
ABSTRACT
Purpose
The present work reports rationalized development and characterization of solidified self-nanoemulsifying drug delivery system for oral delivery of combinatorial (tamoxifen and quercetin) therapeutic regimen.
Methods
Suitable oil for the preparation of liquid SNEDDS was selected based on the maximum saturation solubility of both the drugs while surfactant and co-surfactant were selected based on their emulsification ability. Extreme vertices mixture design and 32 full factorial design were implemented for optimization of liquid SNEDDS and concentration of solid carrier in lyophilization mixture. Finally, extensive characterization of the developed formulation was performed and in vitro cellular uptake was evaluated in Caco-2 cell culture model.
Results
Extreme vertices mixture design indicated the desirability of 0.663, corresponded to 40:30:30 w/w as optimum ratio of oil (Capmul® MCM), surfactant (Cremophor RH 40) and co-surfactant (Labrafil 1944CS) in liquid SNEDDS, which solubilized high amount of tamoxifen (10 mg/g) and quercetin (19.44 mg/g). A, 32 full factorial design revealed the optimum concentration of the selected solid carrier (Aerosil 200) of 5.24% w/w and 1.61, when measured in terms of total solid content and liquid SNEDDS: Aerosil 200 ratio, respectively. The developed formulation revealed instantaneous emulsification (in < 2 min), while maintaning all the quality attributes even after storage at accelerated stability condition for 6 months. Finally, the developed formulation revealed 9.63-fold and 8.44-fold higher Caco-2 uptake of tamoxifen and quercetin, respectively in comparison with free drug counterparts.
Conclusions
The developed formulation strategy revealed a great potential for oral delivery of combination drugs having utmost clinical relevance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Tamoxifen (Tmx), a selective estrogen receptor antagonist, is a drug of choice for the treatment of hormone receptor positive breast cancer at the multiple stages (1). Quercetin (QT), a pentaflavanol, acts as potential free radical scavenger (2,3) as well as an anti-proliferative agent (4–7). As a free radical scavenger it enhance antioxidant enzyme activity, inhibit mitochondrial lipid, and protein oxidation by scavenging the reactive oxygen species (ROS) (8). QT has been extensively explored to combat the liver damage against drug or chemical induced toxic free radicals (9–12). In addition, QT also possess anticancer properties owing to multiple mechanisms such as activation of carcinogenesis, modification of signal transduction pathways and inducing the apoptosis (13,14). Considering the dissimilar site of action of both these agents (15) a synergistic cytotoxicity profile could be sought for and the combination have been tested in hopes of exploiting the improved anticancer efficacy and reduced tumor angiogenesis (14).
Nevertheless, the complete therapeutic potential of Tmx-QT combination has not been yet explored owing to the poor biopharmaceutical properties of both the drugs. Oral deliverability of Tmx is limited by the precipitation of citrate salt in acidic pH of stomach, hepatic first pass extraction and P-gp efflux in the GIT tract (7,16,17). Extensive first pass hepatic metabolism of Tmx in liver leads to the generation of genotoxic metabolites and free radicals which ultimately lead to multiple symptoms of hepatotoxicity (18,19). Similarly, QT possess poor aqueous solubility and extensively metabolized in various segments of GI tract, which ultimately hampers its oral bioavailability and necessitate the higher dose to be administer (250 to 500 mg for three times/day) (20). In addition, the combination therapy is often associated with potential drug interactions, which could result into, altered pharmacokinetic/pharmacodynamics profiles of drug substances (21,22). Furthermore, the co-administration in different dosage forms may not end into similar biodistribution profile resulting into unpredictable and variable levels of drugs in tissue microenvironment. Hence, there lies a strong need to formulate a nanocarrier capable of co-encapsulating both the drugs in clinically relevant composition. Recently, a variety of novel drug delivery technologies are gaining increasing attention for co-encapsulation of cytotoxic drugs along with antioxidant (23), which include liposomes (24,25), polymeric nanoparticles (26–29), polymer drug conjugates (30,31), mesoporous silica nanoparticles (32) and dendrimers (33). Recently, we have reported the oral delivery of Tmx-QT, co-encapsulated in PLGA-NPs and found significant appreciation in oral bioavailability, antitumor efficacy and safety profile of Tmx (34). However, the efficient use of these systems is limited due to their complex manufacturing steps, high production cost and poor drug loading capacity, especially of antioxidants.
