
Abstract
Purpose
The purpose of this study was to evaluate the specifically targeted efficiency of budesonide loaded PLGA nanoparticles for the treatment of inflammatory bowel disease (IBD).
Methods
The nanoparticles were prepared by an oil/water (O/W) emulsion evaporation technique. The nanoparticles were characterized for their size, shape and in vitro drug release profile. Solid state characterization was carried out by differential scanning calorimetry (DSC) and X-ray Power diffraction (XPRD). In order to evaluate the targeted efficiency of nanoparticles, a particle localization study in the healthy and in the inflamed colon was determined in vivo. These data were complemented by cryo-sections.
Results
Nanoparticles were 200 ± 05 nm in size with a smooth and spherical shape. The encapsulation efficiency was around 85 ± 3.5%, which was find-out by both, direct and indirect methods. Release of budesonide from the nanoparticles showed a biphasic release profile with an initial burst followed by sustained release. XPRD data revealed that the drug in the polymer matrix existed in crystalline state. Nanoparticles accumulation in inflamed tissues was evaluated by in-vivo imaging system and it was found that particles are accumulated in abundance at the site of inflammation when compared to the healthy group.
Conclusion
The study demonstrates that the budesonide loaded PLGA nanoparticles are an efficient delivery system for targeted drug delivery to the inflamed intestinal mucosa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) comprises a group of chronic and idiopathic inflammation conditions of the gastrointestinal tract (GIT). The two most commonly studied types are Crohn’s disease (CD) and ulcerative colitis (UC). Both, CD and UC are chronic and relapsing inflammatory bowel diseases. CD can occur in any part of the GIT but most commonly it has been diagnosed in the terminal ileum and colon, whereas, UC is restricted to the descending colon and transverse colon (1–3). The pathogenesis of IBD is not yet fully understood; however, various environmental and genetic factors play an important role (4,5). Incidence rates for IBD are higher in North America and Europe compared to Asia and Africa (6). The main aim of the therapy in IBD is to induce a clinical remission and to maintain this state for an extended period of time. The chosen treatment depends on the severity and location of the disease. The available therapeutic options, for example 5-amino-salicylic acid and corticosteroids are commonly used to control the disease and to maintain its remission (7,8).
In contrast to other corticosteroids, budesonide is the drug of choice for inflammatory disorders such as IBD, asthma and arthritis (9). In mild to moderate cases the standard therapeutic dose of budesonide consists of 9 mg per day (10). However, budesonide has demonstrated stronger adverse effects compared to other corticosteroids (11), which can be minimized by selective delivery of drug to its site of action. Budesonide can be given either by oral, rectal or in some cases by intravenous route. The oral route has some limitations as the drug has to face the harsh environment of the GIT. For this reason higher doses of corticosteroids will be required which tend to evoke systemic adverse effects. The rectal route has considered being effective when the inflammation is located at the distal part of the colon mainly in UC (12), but this route is not efficient when the inflammation occurs in the upper part of the colon. Many drug delivery strategies such as pro drug, enteric coated capsule or tablets, granules or pellets have been tried to reduce the toxicity and improve therapeutic efficacy. But these approaches showed limited bioavailability and are quickly excreted via diarrhea, a common symptom of IBD (13).
A carrier system capable to deliver the drug exclusively to the target site would be a most desirable alternative. In this context, nanoparticle-based drug delivery offers a promising and encouraging approach for the selective IBD treatment. Further, nanoparticles can accumulate to a greater extent in the inflamed intestinal mucosa and thus prolong the residence time at the site of inflammation (14,15). Moreover, it has been reported that nanocarriers can be taken up to a significant extent by the immune regulating cells in IBD. By the degradation of the nanocarriers, the encapsulated drug is then released at the desired site (16). This targeted approach was successfully applied for specific delivery of various drugs for the treatment of IBD, using different nanocarriers and technologies (17–19). On the basis of the aforementioned research studies, a carrier system for the delivery of budesonide to the inflamed intestinal mucosa was designed. Budesonide was loaded into fluoresceinamine labeled poly (L-lactide-co-glycolide) (PLGA) nanoparticles and was characterized for their size, shape, morphology and drug release. After pharmaceutical characterization, the nano formulation was studied with regard to their localization in inflamed and healthy mice model to further confirm their local action. In a previous study (20) we have shown the effectiveness and localized action of budesonide loaded PLGA nanoparticles in different colitis mice models. The focus of the present study is therefore on an in-depth pharmaceutical characterization of budesonide loaded PLGA nanoparticles as would be necessary to take such formulation into subsequent clinical trials. Moreover, we provide additional evidence for the accumulation of such nanocarriers at the site of inflammation by in-vivo fluorescence imaging in a relevant mouse model.
