until E16), suggesting an additional role for Prop1 in regulating Hesx1 expression [61]. Additionally, a role for Lhx3 in the maintenance of Hesx1 expression has been suggested from the analysis of Lhx3 null mutant mice [54]. These data, therefore, suggest that Hesx1 is essential for the initial organ commitment and proliferation events during pituitary morphogenesis.
The phenotype of Hesx1 null mice is reminiscent of that observed in SOD in humans. In light of this observation we investigated the role of the human homologue of HESX1 (OMIM 601802) in patients with SOD. HESX1 maps to chromosome 3p21.1, and the coding region spans 1.7 kb consisting of four coding exons encoding a 185-amino acid open reading frame (ORF) that is highly conserved compared with the mouse and Xenopus, particularly across the homeodomains that share 95 and 80% homology, respectively. The highly conserved eh-1 domain, located near the amino-terminus, is also present in other homeodomain repressors including engrailed and goosecoid.
A homozygous mis-sense mutation in the homeobox of HESX1 was initially identified in a highly consanguineous family in which two affected siblings presented with optic nerve hypoplasia, absence of the corpus callosum, and hypoplasia of the anterior pituitary gland with an undescended/ectopic posterior pituitary and consequent panhypopituitarism [30, 37]. The mutation was identified in the two affected siblings and resulted in the substitution of a highly conserved arginine at residue 160 (position 53 of the homeodomain) by cysteine which leads to a loss of DNA binding by the mutant protein. The parents were heterozygous for the mutation and phenotypically normal. Screening of extended members of the family revealed a further nine phenotypically normal heterozygotes within this highly consanguineous pedigree, consistent with an autosomal recessive inheritance. Subsequently, four further homozygous mutations have been identified.
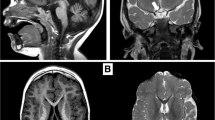
A homozygous substitution (I26T) was identified in a girl presenting with GH and gonadotrophin deficiency, with evolving ACTH and TSH deficiency. MRI revealed anterior pituitary hypoplasia with an undescended posterior pituitary; however she had normal optic nerves and no midline forebrain defects. This mutation occurs in the highly conserved eh-1 domain which is crucial for transcriptional repression [70], and was associated with partial loss of repression [71] (Fig. 1).

MRI findings in HESX1 mutations (from Fig. 1, Development 2001; 128:5189–5199) (A) Sagittal MRI scan of the head of a normal child showing the corpus callosum (CC), the optic chiasm (OC), the anterior pituitary (AP), the pituitary stalk (PS) and posterior pituitary (PP) in the normal sella turcica. Note the well-formed corpus callosum, optic chiasm, and the posterior pituitary which appears as a bright spot within the sella turcica. (B) Sagittal MRI scan of sibling 2 with a homozygous R160C mutation in HESX1. The corpus callosum (CC) is severely hypoplastic, as is the optic chiasm (OC) and the anterior pituitary (AP) located in a well-formed but empty sella turcica. Note the ectopic posterior pituitary (PP) and the lack of a visible pituitary stalk. (C) Sagittal MRI scan of sibling 1 with a homozygous R160C mutation in HESX1. Note that the splenium of the corpus callosum is more hypoplastic than the rest of the structure and that the sella turcica is shallow as compared with the MRI scan of sibling 2 (B). The posterior pituitary (PP) appears to be partially descended. (D) Sagittal MRI of a patient with S170L mutation in HESX1. Note the atrophic posterior pituitary (PP) that has not descended completely into the fossa and an anterior pituitary (AP) gland that is hypoplastic and does not enhance well. The pituitary stalk is thin, but the optic chiasm (OC) is normal as is the corpus callosum (CC)
More recently a third recessive mutation has been identified in two siblings from a consanguineous family, involving an Alu-element insertion in exon 3 of HESX1, which encodes the homeodomain of the protein [72]. Homozygosity for the mutation was associated with a severe phenotype including undetectable levels of all anterior pituitary hormones as a result of aplasia of the anterior pituitary (as observed in 5% of homozygous null mice), together with hypoplasia of the sella turcica. Conversely, the posterior pituitary and pituitary stalk were both normal. One sibling had a coloboma of the right optic nerve resulting in unilateral blindness, whilst the other had no ophthalmic abnormalities but displayed a left-sided diaphragmatic hernia and aortic coarctation and died shortly after birth.
Sobrier et al. [73] have recently reported two additional patients with novel recessive mutations in HESX1 associated with anterior pituitary aplasia in the absence of an ectopic posterior pituitary or optic nerve abnormalities, features typically associated with HESX1 mutations. Sequencing of HESX1 in the two individuals revealed that one patient carried a homozygous 2 bp deletion (c.449_450delAC) resulting in a frameshift, whilst the other patient was homozygous for a mutation in the splice donor site in intron 2 (c.357 + 2T > C), with both mutations predicted to disrupt the homeodomain of the protein [73] with consequent loss of DNA binding. The patient with the 2 bp deletion had additional midline abnormalities including a thin corpus callosum and hydrocephalus (Fig. 2).

Mutations and polymorphisms within the coding region of HESX1
To date, reports of screening patients with sporadic SOD have yielded six novel heterozygous mutations within HESX1. In an initial series of 228 patients with hypopituitarism, of whom 105 had SOD, three heterozygous mutations were initially identified: Q6H, S170L, and T181A [33]. All three mutations are associated with a milder phenotype than the homozygous R160C substitution, invariably leading to GHD with or without an undescended posterior pituitary. The S170L substitution is located three residues downstream of the carboxy terminus of the homeodomain in a highly conserved region of HESX1 and was first described in two siblings with isolated GHD, one of whom also had optic nerve hypoplasia. Subsequently, the same mutation was found in a child with isolated GHD with an undescended posterior pituitary. The Q6H substitution occurs at a highly conserved residue at the N terminus in a patient who presented with GH and thyrotrophin (TSH) deficiency and an undescended posterior pituitary. The T181A change is located at the carboxy terminus of the protein and was observed in an individual with GHD and absence of the posterior pituitary bright spot. Compound heterozygosity within HESX1 was excluded by performing haplotype analysis, and autosomal dominant inheritance with variable penetrance is the most likely mode of inheritance [33]. Furthermore, a deletion at nucleotide position 1684 (g.1684delG), leading to a frameshift altering the C-terminal portion of the protein [34], and a de novo 2 bp insertion (306/307insAG) [35] have been identified in patients presenting with SOD phenotypes characterized by GH deficiency, hypoplasia of the anterior pituitary and optic nerves, and midline forebrain abnormalities. The patient with the insertion mutation also exhibited evidence of gonadotrophin deficiency and hypothyroidism.