Self-nanoemulsifying drug delivery system (SNEDDS) represents one of the most popular, commercially worthwhile and effortlessly scalable approach to improve the deliverability of a variety of drugs via oral route (35). Drug loaded liquid SNEDDS, following oral administration, are spontaneously emulsified upon contact with gastrointestinal (GI) fluids and finally diluted in the differential micro-environment of gastrointestinal tract (GIT) and form surfactant stabilized oily droplets and micelles (droplet size <100 nm), which increase the solubility of drugs and prevents the pre-systemic metabolism and GI degradation (36). Furthermore chylomicrons assisted intestinal absorption of SNEDDS, drains the encapsulated drugs into the lymphatic vessels rather than directly in central compartment, thereby preventing their hepatic first pass metabolism and increase their oral bioavailability (22,37). Recently, Tmx loaded SNEDDS have been developed and patented by our group, which revealed 3.8-fold enhancement in oral bioavailability and significant appreciation in antitumor efficacy as compared to clinically used tamoxifen citrate (38). On the similar line of action, we have recently revealed about ~5-fold appreciation in bioavailability of QT, upon oral administration of QT-SNEDDS (39).
However, the traditional liquid SNEDDS have some practical limitations like lower drug stability especially in liquid from, unusual drug-drug or drug excipient interaction, interaction between liquid and capsule shell, high production cost and possibility of irreversible drugs/excipients precipitation at the lower storage temperature. Solidification of liquid SNEDDS has been widely implemented to overcome the drawbacks associated with the conventional liquid SNEDDS. Solid SNEDDS (s-SNEDDS) combine the advantages of conventional lipid based drug delivery system (i.e. enhanced solubility and bioavailability) with those of solid dosage forms (e.g. relatively lower production cost, convenience of process control, high stability and reproducibility) (40). Manual mixing of liquid SNEDDS with a solid carrier is widely employed method for the solidification (41). However, the implemented strategy is often associated with poor adsorption and desorption phenomenon owing to the contrasting physicochemical properties of isotropic mixture and solid carrier (42). These limitations can be efficiently circumvented by solidification of liquid SNEDDS in the presence of some solid carrier. Classically, such solidification could be materialized by either spray drying or lyophilization (40). Since spray drying is often associated with the limited yield, extensive process parameters and complexities of handling aqueous solutions, lyophilization has been preferred.
In the present work, a systematic optimization was carried out to prepare the liquid SNEDDS loaded with Tmx and QT (Tmx-QT-SNEDDS). Afterwards, a novel stepwise freeze-drying cycle was implemented for stabilization of liquid nanoemulsion onto an inert solid carrier and the same was achieved using exhaustive statistical optimization. In the course of extensive characterization and stability studies, we have nicely demonstrated that s-Tmx-QT-SNEDDS was able to retain all its quality attributes at various physiological and storage conditions. Finally, we demonstrated the intestinal epithelial permeability of s-Tmx-QT-SNEDDS by conducting cellular uptake studies in Caco-2 cells.
MATERIALS AND METHODS
Tmx (free base and citrate salt), QT (anhydrous), Pluronic F-68, Pluronic F-127, 1, 1-diphenyl-2-picrylhydrazyl (DPPH), mannitol, trehalose, Tween 20, Tween 40, Tween 60, Tween 80, trypsin-EDTA, coumarin-6 (C-6), Sodium dodecyl sulfate (SDS) and triton X-100 were purchased from Sigma, USA. Capmul® MCM EP, Captex® 355, Capmul® MCM-C8, Capmul MCM® C10, Capmul® MCM L8 and Captex® 200 P were procured as generous gift from Abitech Corporation, Janesville, USA. Sucrose, dextrose and isopropyl myristate were procured from Loba Chemie Pvt. Ltd., India. Plurol® Oleique, Paceol™, Gelucire®, Caproyl PGMC, Maisine 35-I and Labrafil® 1944 CS were obtained as gift samples from Gattefosse, France. Aerosil 200, Avicel PH 102 and Cremophor RH 40 were purchased from Evonik Degussa, Pvt. Ltd., India, FMC Bio Polymer, USA and BASF Chemical Company, USA respectively. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), antibiotics (Antibiotic-antimycotic solution) and Hanks’s balanced salt solution (HBSS) were purchased from PAA Laboratories GmbH, Austria. CellTiter 96® AQueous one solution cell proliferation assay kit (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS reagent) was purchased from Promega, USA. Alexa Fluor® 488 Phalloidin, Molecular Probes was procured from Life Technologies, Invitrogen, USA. Ethyl acetate (LR grade), acetonitrile (HPLC grade), methanol (HPLC grade) were purchased from Fischer Scientific, USA. Ultra-pure deionized water (SG water purification system, Barsbuttel, Germany) was used throughout all the experiments. All other reagents used were of analytical grade.