Materials and Methods
Materials
The biodegradable polymer poly(lactic-co-glycolic acid) (PLGA) (50:50 lactide: glycolide) polymer, xResomer® was provided by Boehringer Ingelheim, Germany. Budesonide was received from Sigma-Aldrich, Germany. Poly (vinyl alcohol) (PVA) MW 31000 was obtained from Sigma-Aldrich Mowiol® 4-88, Germany. The organic solvent, ethyl acetate and all other chemical reagents were purchased from Merck, Germany and were of analytical grade. Acetonitrile and phosphate buffer PH 3 (40:60) was used as mobile phase in HPLC were purchased from Sigma-Aldrich, Germany. Deionized water was used throughout the experiment. The in vitro drug release measurements were carried out at pH 6.8 at 37°C in phosphate buffer medium.
Methods
Preparation of PLGA Nanoparticles
Fluoresceinamine labeling of PLGA (F-PLGA) was prepared as described previously (21). Nanoparticles were prepared from F-PLGA polymer, according to an emulsion solvent evaporation method (22). In brief, the methodology was performed as follows: 10 mg of budesonide was dissolved in 5 ml of organic solvent (ethyl acetate) containing 100 mg PLGA at room temperature. Thereafter, this organic solution was slowly added to 2% aqueous solution of PVA, using a gear pump (Gilson Minipuls, France). The emulsion was stirred for 30 min at room temperature before homogenizing at 13,500 rpm for 10 min using an Ultra Turrax T-25 (Janke and Kunkle GmbH KG, Staufen, Germany). Water was added to this emulsion under stirring to improve evaporation. Stirring was continued at room temperature until evaporation of organic solvents was completed.
Nanoparticles Characterization
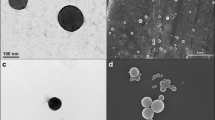
The size and the zeta potential of the particles were measured on a Malvern zetasizer 2000 HS (Malvern, UK). Size measurement was performed in triplicate at room temperature and results were expressed in mean ± standard deviation (SD) (Table I). The Zeta potential (ζ) was also measured in triplicate using a disposable zeta cuvette. The external morphology of NPs was analyzed by AFM (atomic force microscopy) Nanoscope IV Bioscope™ (Digital Instruments, Veeco) in a tapping mode using a Si3N4 cantilever. The nanoparticles were also analyzed by scanning electron microscope (SEM). A drop of nanoparticles was spread on a silicon wafer fixed with a sample holder for SEM and coated afterwards with gold using gold sputter in a high vacuum evaporator and observed on SEM JEOL 7000F (Tokyo, Japan) (Fig. 1).

Atomic force microscopy and scanning electron microscopy images of nanoparticles prepared by emulsion evaporation.
Determination of Drug Encapsulation Efficiency
The drug entrapped in nanoparticles was determined by indirect as well as by direct method (Fig. 2). Indirect method means the amount of drug content measured in the supernatant after washing the NPs three times with distilled water, using a tangential flow apparatus. The samples were then analyzed by HPLC (UltiMate® 3000 from Dionex). A reversed phase C18 Column (4.6 × 250 mm, pore size 5 μm) (Merck KGaA, Germany) was used. A mixture of phosphate buffer (pH 3) and acetonitrile (60:40) was used as a mobile phase and delivered at a flow rate of 1.900 ml/min. Retention time for each sample was 6 min. The method was linear (r2 = 0.9997) over a range from 20 ng/ml to 500 μg/ml. The limit of detection was 0.01 μg/ml and the limit of quantification was 0.5 μg/ml. The encapsulation efficiency was calculated with the following formula. After all, the nanoparticle suspension was then freeze-dried for 72 h (GFL Gesellschaft für Labortechnik mbH, Germany) to obtain a fine powder of nanoparticles.