More recently, we have identified a mis-sense mutation in HESX1, E149K, that is associated with impaired repression of PROP1 activation, in a patient with GHD and an undescended posterior pituitary [27]. Again the penetrance is variable, with the son of the proband being an unaffected carrier of the mutation. To date, we have now screened over 800 patients with SOD and hypopituitarism, and identified mutations in less than 1% of individuals confirming the rarity of HESX1 mutations [27]. As a result of this screening we have identified a number of polymorphisms, including a change of unknown function in a highly conserved base in the 5′ region of HESX1. Whether these polymorphisms contribute to the phenotype remains to be proven. The overall frequency of HESX1 mutations is low suggesting that mutations in other known or unknown genes contribute to this complex disorder, together with a likely contribution from environmental factors [8, 74].
SOX3
SOX3 (OMIM 313430) is a member of the SOX (SRY-related HMG box) family of transcription factors, which were initially identified based on homology to a conserved DNA binding motif of the high mobility group (HMG) class, present in the mammalian sex determining gene, SRY [75, 76]. Approximately 20 different SOX genes have been identified in mammals, and have also been identified in a wide range of species including birds, fish, reptiles, amphibians and insects [77, 78]. The HMG domains of SOX proteins exhibit at least 60% identity to that of SRY; however variation in homology exhibited within the HMG box between different members allows them to be grouped into different subfamilies [79]. SOX3 was among the first of the SOX genes to be cloned, and together with SOX1 and SOX2, belongs to the SOXB1 subfamily exhibiting the highest degree of similarity to SRY [75, 76, 80]. Members of this family of genes are expressed throughout the developing central nervous system and are some of the earliest neural markers that are believed to play a role in neuronal determination.
SOX3 is encoded by a single exon producing a transcript with a coding region of approximately 1.3 kb, and maps to chromosome Xq27. The SOX3 protein consists of a short 66 amino acid N-terminal domain of unknown function, the 79 amino acid DNA binding HMG domain and a longer C-terminal domain, containing four poly-alanine stretches, shown to be involved in transcriptional activation [76, 81]. The HMG domains of SOX proteins share similar DNA binding properties, recognizing a consensus 6–7 base pair motif (A/T A/T CAA A/T G) [82, 83]. SOX3 is highly conserved among mammals, and of all the SOXB1 genes, it shows the greatest sequence homology to SRY within the HMG domain (90% at the protein level) leading to the suggestion that SRY may have evolved from SOX3 [76, 82, 84].
The expression domains of SOXB1 genes have a strong tendency to overlap during development [85–88]. Sox3 is expressed in the earliest stages of development, with its main site of expression within the central nervous system (CNS), and has been strongly implicated in neurogenesis [89]. In the mouse, Sox3 begins to be expressed at 6.5 dpc (days post coitum), prior to the appearance of the primitive streak, throughout the epiblast and in a band of extraembryonic ectoderm at the boundary of the embryonic and extraembryonic tissues [85]. During early gastrulation, expression is rapidly downregulated in extraembryonic tissue and becomes restricted to the anterior ectoderm of the epiblast and to a posterior domain adjacent to the primitive streak. Later expression extends into the posterior region of the epiblast and from the onset of somite formation, expression is restricted to the presumptive neuroectoderm [85]. Subsequently, Sox3 is expressed along the full length of the developing CNS, including the brain and spinal cord, in actively dividing undifferentiated neural progenitor cells where expression is maintained throughout development of the CNS [90]. High levels of expression have also been noted in the ventral diencephalon, including the infundibulum and presumptive hypothalamus [91].
Targeted disruption of Sox3 in mice has been performed in two independent laboratories revealing that the Sox3 mutants have a variable and complex phenotype including craniofacial abnormalities, hypopituitarism, midline CNS defects, and a reduction in size and fertility [91, 92]. Sox3 mutant mice of both sexes are born with expected frequency showing no evidence for embryonic lethality, and approximately one-third of mutant mice are viable and fertile with no gross abnormalities. Heterozygous females are mosaic with respect to the mutation due to X inactivation and generally appear normal, although some displayed a mild craniofacial phenotype. However, approximately 43% of Sox3 null mice did not survive to weaning, and the most severely affected mice exhibited profound growth insufficiency and general weakness with craniofacial defects including overgrowth and misalignment of the front teeth and abnormality of the shape of the pinna which was completely absent in some animals [91]. Despite the high sequence similarity between Sox3 and Sry, the sex ratio of Sox3 mutants did not deviate from that expected, suggesting that Sox3 is not required for sex determination. No reduction in fertility was observed in mice with a milder phenotype. However, severely affected males were hypogonadal with reduced testis weight and abnormal germ cell development; seminiferous tubules were small and irregular with some tubules showing complete absence of germ cells. The ovaries of severely affected Sox3 null female mice were small with numerous atretic follicles and harvesting of oocytes following induction of ovulation showed a greater proportion of dead or malformed oocytes in Sox3 mutants compared with wild-type mice [92].