Solubility Studies
Saturation solubility of Tmx and QT in different oils was evaluated by the shake flask method (43). Briefly, an excess amount of drugs were added to different vials containing 0.5 g of oil and vortexed for 2 min to obtain the homogenous mixture. All the mixtures were incubated in shaker water bath (Lab Tech, Korea) operated at 50 strokes/min for 72 h at 37°C to attain the equilibrium. The resultant mixtures were centrifuged at 5,000 rpm for 5 min and supernatant was diluted with ethyl acetate followed by methanol and further analyzed for amount of drugs (Tmx and QT) by in-house developed and validated HPLC method. The mixture of acetonitrile, ammonium acetate buffer (10 mM pH 3.6) and methanol (32:48:20 v/v) was employed as the mobile phase, while the analysis was carried out by injecting 20 μl of samples to Shimadzu HPLC system equipped with Symmetry RP-18 column and SPD M-20A detector. Based on the solubility studies, Capmul MCM EP was selected as suitable oil for preparation of liquid SNEDDS.
Selection of Surfactants and Co-surfactant
Surfactants were screened for their emulsification ability according to the protocol reported in the literature with slight modifications (44). Briefly, the Capmul MCM EP (0.5 g) was mixed with equal weight of surfactants and vortexed for 2 min followed by warming at 40–45°C for 30 s. The resultant isotropic mixtures (200 mg) were diluted with deionized water (50 ml) to yield the fine emulsion. The emulsification ability of different surfactants was observed based on emulsification time required to obtain the fine emulsion. All the mixtures were allowed to equilibrate for 2 h and evaluated for their droplet size and polydispersity index (PDI) using Zeta Sizer (Malvern Instruments, UK) and % transmittance at 638 nm using microplate spectrophotometer; Bio-TEK, USA. Subsequently, screening of co-surfactant was carried out using the same method as described above. Briefly, the selected surfactant (0.3 g) was mixed with different co-surfactants (0.2 g) followed by the addition of oily phase (0.5 g). Later, screening of a specific co-surfactant was carried using the similar protocol as described above. Based on the above mentioned studies, Cremophor RH 40 and Labrafil 1944 CS were selected as suitable surfactant and co-surfactant, respectively.
Construction of Ternary Phase Diagrams
Different ratios of Capmul MCM EP, Cremophor RH 40 and Labrafil 1944 CS were plotted on ternary phase diagram in order to identify the self-emulsification region. In the ternary mixtures, the concentration of Capmul MCM EP, Cremophor RH 40 and Labrafil 1944 CS was varied from 20 to 70% (w/w), 30–80% (w/w) and 0–30% (w/w), respectively. Various compositions were prepared by altering the Labrafil 1944 CS concentration in the order of 5% while fixing the concentration of Capmul MCM EP and sufficient Cremophor RH 40 was added to the resulting mixture to make it 100%. The prepared compositions were then subjected to dilution (200 mg in 50 ml) with distilled water and evaluated for % transmittance, droplet size and PDI. The compositions which formed submicron emulsions with droplet size <100 nm and PDI <0.2, were plotted on the pseudoternary phase diagram and considered as to be the part of self-emulsification region.