Direct and indirect encapsulation efficiency of the budesonide loaded PLGA nanoparticles.
Determination of Drug Content
The results of this indirect entrapment assay were then compared to the results obtained by the direct entrapment assay (Fig. 2) and a good correlation was found for both methods. In a word, 5 mg of the freeze-dried nanoparticles were dissolved in ethyl acetate under stirring for 4 to 5 h. Then after complete evaporation of ethyl acetate the residue was then dissolved in 5 ml of acetonitrile and phosphate buffer (4:6) pH 3 for 2 h. Afterwards, the solution was filtered, using a disposable syringe filter paper (CHROMAFIL GF/PET 45/25) pore size 0.45 μm. From the clear filtrate 1 ml sample was collected and analyzed on HPLC, as described above and calculated with the following formula.
Percent Yield
The percent yield of the freeze dried nanoparticles was calculated (Table I) by weighing the total amount of the purified and freeze dried nanoparticles and calculated by the following formula.
In-Vitro Drug Release
Budesonide release kinetics from F-PLGA nanoparticles was studied in phosphate buffer saline (PBS) solution pH 6.8 (Fig. 3). Briefly, 5 mg of the nanoparticles were dispersed in 30 ml of PBS solution. Then 1.5 ml from this suspension were collected at a pre-determined time interval (1, 2 4 6, 24, 48 and 72 h) and centrifuged at 244,000 g for 20 min at 25°C. 1 ml sample from the supernatant was collected for HPLC analysis and was replaced by an equal amount of fresh PBS solution.

In-vitro release of budesonide from PLGA nanoparticles in PBS pH 6.8 (mean, n = 3 ± SD).
Differential Scanning Calorimetry
The crystalline state of the drug in the polymer matrix was evaluated in freeze-dried nanoparticles by differential scanning calorimetry (DSC) (Fig. 4). Thermograms were recorded using a Perkin Elmer (DSC-7) instrument with a standard aluminum pan. Nitrogen was the sweeping gas, and the heating rate was 10°C/min. Samples (2–8 mg) were loaded in an aluminium pan and sealed. The initial and final temperatures were 50°C and 400°C, respectively.

DSC thermogram of budesonide loaded PLGA nanoparticles, pure budesonide and mixture of PLGA polymer and budesonide.
XPRD (X-ray Powder Diffraction)
An automatic X-ray power diffractometer (Philips PW 3710; Eindhoven, The Netherlands) equipped with a PW R30 x-ray generator was used for the XRD study. Nickel-filtered Cu kα1 radiation, having a wavelength of 1.5106 Å was used. The instrument was operated at 35 kW and 20 mA in the range (2θ) of 5 to 50° (Fig. 5). The data were obtained over an angular range from 5 to 50° 2θ using a step size of 0.02° 2θ and step-time of 10 s, with a non-stop mode.

XRPD spectra of budesonide loaded PLGA nanoparticles, pure budesonide, mixture of PLGA polymer and budesonide, pure PLGA polymer and pure budesonide.
Animals
BALB/c mice at the age of 8 to 12 weeks were used for experimental colitis model. All animals were housed under specific pathogen-free conditions and experiments were performed in accordance with institutional guidelines.
Experimental Colitis Model
Oxazolone-mediated experimental colitis was induced using a modification of previously described method (23). In brief, mice were sensitized by epicutaneous application of 150 μl of 3% oxazolone in acetone/oil suspension (3:1) on day 0 followed by intrarectal administration of 100 μl of 1% oxazolone solution in 50% ethanol to anesthetized mice on day 7. Animals received 50% ethanol alone using the same technique served as controls.