Rizzoti et al. [91] analysed the pituitary gland and brain of Sox3 mutant mice in detail, revealing the mutants to have a variable endocrine deficit, the extent of which was correlated with body weight. Pituitary levels of GH, luteinizing hormone, follicle stimulating hormone and TSH were all lower in mutant compared to wild-type mice at 2 months of age. Histological analysis of the pituitary gland at this stage showed that all three lobes of the pituitary were present, however the anterior lobe was smaller with the presence of an additional abnormal cleft disrupting the boundary between the anterior and intermediate lobes. Further examination of Sox3 mutant embryos revealed that normal pituitary development was disrupted. Rathke’s pouch, the embryonic primordium of the anterior pituitary, displayed an abnormally expanded and bifurcated appearance in mutant embryos which possibly results in the additional cleft observed at later stages of development and in the adult pituitary. Sox3 is not expressed in Rathke’s pouch; however it is expressed at high levels in the ventral diencephalon including the infundibulum which provides necessary inductive signals for the formation of the anterior pituitary [93]. In Sox3 mutants, the evagination of the infundibulum was less pronounced than observed in wild-type mice and the presumptive hypothalamus thinner and shorter with loss of Sox3 resulting in a marked reduction of proliferation in cells that would normally express the gene in these tissues [91]. This suggests that the hypopituitary phenotype observed in mutant mice arises as a secondary consequence of the absence of Sox3 in the ventral diencephalon. Furthermore, two of the signals required for the induction of pituitary morphogenesis and the development of Rathke’s pouch, Fgf8 and Bmp4 [46, 94], show expanded domains of expression in the absence of Sox3. Additionally, experiments in Xenopus have shown that overexpression of xSox3 inhibits Wnt signalling pathways [95] which have an important role in pituitary development [46, 96, 97]. Therefore, although pituitary development is abnormal in Sox3 null mice, expression of the gene is required in the ventral diencephalon for normal development of the hypothalamus and infundibulum and consequently for the induction of Rathke’s pouch morphogenesis. Furthermore the observed hypopituitary defect is probably the result of additional defects at the level of hypothalamic control of the pituitary [91].
Several pedigrees have been described with variable hypopituitarism and mental retardation mapping to the X chromosome. The observation of affected males carrying two copies of a linked microsatellite marker suggested duplication of the region; subsequently tandem duplications involving chromosome Xq26–27 have been identified in several of these pedigrees [98–101]. By using array comparative genomic hybridisation, Solomon et al. [101] identified additional duplications and, by combining data from five unrelated pedigrees with X linked hypopituitarism, were able to define a critical duplication region of 3.9 Mb between Xq26.1 and Xq27.3 containing 18 annotated transcripts including SOX3. The phenotypes of affected males with X linked hypopituitarism involving duplications within this region are variable. All affected males manifest GH deficiency and varying degrees of developmental delay or mental retardation [99, 101]. Some individuals have been reported to have varying combinations of deficiencies of other hormones including adrenocorticotrophin, thyroid stimulating hormone (TSH) or gonadotrophins, and complete panhypopituitarism has been documented in some cases [100]. A single family has been reported with duplication of Xq26–q27 associated with hypopituitarism and spina bifida; however the duplication in this family is large (13 Mb), and the authors concluded that the spina bifida is most likely to result from the disruption of a gene distinct from that responsible for the hypopituitary phenotype [100]. Unaffected carrier females in these pedigrees show preferential inactivation of the duplicated X chromosome. However, a rare family with five affected females presenting with short stature secondary to hypopituitarism, speech and language problems, hearing impairment and facial dysmorphism has also been reported with a 7.5 Mb duplication of chromosome Xq26.2–q27.1 [102]. The authors suggested that the duplication may disrupt SOX3 resulting in hemizygosity in affected females, although this was not confirmed at the molecular level. However, it is of interest that no male offspring within the three generation pedigree carried the duplication and the maternal grandmother of the proband was noted to have had three miscarriages.
Woods et al. [103] described a pedigree with two half brothers manifesting evidence of X linked hypopituitarism harbouring a submicroscopic duplication on chromosome Xq27.1, further refining the critical interval to approximately 690 kb. The phenotype was variable between the two siblings, possibly reflected by differences in genetic background from their different fathers. The first child manifested growth hormone (GH) deficiency, with borderline low FT4 concentrations, a possible indicator of evolving TSH deficiency, with hypoplasia of the lower half of the infundibulum and an abnormal corpus callosum which contained a cyst within the splenium. The second sibling manifested a more severe phenotype of combined pituitary hormone deficiency, with complete absence of the infundibulum and hypoplastic genitalia; however his corpus callosum appeared normal. Both patients had anterior pituitary hypoplasia and an undescended posterior pituitary as revealed by MRI. There was no evidence of mental retardation or craniofacial dysmorphism in either individual, unlike individuals who had previously been shown to be duplicated. The unaffected mother of the patients was also shown to carry the duplication, however no skewing of X inactivation was observed. The duplication identified in this family is the smallest described to date encompassing SOX3 and two additional transcripts of unknown function, neither of which are expressed in the developing infundibulum [103] suggesting that the phenotype in these patients is due to the presence of an additional copy of SOX3.
Further implication of SOX3 in X linked hypopituitarism comes from the identification of patients harbouring an expansion of one of the polyalanine tracts within the gene [103, 104]. Laumonnier et al. [104] identified an in frame duplication of 33 bp occurring between nucleotides 711–743 and co-segregating in affected males in a large family with X linked mental retardation and GH deficiency. This duplication encodes an additional 11 alanine residues and is predicted to cause expansion of the normal polyalanine tract from 15 to 26 residues and was associated with a phenotype of short stature, isolated GH deficiency and mental retardation, with facial anomalies in some, but not all, patients. Additionally, a second novel expansion of seven alanine residues within the same tract has been identified in three siblings of a consanguineous pedigree presenting with profound and complete panhypopituitarism in association with anterior pituitary hypoplasia, an absent or hypoplastic infundibulum and an undescended posterior pituitary. There was no evidence of mental retardation or craniofacial dysmorphism in these individuals. Additionally, a fourth sibling had GH and possible gonadotrophin deficiencies, with similar neuroradiological findings, although genetic testing was not performed. It is likely that this fourth child also harbours the same polyalanine expansion [103]. A deletion resulting in contraction of the same polyalanine repeat by nine residues has also been reported in two brothers with mental retardation; however the significance of this finding remains unknown as functional studies have not been performed and the deletion was also present in the maternal grandfather of the patients who was clinically unaffected [104]. No mutations involving SOX3 have been found in patients with sex reversal, gonadal dysgenesis or infertility [105, 106].
In vitro analysis of the SOX3 +7 alanine expansion identified by Woods et al. [103], revealed that the expansion leads to partial loss of function possibly due to impairment of nuclear localization as the mutant protein was largely excluded from the nucleus, compared to wild-type SOX3 which is predominantly localized within the nucleus of the cell. Similar findings have also been shown for the mutant SOX3 protein containing the +11 alanine expansion which forms aggregates within the cytoplasm [107]. Furthermore, expansion mutations in HOXA13 and RUNX2 show a similar effect, suggesting that expanded polyalanine tract mutations in transcription factors are associated with loss of function as a result of cytoplasmic aggregation of the mutant protein [107].