Formulation Optimization of Tmx-QT-SNEDDS
A mix D-optimal mixture design of constrained region was employed to optimize the liquid Tmx-QT-SNEDDS composition, which was selected based on maximum loading of QT and droplet size and PDI of submicron emulsion resulted after dilution. A methodical optimization of various dependent variables (QT loading, droplet size and PDI of diluted emulsion) was performed by varying the percentage of ternary component in liquid SNEDDS, which was considered as the independent variables. The levels of the three independent variables and content of Tmx were selected based on the preliminary studies carried out before applying the experimental design. The response surface methodology of three component system was performed with the constraints (0.2 ≤ C (Capmul MCM EP) ≤0.7; 0.3 ≤ A (Cremophor RH 40) ≤0.8; 0.0 ≤ B (Labrafil 1944 CS) ≤0.3) of the all the independent variables. Design Expert software (Version 7.1, 2007, Stat-Ease Inc., Minneapolis, MN, USA) was employed for the optimization study and contours plots of all the 3 responses (droplet size, PDI and QT content) were also constructed. The responses of all the 16 runs were fitted in the quadratic polynomial model. The polynomial equations were generated for each response using the Design Expert software. The appropriate fitting model for each response was selected based on the comparison of various statistical parameters such as R2, Sequential Model Sum of Squares, Lack of fit and partial sum of square was provided by the analysis of variance (ANOVA).
Preparation of s-Tmx-QT-SNEDDS
s-Tmx-QT-SNEDDS was prepared by lyophilization of Tmx-QT-SNEDDS in the presence of a solid carrier. Briefly, Tmx-QT-SNEDDS was diluted in the minimum quantity of deionized water and thoroughly mixed with a suitable solid carrier. The resultant mixture was then allowed to stand for 15 min to attain the equilibrium and subsequently lyophilized by the step-wise freeze-drying cycle previously patented by our group (45).
Selection of Solid Carrier
Tmx-QT-SNEDDS was diluted with minimum volume of deionized water and thoroughly mixed with different solid carriers (sucrose, trehalose, mannitol, Aerosol 200, Avicel). Resultant mixtures were allowed to stand for 15 min and finally lyophilized by step-wise freeze drying cycle. Suitable solid carrier for lyophilization of Tmx-QT-SNEDDS was selected based on the characteristics of lyophilized SNEDDS upon dilution with deionized water, which were evaluated as a function of reconstitution time, droplet size, PDI of resultant submicron emulsion and Sf/Si ratio (ratio of droplet size after and before reconstitution). During the selection of a solid carrier, total solid content and ratio of solid carrier: liquid SNEDDS were fixed at 7.5% and 1:2 w/w respectively.
Optimization of Concentration of Solid Carrier
Response surface methodology (RSM) was employed to optimize the concentration of solid carrier for lyophilization of Tmx-QT-SNEDDS. A 32 full factorial design was applied to design the experiments. Concentration of solid carrier in lyophilization mixture was optimized as a function of total solid content (%) and weight ratio of Tmx-QT-SNEDDS: solid carrier, which was selected as the independent variables while time to release 75% of drugs (TTmx75 and TQT75), droplet size and PDI after reconstitution were kept as dependent variables. A series of nine (S1 to S9) lyophilization mixtures (See Supplementary Material Table S I) were lyophilized using a method as described above. The responses of time to release 75% of drugs (TTmx75 and TQT75) from lyophilized SNEDDS was evaluated by performing in vitro drug release in shaker water bath operated at 37°C and 100 strokes/min in 25 ml of deionized water containing Tween 80 (5% w/v). Dialysis bag of MWCO 12 kD was employed to study the in vitro release pattern of various samples and an in-house validated HPLC method was employed to determine the amount of both the drugs in the release medium. Additionally, responses of droplet size and PDI of resultant emulsions obtained after dilution of lyophilized SNEDDS were also evaluated. Polynomial equations and contour plots were generated for each response using the Design Expert Software (Stat Ease Inc. USA, version 7.1.4). Finally, the optimum composition of lyophilization mixture was selected based on desirability value.
Characterization of s-Tmx-QT-SNEDDS
Reconstitution Behavior
The reconstitution behavior of s-Tmx-QT-SNEDDS was evaluated as a function of reconstitution time upon dilution with simulated gastric fluid (SGF; pH 1.2), droplet size and PDI of diluted emulsion. Briefly, SGF (pH 1.2) (50 ml) was added to s-Tmx-QT-SNEDDS (350 mg) and time required for the formation of submicron emulsion was evaluated. Further, the same mixture was allowed to stand for 2 h to attain equilibrium and passed through 0.22 μm membrane filter to obtain the clear nanoemulsion, which was finally evaluated for droplet size and PDI using the Zeta sizer (Malvern Instrument, UK).