Localization of Nanoparticles in the Murine Gut
To investigate the localization of F-PLGA nanoparticles in the colon of healthy and inflamed mice, an In-Vivo-Imaging System (Maestro, CRI Inc., Woburn, MA, USA) was used. After the induction of colitis, all animals received an equal dose (42 μg/150 μl) of nanoparticles suspension by oral gavage at day 7. Then, the mice were anesthetized and the abdominal area was analyzed with regard to fluorescence signals from FITC-labeled nanoparticles after 18 h. Additionally, cryo-sections from colon tissue were performed and cell cores were stained with DAPI (blue fluorescence). Confocal laser scanning microscopy (Carl Zeiss) was used to further confirm the location of green fluorescent nanoparticles in the colon tissue. The experiment was done twice and representative images were shown. The green positive signals in cryo-sections were further quantified with Image J software (version 1.46r software, NIH, USA).
Statistical Analysis
Statistical analysis of the in-vitro and in-vivo data was performed using student t-test with SigmaPlot (SigmaPlot Software, San Jose, California).
Results
Nanoparticles Preparation and Characterization
Drug loaded PLGA nanoparticles were prepared by an O/W emulsion evaporation techniques. The particle size was measured in triplicate at room temperature. The size of the nanoparticles was approximately 200 ± 05 nm and was mono dispersed. The zeta potential of the nanoparticles was nearly zero and the poly-dispersity index (PDI) was 0.05 ± 0.02 (Table I). The prepared nano-emulsion was milky white in appearance and was stable at room temperature. The morphology of the nanoparticle was studied by SEM and AFM (Fig. 1). It was observed that the nanoparticles are spherical in shape and have a smooth surface. The entrapment efficiency was about 85 ± 3.5%, quantified by both direct and indirect assays (Fig. 2) with a production yield of 94%. In-vitro drug release profile was measured in PBS at pH 6.8 for 72 h (Fig. 3). The budesonide loaded PLGA nanoparticles showed an initial burst release followed by a slow and sustained drug release over an extended period of time. The initial burst release was around 52% in the first hour of the release profile and approximately 58% of drug release was achieved after 72 h.
Differential Scanning Calorimetry (DSC) Measurements
Thermal analysis of the budesonide loaded PLGA nanoparticles, pure drug and their physical mixture were performed using DSC (Fig. 4). A physical mixture (1:1 w/w) of budesonide and PLGA was prepared and completely mixed before measurement. Sample of 5 mg were sealed in an aluminum pan and heated with a scan rate of 10°C/min under a nitrogen supply at a rate of 10 ml/min. The obtained results were then analyzed using Universal Analysis 2000 software (Version 4.5a TA Instruments, New Castle, USA). Pure budesonide shows a clear melting peak, whereas in the combination of PLGA and budesonide (in a physical mixture as well as in nanoparticles) such peaks cannot be detected.
X-ray Powder Diffraction (XRPD) Measurements
XRPD diffractograms for pure budesonide showed a peak patterns between 10 and 25°, which corresponds to the crystalline state of budesonide. In contrast, no peak was observed for pure PLGA, which indicates that PLGA is an amorphous (Fig. 5). For the physical mixture as well as for the nanoparticles, the unique peak patterns of budesonide can be detected. This, it can be concluded that the drug within the nanoparticles is in crystalline state.
Localization of PLGA Nanoparticles in Murine Gut
Murine colon tissues were used to investigate the localization of budesonide loaded PLGA nanoparticles in inflamed and healthy groups using an In-Vivo Imaging System. Both, the healthy and the inflamed mice received an equal dose of budesonide loaded PLGA nanoparticles by oral gavage with a final volume of 40 μg/150 μl solution. 18 h after administration, the mice were anesthetized and their abdomens were analyzed with regard to green fluorescence signals from nanoparticles. In this study, it was observed that nanoparticles were spread in the whole gut in the healthy mice group, whereas in the inflamed group nanoparticles were more abundant at the site of inflammation (Fig. 6a). These findings were further confirmed by a cryo-sectional study of the colon mucosa (Fig. 6b) for which a confocal laser scanning microscopy was used. The result showed that the nanoparticles were found in greater quantities at the site of inflammation in the inflamed group as compared to the healthy group. Furthermore, the results were quantified and significantly high fluorescent positive signals in the inflamed colon were observed (Fig. 6c). To sum up, the obtained results showed that particles were found in abundance at the inflammation site and exhibited a statistically significant difference (p < 0.001) as compared to the healthy mice.