Bowl et al. [108] recently reported a case of X linked hypoparathyroidism previously mapped to Xq27. Molecular analyses revealed a novel deleted region of 23–25 kb which had been replaced by a 340 kb insertion originating from chromosome 2p25, which contained a segment of the SNTG2 gene but lacked an open reading frame. The deleted region of Xq27 did not contain any known genes but the breakpoints were situated approximately 67 kb downstream of SOX3. Patients with X linked hypopituitarism have not been reported to suffer from hypoparathyroidism. However Sox3 expression has been detected in the second and third pharyngeal pouches, the developing parathyroids and thymic and thyroid rudiments in the mouse [108]. It is possible that the deleted region of Xq27 identified in the patients contains long-range cis-acting regulatory elements for SOX3. The authors postulated that disruption of such putative regulatory sequences may result in mis-regulation of the gene leading to parathyroid agenesis and hypoparathyroidism.
Duplications of Xq27 encompassing SOX3 and loss-of-function polyalanine expansion mutations are essentially associated with similar phenotypes, predominantly infundibular hypoplasia. As suggested by, and consistent with, observations from the mouse, anterior pituitary hypoplasia with associated deficiency of anterior pituitary hormones is most likely a secondary effect of the disruption of inductive signalling mechanisms from the developing hypothalamus and infundibulum which are important for normal development of the anterior pituitary. Significantly, it appears that gene dosage of SOX3 is critical for normal development of the diencephalon and infundibulum and, consequently the anterior pituitary, as both over- and under-dosage of SOX3 are associated with infundibular hypoplasia and variable hypopituitarism with variable effects on the corpus callosum. At present, no obvious genotype–phenotype correlations can be made, particularly given the variability of the phenotypes within pedigrees [103].
SOX2
SOX2 (OMIM 184429) is also a member of the SOXB1 subfamily with SOX1 (OMIM 602148) and SOX3. In the mouse, initial expression of Sox2 is detected at 2.5 dpc at the morula stage, and then in the inner cell mass of the blastocyst at 3.5 dpc. Later expression of Sox2, following gastrulation, is restricted to the presumptive neuroectoderm and by 9.5 dpc it is expressed throughout the brain, CNS, sensory placodes, branchial arches, gut endoderm and the esophagus and trachea [85, 109]. Homozygous loss of Sox2 results in peri-implantation lethality, whereas Sox2 heterozygous mice appear relatively normal but show a reduction in size and male fertility [110]. By introducing a regulatory mutation in Sox2 (Sox2ΔENH) in addition to the Sox2βgeo null allele, Ferri et al. could access some later functions of SOX2 in the CNS [111]. Compound heterozygosity for both alleles reduced levels of Sox2 expression to 25–30% of wild-type levels within specific regions of the CNS. This resulted in a reduction in both numbers of pups being born, and in their post-natal survival, with surviving mice showing severe abnormalities including growth retardation, although this was usually compensated by 6 weeks of age, dystonic reactions, epileptic spikes in the cortex and hippocampus, and circling behaviour. These data support a role for SOX2 in neural progenitors, as has been suggested from studies in other model systems [89], as well as highlighting its importance for the maintenance of specific neuronal populations.
Further studies using conditional and hypomorphic alleles of Sox2 to create a gene dosage series of mutant mice with expression ranging from 50% to 15–30% have been able to investigate the role of Sox2 in eye development. Reduction of less than 40% of normal Sox2 levels results in a range of eye phenotypes with severity directly related to the level of expression [112]. Additionally, two regulatory mutations have been reported in mice resulting in sensorineural deafness or severe hearing impairment as a result of the complete absence or reduced expression of Sox2 specifically within the developing inner ear [113].
Given the observation of growth retardation and reduced fertility, Kelberman et al. [114] investigated the role of Sox2 in murine pituitary development, showing that a proportion of heterozygous animals manifested a variable hypopituitary phenotype, with hypoplasia and abnormal morphology of the anterior pituitary gland with concomitant reduction in levels of GH, LH, ACTH and TSH. Like its murine counterpart, the human SOX2 gene is a single exon gene encoding a 317 amino acid protein containing an N-terminal domain of unknown function, a DNA binding HMG domain and a C-terminal transcriptional activation domain. Twelve heterozygous de novo mutations in SOX2 were previously reported in 14 human patients associated with bilateral anophthalmia or severe microphthalmia with additional abnormalities reported including developmental delay, learning difficulties, esophageal atresia and genital abnormalities [109, 115–118]. Additional phenotypic abnormalities include developmental delay, learning difficulties, esophageal atresia, sensori-neural hearing loss and genital abnormalities. All of these mutations occurred de novo and included five nonsense, four frameshift, one deletion, and two mis-sense mutations. More recently, Kelberman et al. [114] reported six patients harbouring de novo heterozygous mutations in SOX2 resulting in loss-of-function of the mutant protein, four of which were previously unreported (c.60insG, c.387delC, Y160X and c.479delA; Fig. 3). Clinical evaluation revealed that in addition to anophthalmia/microphthalmia, SOX2 mutations were also associated with anterior pituitary hypoplasia and hypogonadotropic hypogonadism, which resulted in the absence of puberty in all six patients and genital abnormalities in males. All affected individuals exhibited learning difficulties with other variable manifestations including hippocampal abnormalities, defects of the corpus callosum, esophageal atresia and sensorineural hearing loss [114]. The mutations were associated with significant loss of function that included loss of DNA binding, nuclear localization, and transcriptional activation, suggesting these phenotypes arise as a result of haploinsufficiency of SOX2 in development. It is unusual to observe a normal GH response to provocation in the face of abnormal gonadotrophin secretion in patients with hypopituitarism. Additionally, other pituitary hormone axes appeared to be clinically normal in patients despite structural hypoplasia of the gland. Dysregulation of hypothalamic secretion associated with SOX2 mutations could lead to endocrine deficits; however this would have to be more severe for neurons secreting gonadotropin-releasing hormone (GnRH). It is also possible, though unlikely, that a primary abnormality in the formation of Rathke’s pouch selectively affects the gonadotrope cell lineage during pituitary development in humans. Given the widespread expression of Sox2 in the CNS in mice during development, the phenotype of gonadotropin deficiency may be due to a loss of function at an earlier stage, for example in determining the normal number and/or specification of GnRH neurons in the developing hypothalamus.