Drug Content
A, 10 ml of methanol was added to 500 mg of the s-Tmx-QT-SNEDDS and the mixture was vortexed for 15 min to allow complete extraction of drugs in methanol. The resultant mixture was centrifuged at 10,000 rpm for 10 min. The supernatant was suitably diluted with methanol and analyzed by the in-house validated HPLC method.
Surface Morphology
Scanning electron microscope (SEM, S-3,400 N, Hitachi, Japan) was employed for evaluating the surface morphology of s-Tmx-QT-SNEDDS. Briefly, samples (s-Tmx-QT-SNEDDS, pure solid carrier, free Tmx base and free QT) were placed on double-sided bi-adhesive carbon tape previously adhered to metallic stub. Subsequently, samples were gold coated using the Hitachi Ion Sputter for 30 s at 15 mA to obtain a conducting surface and finally visualized under scanning electron microscopy (SEM).
Solid State Characterization
Differential Scanning Calorimetry (DSC)
The thermal characteristics and nature of the drugs in s-Tmx-QT-SNEDDS were investigated by DSC. Analysis was carried out with pure Tmx base, pure QT, solid carrier, s-Tmx-QT-SNEDDS and physical mixture using the Mettler Toledo differential scanning calorimeter calibrated with indium standard. All the measurements were performed at the heating rate of 10°C/min from 0 to 350°C.
X-ray Diffraction (XRD)
The powder crystallinity of s-Tmx-QT-SNEDDS was investigated by performing the XRD analysis of pure Tmx, pure QT, physical mixture, solid carrier and s-Tmx-QT-SNEDDS using the X-ray diffractometer (Bruker D8 advance, Bruker, Germany). Measurements were performed at a voltage of 40 kV and 25 mA. The scanned angle was set from 3° ≤ 2θ ≥ 40°, and samples were scanned at a rate of 2° min−1.
Stability Studies
Stability in Simulated GI Fluids
s-Tmx-QT-SNEDDS was diluted with simulated gastric fluid (SGF; pH 1.2) and simulated intestinal fluid (SIF; pH 6.8) to assess their stability and possibility of drugs precipitation at different pH and enzymatic condition of GI tract. Briefly, 500 mg of s-Tmx-QT-SNEDDS was added into the 50 ml of simulated GI fluids and the mixture was incubated for 2 h in case of SGF (pH 1.2) while for 6 h in case of SIF (pH 6.8). Following the completion of incubation period, diluted formulation was evaluated for the droplet size and PDI using the Zeta sizer (Malvern, UK).
Stability After Dilution to Different Folds
s-Tmx-QT-SNEDDS was evaluated for the robustness to dilution at different folds (200, 400, 600 and 800 fold) with simulated gastric fluid (SGF). Briefly, 500 mg of s-Tmx-QT-SNEDDS was diluted with SGF to different folds (200, 400, 600 and 800 fold). All the diluted samples were kept for 2 h to attain the equilibrium and droplet size and PDI of the nanoemulsion was subsequently evaluated by Zeta sizer (Malvern, UK).
Accelerated Stability Studies
s-Tmx-QT-SNEDDS was evaluated for the accelerated stability for 6 month according to the ICH guideline (46). Briefly, different samples of s-Tmx-QT-SNEDDS were transferred to 5 ml glass vials sealed with plastic caps and were kept in stability chamber maintained at temperature and relative humidity of 40°C and 75%, respectively. Different samples were taken out at the predetermined time interval (1, 3 and 6 months) and characterized for physical appearance, drug content and reconstitution behavior.