(a) In vivo localization of nanoparticles in the colon of healthy control and inflamed control group. The negative control received no treatment. The green fluorescent dye is due to the FITC labeled plain PLGA nanoparticles. The yellow color is caused by auto-fluorescence by intestinal bacteria. (b) Cryo-sections of colon tissue from both groups were stained with DAPI as core dye. Green fluorescent signals can be identified as nanoparticles in the tissue and more positive spots are located in tissue from inflamed mice. (c) Green positive signals were quantified in HPF (high power field) by microscopy and statistically analyzed. The values are reported as mean ± SD (***) p < 0.001 indicated significant differences from healthy control group.
Discussion
Nanoparticles have been in use for drug delivery applying active as well as passive targeting to the site of action and moreover, nanoparticles have been shown to be a promising approach for the treatment of IBD (14,24). Therefore, the primary aim of this study was to formulate a nanocarrier system that could deliver the drug to the site of action in the intestine to minimize the adverse effect and to improve the therapeutic efficiency. The PLGA nanoparticles presented an ideal matrix system for incorporation of hydrophobic drugs. In this study, budesonide loaded PLGA nanoparticles were prepared and were fully characterized. Budesonide has a strong affinity for glucocorticoid receptors. This substance, however, faces an extensive metabolism by cytochrome P450 enzyme in the liver (25), thus a limited amount of the drug reaches the systemic circulation, which is not sufficient for the required therapeutic effect. Therefore, a carrier system that should deliver the drug to the site of action was needed. In this study, the prepared nanoparticles exhibited a smooth and spherical surface with a size range of 200 ± 05 nm approximately. The production yield was around 94% that illustrates that the selected formulation method was appropriate for the production of budesonide loaded PLGA nanoparticles. Moreover, The PLGA polymer provided an efficient carrier system for the delivery of hydrophobic drugs, as it has already been reported in the literature (26).
The drug release from this formulation showed a biphasic release profile with an initial burst release followed by slow and continuous release (Fig. 3). This initial burst release might be due to drug molecules, which are not completely encapsulated in the polymer core but loosely attached to the surface of nanoparticles, thus being more accessible to the release medium these drug molecules rapidly released. After the initial burst release, the drug release profile showed a slow and sustained drug release profile and approximately 58% drug release was achieved after 72 h. This slow drug release was due to the diffusion of drug molecules across the polymer matrix.
Generally, the solid-state characterization with DSC (Fig. 4) and XRPD (Fig. 5) was carried out to determine whether the incorporated drug in the nanoparticulate system appears in crystalline, amorphous, or bound form. The DSC thermograms for pure budesonide, budesonide loaded PLGA nanoparticles and the physical mixture of budesonide and PLGA polymer were recorded and shown in Fig. 4. The DSC analysis confirms the crystalline character of budesonide that exhibited a characteristic melting endotherm at 260.3°C. This result was found in a good correlation with the reported literature data (27,28). In comparison, a physical mixture of budesonide and PLGA polymer (ratio 1:1 w/w) and the drug-loaded nanoparticles were also analyzed with DSC and such peaks could not be detected. As budesonide is soluble in molten PLGA, which is melting at lower temperature than budesonide, DSC is not suitable to analyze the physical state (crystalline/amorphous) of the drug in a mixture with PLGA. To overcome this analytical issue, XRPD was used to investigate if budesonide in the nanoparticles is crystalline or amorphous (Fig. 5). The XRPD diffractogram of the pure drug showed the full crystalline peak patterns. For the physical mixture as well as for the nanoparticles, the unique peak patterns of budesonide can also be detected, even though due to the lower concentration the peak intensity is reduced. Thus, it can be concluded that the drug within the nanoparticles is in crystalline state.