Mutations/polymorphisms within the coding region of SOX2
Conclusions
SOD is now known to be due to disordered development of the forebrain and related structures. It has previously been speculated that SOD is due to an environmental effect. Mutations in developmental genes have been shown to play a role in the pathogenesis of a number of conditions in man. A role for the homeobox gene HESX1 in rare familial forms of SOD has been demonstrated [37, 71–73] in addition to a suggestive role for this gene in some cases of the more frequently occurring sporadic form of SOD [33, 70]. Nevertheless, the majority of patients screened in our studies show no mutation of HESX1. There may be several explanations for this observation; variable phenotypes may actually reflect the heterogeneity of multiple conditions with different pathways affecting forebrain/midbrain development. Alternatively, the variability could theoretically reflect a single SOD phenotype with the same genetic defect (e.g., HESX1), but which is variably penetrant. Alterations in regulatory regions may also contribute to the etiology of this condition, as forebrain and pituitary-specific regulatory regions have been identified for some genes and mutations in these regions may lead to a principally forebrain or pituitary phenotype.
Additionally, as with holoprosencephaly, SOD may have a multigenic basis, and mutations in as yet unidentified genes could contribute to some cases. Mutations identified in SOX2 and SOX3 are associated with rarer variants of SOD that include severe bilateral eye defects and abnormalities of the corpus callosum, respectively. To date, the targets and partners of these transcriptional factors remain unknown. These may shed further light on the molecular basis of not only SOD, but of various other pituitary disorders. The variability of the phenotypes within a single SOD pedigree may also suggest a complex interaction between genetics and the environment, and at present, the understanding of these conditions is rudimentary. Study of the interactions between these critical transcription factors may shed further light on the aetiology of this complex disorder.
References
De Morsier G (1956) [Studies on malformation of cranio-encephalic sutures. III. Agenesis of the septum lucidum with malformation of the optic tract.]. Schweiz Arch Neurol Psychiatr 77(1–2):267–292
Reeves DL (1941) Congenital absence of the septum pellucidum. Bull Johns Hopkins Hosp 69:61–71
Hoyt WF, Kaplan SL, Grumbach MM, Glaser JS (1970) Septo-optic dysplasia and pituitary dwarfism. Lancet 1(7652):893–894
Arslanian SA, Rothfus WE, Foley TP Jr, Becker DJ (1984) Hormonal, metabolic, and neuroradiologic abnormalities associated with septo-optic dysplasia. Acta Endocrinol (Copenh) 107(2):282–288
Izenberg N, Rosenblum M, Parks JS (1984) The endocrine spectrum of septo-optic dysplasia. Clin Pediatr (Phila) 23(11):632–636
Roessmann U (1989) Septo-optic dysplasia (SOD) or DeMorsier syndrome. J Clin Neuroophthalmol 9(3):156–159
Stanhope R, Preece MA, Brook CG (1984) Hypoplastic optic nerves and pituitary dysfunction. A spectrum of anatomical and endocrine abnormalities. Arch Dis Child 59(2):111–114
Patel L, McNally RJ, Harrison E, Lloyd IC, Clayton PE (2006) Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J Pediatr 148(1):85–88
Kuriyama M, Shigematsu Y, Konishi K, Konishi Y, Sudo M, Haruki S, Ito H (1988) Septo-optic dysplasia with infantile spasms. Pediatr Neurol 4(1):62–65
Miller SP, Shevell MI, Patenaude Y, Poulin C, O’Gorman AM (2000) Septo-optic dysplasia plus: a spectrum of malformations of cortical development. Neurology 54(8):1701–1703
Brodsky MC, Glasier CM (1993) Optic nerve hypoplasia. Clinical significance of associated central nervous system abnormalities on magnetic resonance imaging [published erratum appears in Arch Ophthalmol 1993 Apr;111(4):491]. Arch Ophthalmol 111(1):66–74
Zeki SM, Hollman AS, Dutton GN (1992) Neuroradiological features of patients with optic nerve hypoplasia. J Pediatr Ophthalmol Strabismus 29(2):107–112
Shammas NW, Brown JD, Foreman BW, Marutani DR, Maddela D, Tonner D (1993) Septo-optic dysplasia associated with polyendocrine dysfunction. J Med 24(1):67–74
Willnow S, Kiess W, Butenandt O, Dorr HG, Enders A, Strasser-Vogel B, Egger J, Schwarz HP (1996) Endocrine disorders in septo-optic dysplasia (De Morsier syndrome) – evaluation and follow up of 18 patients. Eur J Pediatr 155(3):179–184
Acers TE (1981) Optic nerve hypoplasia: septo-optic-pituitary dysplasia syndrome. Trans Am Ophthalmol Soc 79:425–457
Birkebaek NH, Patel L, Wright NB, Grigg JR, Sinha S, Hall CM, Price DA, Lloyd IC, Clayton PE (2003) Endocrine status in patients with optic nerve hypoplasia: relationship to midline central nervous system abnormalities and appearance of the hypothalamic-pituitary axis on magnetic resonance imaging. J Clin Endocrinol Metab 88(11):5281–5286
Cameron FJ, Khadilkar VV, Stanhope R (1999) Pituitary dysfunction, morbidity and mortality with congenital midline malformation of the cerebrum. Eur J Pediatr 158(2):97–102
Costin G, Murphree AL (1985) Hypothalamic-pituitary function in children with optic nerve hypoplasia. Am J Dis Child 139(3):249–254
Freude S, Frisch H, Wimberger D, Schober E, Husler G, Waldhauser F, Aichner F (1992) Septo-optic dysplasia and growth hormone deficiency: accelerated pubertal maturation during GH therapy. Acta Paediatr 81(8):641–645
Huseman CA, Kelch RP, Hopwood NJ, Zipf WB (1978) Sexual precocity in association with septo-optic dysplasia and hypothalamic hypopituitarism. J Pediatr 92(5):748–753
Hanna CE, Mandel SH, LaFranchi SH (1989) Puberty in the syndrome of septo-optic dysplasia. Am J Dis Child 143(2):186–189
Lam KS, Wang C, Ma JT, Leung SP, Yeung RT (1986) Hypothalamic defects in two adult patients with septo-optic dysplasia. Acta Endocrinol (Copenh) 112(3):305–309