In Vitro Release Studies
In vitro release profile of s-Tmx-QT-SNEDDS was investigated by dialysis bag method using the two dialysis bags of different molecular weight cut off (1 kD and12 kD) (39,47). In vitro release of s-Tmx-QT-SNEDDS and equivalent free drug combination was carried out for 2 h in SGF followed by 6 h in SIF. s-Tmx-QT-SNEDDS and free drug mixture equivalent to 2 mg of Tmx/4 mg of QT were dispersed in 1 ml of SGF (pH 1.2) and filled in dialysis bags. The release of Tmx and QT was performed in shaker water bath operated at 37°C and 100 strokes/min in 25 ml of release media containing 2.5% w/v Tween 80. Samples (1 ml) were withdrawn at the predetermined time intervals and replaced with equal volume of fresh medium each time to maintain the sink condition. The cumulative amount of drugs released was analyzed by the in-house validated HPLC method.
Caco-2 Cell Culture Experiments
Cell Culture
Caco-2 cells (ATCC, Manassas, VA, USA) were grown in tissue culture flasks (75 cm2) and maintained under 5% CO2 atmosphere at 37°C. The growth medium comprised of Dulbecco’s Modified Eagle’s culture medium (DMEM), 20% fetal bovine serum (FBS), 100 IU/ml penicillin and 100 μg/ml streptomycin (PAA, Austria). The growth medium was changed on every alternate day. The cultured cells were trypsinized with 0.25% trypsin-EDTA solution (Sigma, USA) once 90% confluent. The harvested cells were then used for subsequent studies.
Qualitative Uptake
The harvested Caco-2 cells were seeded at a density of 3,00,000 cells/well in 6 well plate (Costars, Corning Inc., NY, USA), incubated overnight for cell attachment and employed for qualitative uptake studies. Briefly, C-6, Tmx and QT loaded solid SNEDDS (s-C6-Tmx-QT-SNEDDS) was prepared by solubilizing 1 mg of C-6 in liquid Tmx-QT-SNEDDS, which were then lyophilized following the same protocol as described earlier. s-C6-Tmx-QT-SNEDDS was suitably reconstituted with deionized water and allowed to stand for 2 h and filter sterilized. Formulations equivalent to 1 μg/ml of C-6 were added to each plate and incubated for 1 h. Following the incubation period, medium was removed and cells were washed twice with the PBS (pH 7.4) and observed under the confocal laser-scanning microscope (CLSM) (Olympus FV1000).
Quantitative Cell Uptake
Caco-2 cells were seeded at a density of 1,00,000 cells/well in 24 well plate containing cell culture inserts filter paper pore size 0.4 μm (Millicell®, Millipore®, USA) allowed to grow into monolayers over a period of 21 days. The growth medium form the basolateral side was changed on every alternate day. Transepithelial electrical resistance (TEER) values were also measured on every second day of media replacement as a marker of monolayer integrity using Millicell ERS-2 Volt-Ohm Meter, Merck, USA. Apical uptake of free drugs (free Tmx and free QT), their combination (free Tmx + QT 1:2 w/w) and diluted s-Tmx-QT-SNEDDS was carried out in the time and concentration dependent manner. Briefly, confluent Caco-2 monolayer was exposed apically to fresh medium supplemented with varying concentrations of formulations (equivalent to 0.5, 1, 5, 10 μg/ml Tmx) and further incubated for 1 h. Subsequently, cell culture medium from the apical chamber was removed and monolayers washed twice with PBS (pH 7.4). Cells on the filter paper were then scrapped and lysed with the 0.1% Triton X-100 followed by washing with methanol to completely solubilize the internalized drug. The cell lysate was centrifuged at 21,000 rpm for 10 min and obtained supernatant was subjected to HPLC analysis for quantification of internalized drugs. Similarly, time dependent apical uptake was also carried out by exposing formulations (equivalent to 1 μg/ml Tmx) to the apical surface of Caco-2 monolayers and incubated for varying time (0.5, 1 and 2 h).