In a previous study with an experimental colitis model (20), particles tend to preferentially accumulate in inflamed tissues, thus avoiding rapid elimination by diarrhea. In this study, the previously raised results can be confirmed with in-vivo imaging in (Fig. 6a). After 18 h of the administration of fluorescent labeled nanoparticles they can be found in higher quantities at the site of inflammation in the colitogenic group as compared to the healthy control group. This can further support the hypothesis that small sized particles can accumulate to a greater extent at the site of inflammation. These findings were further underlined by confocal fluorescence analysis of colon sections in (Fig. 6b). Similarly, the statistically analysis showed significant amount of nanoparticles in the inflamed areas. In the colon tissue of healthy mice, it was observed that the nanoparticles are dispersed in the whole GI tract; while in inflamed mice the particles were found to a greater extent at the site of inflammation. These data highlight the potential of nanoparticles for targeted mucosal drug delivery in the treatment of IBD.
Conclusion
The developed budesonide-loaded PLGA nanoparticles exhibited appropriate particles size and a biphasic release profile, resulting from efficient encapsulation of the active by the polymer in crystalline form. Accumulation of such particles at the site of inflammation as visualized by in vivo fluorescence imaging in a relevant mouse model, underscores the potential of nanoparticles for an improved therapy of IBD. As all materials and methods are well characterized and safe, such formulation appears mature enough for some subsequent clinical trial in IBD patients.
Abbreviations
- AFM:
-
Atomic force microscopy
- CD:
-
Crohn’s disease
- DSC:
-
Differential scanning calorimetry
- GIT:
-
Gastrointestinal tract
- IBD:
-
Inflammatory bowel disease
- O/W:
-
Oil/water
- PBS:
-
Phosphate buffer saline
- PDI:
-
Poly-dispersity index
- PLGA:
-
Poly(lactic-co-glycolic acid)
- PVA:
-
Poly-vinyl alcohol
- SD:
-
Standard deviation
- SEM:
-
Scanning electron microscope
- UC:
-
Ulcerative colitis
- XRD:
-
X-ray diffraction
References
Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347(6):417–29.
Head KA, Jurenka JS. Inflammatory bowel disease part 1: ulcerative colitis—pathophysiology and conventional and alternative treatment options. Altern Med Rev. 2003;8(3):247–83.
Shanahan F. Crohn’s disease. Lancet. 2002;359(9300):62–9.
Gramlich T, Petras RE. Pathology of inflammatory bowel disease. Semin Pediatr Surg. 2007;16(3):154–63.
Koutroubakis I, Manousos ON, Meuwissen SG, Pena AS. Environmental risk factors in inflammatory bowel disease. Hepatogastroenterology. 1996;43(8):381–93.
Nguyen GC, Torres EA, Regueiro M, Bromfield G, Bitton A, Stempak J, et al. Inflammatory bowel disease characteristics among African Americans, hispanics, and non-Hispanic Whites: characterization of a large north American cohort. Am J Gastroenterol. 2006;101:1012–23.
Fiorino G, Fries W, De La Rue SA, Malesci AC, Repici A, Danese S. New drug delivery systems in inflammatory bowel disease: MMX and tailored delivery to the gut. Curr Med Chem. 2010;17(17):1851–7.
Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369(9573):1641–57.
Kompella UB, Bandi N, Ayalasomayajula SP. Subconjunctival nano- and microparticles sustain retinal delivery of budesonide, a corticosteroid capable of inhibiting VEGF expression. Invest Ophthalmol Vis Sci. 2003;44:192–201.
Greenberg GR, Feagan BG, Martin F, Sutherland LR, Thomson ABR, et al. Oral budesonide for active Crohns-disease. N Engl J Med. 1994;331:836–41.
Lofberg R, Rutgeerts P, Malchow H, Lamers C, Danielsson A, Olaison G, et al. Budesonide prolongs time to relapse in ileal and ileocaecal Crohn’s disease. A placebo controlled one year study. Gut. 1996;39:82–6.