Yukizane S, Kimura Y, Yamashita Y, Matsuishi T, Horikawa H, Ando H, Yamashita F (1990) Growth hormone deficiency of hypothalamic origin in septo-optic dysplasia. Eur J Pediatr 150(1):30–33
Roessmann U, Velasco ME, Small EJ, Hori A (1987) Neuropathology of “septo-optic dysplasia” (de Morsier syndrome) with immunohistochemical studies of the hypothalamus and pituitary gland. J Neuropathol Exp Neurol 46(5):597–608
Masera N, Grant DB, Stanhope R, Preece MA (1994) Diabetes insipidus with impaired osmotic regulation in septo-optic dysplasia and agenesis of the corpus callosum. Arch Dis Child 70(1):51–53
Morishima A, Aranoff GS (1986) Syndrome of septo-optic-pituitary dysplasia: the clinical spectrum. Brain Dev 8(3):233–239
McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou A, Keller E, Keller A, Haufs N, Krude H, Shalet SM, Dattani MT (2006) HESX1 mutations are an uncommon cause of septo-optic dysplasia and hypopituitarism. J Clin Endocrinol Metab 92(2):691–697
Zaias B, Becker D (1978) Septo-optic dysplasia: developmental or acquired abnormality? A case report. Trans Am Neurol Assoc 103:273–277
Thomas P, Brickman JM, Popperl H, Krumlauf R, Beddington RS (1997) Axis duplication and anterior identity in the mouse embryo. Cold Spring Harb Symp Quant Biol 62:115–125
Wales JK, Quarrell OW (1996) Evidence for possible Mendelian inheritance of septo-optic dysplasia. Acta Paediatr 85(3):391–392
Blethen SL, Weldon VV (1985) Hypopituitarism and septooptic “dysplasia” in first cousins. Am J Med Genet 21(1):123–129
Benner JD, Preslan MW, Gratz E, Joslyn J, Schwartz M, Kelman S (1990) Septo-optic dysplasia in two siblings. Am J Ophthalmol 109(6):632–637
Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F, Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS (2001) Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet 10(1):39–45
Cohen RN, Cohen LE, Botero D, Yu C, Sagar A, Jurkiewicz M, Radovick S (2003) Enhanced repression by HESX1 as a cause of hypopituitarism and septooptic dysplasia. J Clin Endocrinol Metab 88(10):4832–4839
Tajima T, Hattorri T, Nakajima T, Okuhara K, Sato K, Abe S, Nakae J, Fujieda K (2003) Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and optic nerve hypoplasia and combined pituitary hormone deficiency in a Japanese patient. J Clin Endocrinol Metab 88(1):45–50
Beddington RS, Robertson EJ (1999) Axis development and early asymmetry in mammals. Cell 96(2):195–209
Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JKH, Hindmarsh PC, Krauss S, Beddington RSP, Robinson ICAF (1998) Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet 19(2):125–133
Shawlot W, Behringer RR (1995) Requirement for Lim1 in head-organizer function. Nature 374(6521):425–430
Ang SL, Jin O, Rhinn M, Daigle N, Stevenson L, Rossant J (1996) A targeted mouse Otx2 mutation leads to severe defects in gastrulation and formation of axial mesoderm and to deletion of rostral brain. Development 122(1):243–252
Rubenstein JL, Shimamura K, Martinez S, Puelles L (1998) Regionalization of the prosencephalic neural plate. Annu Rev Neurosci 21:445–477
Couly G, Le Douarin NM (1988) The fate map of the cephalic neural primordium at the presomitic to the 3-somite stage in the avian embryo. Development 103(Suppl):101–113
Eagleson GW, Harris WA (1990) Mapping of the presumptive brain regions in the neural plate of Xenopus laevis. J Neurobiol 21(3):427–440
Jacobson AG, Miyamoto DM, Mai SH (1979) Rathke’s pouch morphogenesis in the chick embryo. J Exp Zool 207:351–366
Dasen JS, Rosenfeld MG (1999) Signaling mechanisms in pituitary morphogenesis and cell fate determination. Curr Opin Cell Biol 11(6):669–677
Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ (1996) The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 10(1):60–69
Treier M, Gleiberman AS, O’Connell SM, Szeto DP, McMahon JA, McMahon AP, Rosenfeld MG (1998) Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev 12(11):1691–1704
Takuma N, Sheng HZ, Furuta Y, Ward JM, Sharma K, Hogan LM, Pfaff SL, Westphal H, Kimura S, Mahon KA (1998) Formation of Rathke’s pouch requires dual induction from the diencephalon. Development 125(23):4835–4840
Oliver G, Mailhos A, Wehr R, Copeland NG, Jenkins NA, Gruss P (1995) Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 121(12):4045–4055
Walther C, Gruss P (1991) Pax-6, a murine paired box gene, is expressed in the developing CNS. Development 113(4):1435–1449
Thomas PQ, Johnson BV, Rathjen J, Rathjen PD (1995) Sequence, genomic organization, and expression of the novel homeobox gene Hesx1. J Biol Chem 270(8):3869–3875
Hermesz E, Mackem S, Mahon KA (1996) Rpx: a novel anterior-restricted homeobox gene progressively activated in the prechordal plate, anterior neural plate and Rathke’s pouch of the mouse embryo. Development 122(1):41–52
Zhadanov AB, Bertuzzi S, Taira M, Dawid IB, Westphal H (1995) Expression pattern of the murine LIM class homeobox gene Lhx3 in subsets of neural and neuroendocrine tissues. Dev Dyn 202(4):354–364
Bach I, Rhodes SJ, Pearse RV, Heinzel T, Gloss B, Scully KM, Sawchenko PE, Rosenfeld MG (1995) P-Lim, a LIM homeodomain factor, is expressed during pituitary organ and cell commitment and synergizes with Pit-1. Proc Natl Acad Sci USA 92(7):2720–2724
Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, Westphal H (1997) Multistep control of pituitary organogenesis. Science 278(5344):1809–1812
Sheng HZ, Zhadanov AB, Mosinger B, Fujii T, Bertuzzi S, Grinberg A, Lee EJ, Huang SP, Mahon KA, Westphal H (1996) Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science 272(5264):1004–1007
Lamonerie T, Tremblay JJ, Lanctot C, Therrien M, Gauthier Y, Drouin J (1996) Ptx1, a bicoid-related homeo box transcription factor involved in transcription of the pro-opiomelanocortin gene. Genes Dev 10(10):1284–1295
Szeto DP, Ryan AK, O’Connell SM, Rosenfeld MG (1996) P-OTX: a PIT-1-interacting homeodomain factor expressed during anterior pituitary gland development. Proc Natl Acad Sci USA 93(15):7706–7710
Simmons DM, Voss JW, Ingraham HA, Holloway JM, Broide RS, Rosenfeld MG, Swanson LW (1990) Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes Dev 4(5):695–711
Japon MA, Rubinstein M, Low MJ (1994) In situ hybridization analysis of anterior pituitary hormone gene expression during fetal mouse development. J Histochem Cytochem 42(8):1117–1125
Li S, Crenshaw EB III, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG (1990) Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature 347(6293):528–533
Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, O’Connell SM, Gukovsky I, Carriere C, Ryan AK, Miller AP, Zuo L, Gleiberman AS, Andersen B, Beamer WG, Rosenfeld MG (1996) Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature 384(6607):327–333
Wu W, Cogan JD, Pfaffle RW, Dasen JS, Frisch H, O’Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips JA III, Rosenfeld MG (1998) Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet 18(2):147–149
Artman HG, Boyden E (1990) Microphthalmia with single central incisor and hypopituitarism. J Med Genet 27(3):192–193
Brook CG, Sanders MD, Hoare RD (1972) Septo-optic dysplasia. Br Med J 3(830):811–813
Coulter CL, Leech RW, Schaefer GB, Scheithauer BW, Brumback RA (1993) Midline cerebral dysgenesis, dysfunction of the hypothalamic-pituitary axis, and fetal alcohol effects. Arch Neurol 50(7):771–775
Fitz CR (1994) Holoprosencephaly and septo-optic dysplasia. Neuroimaging Clin N Am 4(2):263–281
Thomas P, Beddington R (1996) Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol 6(11):1487–1496
Dasen JS, Barbera JP, Herman TS, Connell SO, Olson L, Ju B, Tollkuhn J, Baek SH, Rose DW, Rosenfeld MG (2001) Temporal regulation of a paired-like homeodomain repressor/TLE corepressor complex and a related activator is required for pituitary organogenesis. Genes Dev 15(23):3193–3207
Dasen JS, Rosenfeld MG (2001) Signaling and transcriptional mechanisms in pituitary development. Annu Rev Neurosci 24:327–355
Brickman JM, Clements M, Tyrell R, McNay D, Woods K, Warner J, Stewart A, Beddington RS, Dattani M (2001) Molecular effects of novel mutations in Hesx1/HESX1 associated with human pituitary disorders. Development 128(24):5189–5199
Carvalho LR, Woods KS, Mendonca BB, Marcal N, Zamparini AL, Stifani S, Brickman JM, Arnhold IJ, Dattani MT (2003) A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor-corepressor interaction. J Clin Invest 112(8):1192–1201
Sobrier ML, Netchine I, Heinrichs C, Thibaud N, Vie-Luton MP, Van Vliet G, Amselem S (2005) Alu-element insertion in the homeodomain of HESX1 and aplasia of the anterior pituitary. Hum Mutat 25(5):503
Sobrier ML, Maghnie M, Vie-Luton MP, Secco A, di Iorgi N, Lorini R, Amselem S (2006) Novel HESX1 mutations associated with a life-threatening neonatal phenotype, pituitary aplasia, but normally located posterior pituitary and no optic nerve abnormalities. J Clin Endocrinol Metab 91(11):4528–4536
Tornqvist K, Ericsson A, Kallen B (2002) Optic nerve hypoplasia: risk factors and epidemiology. Acta Ophthalmol Scand 80(3):300–304
Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Munsterberg A, Vivian N, Goodfellow P, Lovell-Badge R (1990) A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346(6281):245–250
Stevanovic M, Lovell-Badge R, Collignon J, Goodfellow PN (1993) SOX3 is an X-linked gene related to SRY. Hum Mol Genet 2(12):2013–2018
Pevny LH, Lovell-Badge R (1997) Sox genes find their feet. Curr Opin Genet Dev 7(3):338–344
Wegner M (1999) From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res 27(6):1409–1420
Bowles J, Schepers G, Koopman P (2000) Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol 227(2):239–255
Stevanovic M, Zuffardi O, Collignon J, Lovell-Badge R, Goodfellow P (1994) The cDNA sequence and chromosomal location of the human SOX2 gene. Mamm Genome 5(10):640–642
Kamachi Y, Uchikawa M, Collignon J, Lovell-Badge R, Kondoh H (1998) Involvement of Sox1, 2 and 3 in the early and subsequent molecular events of lens induction. Development 125(13):2521–2532
Denny P, Swift S, Brand N, Dabhade N, Barton P, Ashworth A (1992) A conserved family of genes related to the testis determining gene, SRY. Nucleic Acids Res 20(11):2887
Mertin S, McDowall SG, Harley VR (1999) The DNA-binding specificity of SOX9 and other SOX proteins. Nucleic Acids Res 27(5):1359–1364
Foster JW, Graves JA (1994) An SRY-related sequence on the marsupial X chromosome: implications for the evolution of the mammalian testis-determining gene. Proc Natl Acad Sci USA 91(5):1927–1931
Wood HB, Episkopou V (1999) Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Dev 86(1–2):197–201
Collignon J, Sockanathan S, Hacker A, Cohen-Tannoudji M, Norris D, Rastan S, Stevanovic M, Goodfellow PN, Lovell-Badge R (1996) A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development 122(2):509–520
Pevny LH, Sockanathan S, Placzek M, Lovell-Badge R (1998) A role for SOX1 in neural determination. Development 125(10):1967–1978
Zappone MV, Galli R, Catena R, Meani N, De Biasi S, Mattei E, Tiveron C, Vescovi AL, Lovell-Badge R, Ottolenghi S, Nicolis SK (2000) Sox2 regulatory sequences direct expression of a (beta)-geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development 127(11):2367–2382
Pevny L, Placzek M (2005) SOX genes and neural progenitor identity. Curr Opin Neurobiol 15(1):7–13
Bylund M, Andersson E, Novitch BG, Muhr J (2003) Vertebrate neurogenesis is counteracted by Sox1–3 activity. Nat Neurosci 6(11):1162–1168