Hanauer SB. New steroids for IBD: progress report. Gut. 2002;51:182–3.
Meissner Y, Lamprecht A. Alternative drug delivery approaches for the therapy of inflammatory bowel disease. J Pharm Sci. 2008;97(8):2878–91.
Lamprecht A, Ubrich N, Yamamoto H, Schafer U, Takeuchi H, Maincent P, et al. Biodegradable nanoparticles for targeted drug delivery in treatment of inflammatory bowel disease. J Pharmacol Exp Ther. 2001;299(2):775–81.
Lamprecht A. IBD: selective nanoparticle adhesion can enhance colitis therapy. Nat Rev Gastroenterol Hepatol. 2010;7(6):311–2.
Nakase H, Okazaki K, Tabata Y, Uose S, Ohana M, Uchida K, et al. Development of an oral drug delivery system targeting immune-regulating cells in experimental inflammatory bowel disease: a new therapeutic strategy. J Pharmacol Exp Ther. 2000;292:15–21.
Lamprecht A, Yamamoto H, Takeuchi H, Kawashima Y. A pH-sensitive microsphere system for the colon delivery of tacrolimus containing nanoparticles. J Control Release. 2005;104(2):337–46.
Lamprecht A, Ubrich N, Yamamoto H, Schafer U, Takeuchi H, Lehr CM, et al. Design of rolipram-loaded nanoparticles: comparison of two preparation methods. J Control Release. 2001;71(3):297–306.
Lamprecht A, Yamamoto H, Ubrich N, Takeuchi H, Maincent P, Kawashima Y. FK506 microparticles mitigate experimental colitis with minor renal calcineurin suppression. Pharm Res. 2005;22(2):193–9.
Ali H, Weigmann B, Neurath MF, Collnot EM, Windbergs M, Lehr C-M. Budesonide loaded nanoparticles with pH-sensitive coating for improved mucosal targeting in mouse models of inflammatory bowel diseases. J Control Release [Internet]. June 10, 2014 [cited April 7, 2015];183:167–77. Retrieved from: http://www.sciencedirect.com/science/article/pii/S0168365914001849.
Weiss B, Schaefer UF, Zapp J, Lamprecht A, Stallmach A, Lehr C-M. Nanoparticles made of fluorescence-labelled Poly(L-lactide-co-glycolide): preparation, stability, and biocompatibility. J Nanosci Nanotechnol. 2006;6(9–10):3048–56.
Leroux JC, Allemann E, Doelker E, Gurny R. New approach for the preparation of nanoparticles by an emulsification-diffusion method. Eur J Pharm Biopharm. 1995;41:14–8.
Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–6.
Lamprecht A, Schafer U, Lehr CM. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18(6):788–93.
Jonsson G, Astrom A, Andersson P. Budesonide is metabolized by cytochrome P450 3A (CYP3A) enzymes in human liver. Drug Metab Dispos. 1995;23:137–42.
Sahana DK, Mittal G, Bhardwaj V, Kumar M. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J Pharm Sci. 2008;57:1530–42.
Tajber L, Corrigan DO, Corrigan OI, Healy AM. Spray drying of budesonide, formoterol fumarate and their composites—I. Physicochemical characterisation. Int J Pharm [Internet]. February 9, 2009 [cited May 5, 2015];367(1–2):79–85. Retrieved from: http://www.sciencedirect.com/science/article/pii/S0378517308006613.
Velaga SP, Berger R, Carlfors J. Supercritical fluids crystallization of budesonide and flunisolide. Pharm Res. 2002;19(10):1564–71.
ACKNOWLEDGMENTS AND DISCLOSURES
Dr. Salman Khan is thanked here for his technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ali, H., Weigmann, B., Collnot, EM. et al. Budesonide Loaded PLGA Nanoparticles for Targeting the Inflamed Intestinal Mucosa—Pharmaceutical Characterization and Fluorescence Imaging. Pharm Res 33, 1085–1092 (2016). https://doi.org/10.1007/s11095-015-1852-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1852-6