Rizzoti K, Brunelli S, Carmignac D, Thomas PQ, Robinson IC, Lovell-Badge R (2004) SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet 36(3):247–255
Weiss J, Meeks JJ, Hurley L, Raverot G, Frassetto A, Jameson JL (2003) Sox3 is required for gonadal function, but not sex determination, in males and females. Mol Cell Biol 23(22):8084–8091
Rizzoti K, Lovell-Badge R (2005) Early development of the pituitary gland: induction and shaping of Rathke’s pouch. Rev Endocr Metab Disord 6(3):161–172
Ericson J, Norlin S, Jessell TM, Edlund T (1998) Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development 125(6):1005–1015
Zorn AM, Barish GD, Williams BO, Lavender P, Klymkowsky MW, Varmus HE (1999) Regulation of Wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol Cell 4(4):487–498
Douglas KR, Brinkmeier ML, Kennell JA, Eswara P, Harrison TA, Patrianakos AI, Sprecher BS, Potok MA, Lyons RH Jr, MacDougald OA, Camper SA (2001) Identification of members of the Wnt signaling pathway in the embryonic pituitary gland. Mamm Genome 12(11):843–851
Cha KB, Douglas KR, Potok MA, Liang H, Jones SN, Camper SA (2004) WNT5A signaling affects pituitary gland shape. Mech Dev 121(2):183–194
Hamel BC, Smits AP, Otten BJ, van den Helm HB, Ropers HH, Mariman EC (1996) Familial X-linked mental retardation and isolated growth hormone deficiency: clinical and molecular findings. Am J Med Genet 64(1):35–41
Lagerstrom-Fermer M, Sundvall M, Johnsen E, Warne GL, Forrest SM, Zajac JD, Rickards A, Ravine D, Landegren U, Pettersson U (1997) X-linked recessive panhypopituitarism associated with a regional duplication in Xq25-q26. Am J Hum Genet 60(4):910–916
Hol FA, Schepens MT, van Beersum SE, Redolfi E, Affer M, Vezzoni P, Hamel BC, Karnes PS, Mariman EC, Zucchi I (2000) Identification and characterization of an Xq26-q27 duplication in a family with spina bifida and panhypopituitarism suggests the involvement of two distinct genes. Genomics 69(2):174–181
Solomon NM, Nouri S, Warne GL, Lagerstrom-Fermer M, Forrest SM, Thomas PQ (2002) Increased gene dosage at Xq26-q27 is associated with X-linked hypopituitarism. Genomics 79(4):553–559
Stankiewicz P, Thiele H, Schlicker M, Cseke-Friedrich A, Bartel-Friedrich S, Yatsenko SA, Lupski JR, Hansmann I (2005) Duplication of Xq26.2-q27.1, including SOX3, in a mother and daughter with short stature and dyslalia. Am J Med Genet Part A 138A(1):11–17
Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, Wong J, Chong WK, Al Zyoud M, El Ali M, Otonkoski T, Martinez-Barbera JP, Thomas PQ, Robinson IC, Lovell-Badge R, Woodward KJ, Dattani MT (2005) Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet 76(5):833–849
Laumonnier F, Ronce N, Hamel BCJ, Thomas P, Lespinasse J, Raynaud M, Paringaux C, Van Bokhoven H, Kalscheuer V, Fryns JP, Chelly J, Moraine C, Briault S (2002) Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet 71(6):1450–1455
Lim HN, Berkovitz GD, Hughes IA, Hawkins JR (2000) Mutation analysis of subjects with 46, XX sex reversal and 46, XY gonadal dysgenesis does not support the involvement of SOX3 in testis determination. Hum Genet 107(6):650–652
Raverot G, Lejeune H, Kotlar T, Pugeat M, Jameson JL (2004) X-linked sex-determining region Y box 3 (SOX3) gene mutations are uncommon in men with idiopathic oligoazoospermic infertility. J Clin Endocrinol Metab 89(8):4146–4148
Albrecht AN, Kornak U, Boddrich A, Suring K, Robinson PN, Stiege AC, Lurz R, Stricker S, Wanker EE, Mundlos S (2004) A molecular pathogenesis for transcription factor associated poly-alanine tract expansions. Hum Mol Genet 13(20):2351–2359
Bowl MR, Nesbit MA, Harding B, Levy E, Jefferson A, Volpi E, Rizzoti K, Lovell-Badge R, Schlessinger D, Whyte MP, Thakker RV (2005) An interstitial deletion-insertion involving chromosomes 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism. J Clin Invest 115(10):2822–2831
Williamson KA, Hever AM, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, Schneider A, Messina M, Turnpenny PD, Fantes JA, van Heyningen V, FitzPatrick DR (2006) Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet 15(9):1413–1422
Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R (2003) Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17(1):126–140
Ferri ALM, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK (2004) Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131(15):3805–3819
Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH (2006) SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev 20(9):1187–1202
Kiernan AE, Pelling AL, Leung KKH, Tang ASP, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KSE (2005) Sox2 is required for sensory organ development in the mammalian inner ear. Nature 434(7036):1031–1035
Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT (2006) Mutations withinSox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest 116:2442–2455
Fantes J, Ragge NK, Lynch SA, Mcgill NI, Collin JRO, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR (2003) Mutations in SOX2 cause anophthalmia. Nat Genet 33(4):461–463
Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JRO, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, FitzPatrick DR (2005) SOX2 anophthalmia syndrome. Am J Med Genet Part A 135A(1):1–7
Hagstrom SA, Pauer GJT, Reid J, Simpson E, Crowe S, Maumenee IH, Traboulsi EI (2005) SOX2 mutation causes anophthalmia, hearing loss, and brain anomalies. Am J Med Genet Part A 138A(2):95–98
Zenteno JC, Gascon-Guzman G, Tovilla-Canales JL (2005) Bilateral anophthalmia and brain malformations caused by a 20-bp deletion in the SOX2 gene. Clin Genet 68(6):564–566
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kelberman, D., Dattani, M.T. Genetics of septo-optic dysplasia. Pituitary 10, 393–407 (2007). https://doi.org/10.1007/s11102-007-0055-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-007-0055-5