Caco-2 Cells Cytotoxicity
Safety of s-Tmx-QT-SNEDDS and free drugs on monolayers was evaluated as a function of cell viability (MTS assay) and monolayer integrity (TEER measurement)(48,49). Briefly, varying concentration of reconstituted s-Tmx-QT-SNEDDS and free drug compounds (1, 5, 10 μg/ml) were exposed to Caco-2 monolayer apically and incubated for 3 h. Subsequent to the incubation period, monolayers were subjected to cell viability assay using CellTiter 96® AQueous one solution cell proliferation assay kit (Promega, USA). Briefly, 20 μl of the MTS reagent was added to each well and re-incubated for 3–4 h to allow the formation of water-soluble formazan product. The optical density of the resultant supernatant solution was then measured at 490 nm using an ELISA plate reader (BioTek, USA). Cell viability was assessed using following formula:
Concomitantly, TEER values of Caco-2 monolayers were also measured. In separate set of experiments, the effect of formulations on the actin filaments of developed monolayer was also evaluated using Alexa Fluor® 488 phalloidin staining (Invitrogen, USA) following manufacturer’s protocol. Upon completion of the incubation period, monolayers were washed three times with PBS (pH 7.4), fixed with glutaraldehyde (2.5% v/v, 10 min). Monolayers were then washed twice with PBS (pH 7.4) and permeabilized with Triton X-100 (0.1% v/v, 2 min). Finally, washed again with PBS (pH 7.4) and incubated with Alexa Fluor® 488 phalloidin (1 IU/monolayer, 30 min) in dark at room temperature. The stained monolayers were then washed and visualized under the CLSM (Olympus FV1000).
Statistical Analysis
All the results were expressed as mean ± standard deviation (SD). Statistical analysis was performed with Prism Graph software. The level of significance was calculated by ANOVA followed by Tukey–Kramer multiple comparison test. P <0.05 was considered as statistically significant difference.
RESULTS
Solubility Studies
Figure 1 shows the saturation solubility of Tmx and QT in various natural and synthetic oils. Maximum solubility of Tmx was estimated in Capmul MCM C10 (133.32 ± 7.32 mg/g) followed by Capmul MCM EP, whereas QT possess maximum solubility (31.93 ± 2.34 mg/g) in Capmul MCM EP. In view of the limited dose requirement of Tmx (10–20 mg/day) and high dose requirement of QT (100–200 mg/day), Capmul MCM EP was selected as the suitable oil for the preparation of Tmx-QT-SNEDDS.

Solubility studies of tamoxifen-quercetin combination in different oils. Each data point represents mean ± SD (n = 6).
Selection of Surfactants and Co-Surfactant
The emulsification ability of different surfactants and co-surfactants to emulsify for Capmul MCM EP is shown in Table I. Translucent emulsions (transmittance <80%) with droplet size >200 nm was produced in case of Tween 20, Tween 80, Gelucire, Tween 40 and Tween 60. Although Pluronic F-68 and F-127 produced, emulsion having good droplet size (145.8 ± 10.45 nm and 42.43 ± 5.56 nm) and PDI (0.223 ± 0.178 and 0.259 ± 0.158) but it required relatively long time (5–10 min) for self-emulsification. Among all the surfactants, Cremophor RH 40 instantaneously (in < 2 min) resulted into emulsion with desired droplet size, PDI and transmittance (63.28 ± 12.40 nm, 0.205 ± 0.110 and 99.25 ± 0.12%), thus selected for further studies. All the co-surfactants produced transparent submicron emulsions with good droplet size (< 200 nm) with Capmul MCM EP/Cremophor RH 40 based system, but Labrafil 1944 CS produced submicron emulsion with maximum transmittance (98.23 ± 1.34%) and narrow droplet size distribution (0.096 ± 0.023) (Table I). Thus, Labrafil 1944 CS was selected as the suitable co-surfactant for Capmul MCM EP/Cremophor RH 40 based system.
Construction of Ternary Phase Diagrams
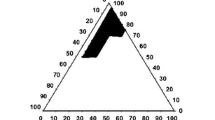
Figure 2a and b represents the explored and obtained self-emulsifying region, respectively in the ternary phase diagram of Capmul MCM EP-Cremophor RH 40-Labrafil 1944 CS based system. Ternary phase diagram of Capmul MCM EP-Cremophor RH 40-Labrafil 1944 CS based system revealed a larger self-emulsification area that occupied nearly entire space within the explored region. Nearly all the compositions in explored concentration range (Capmul MCM EP 20–70%; Cremophor RH 40 30–80%; Labrafil 1944 CS 0–30%) except some which possess Cremophor RH 40 > 80% w/w or Labrafil 1944 CS <10%, produced the nanoemulsions with droplet size < 200 nm and narrow PDI (< 0.2). Furthermore, at a fixed oil concentration (30, 40, 50% w/w), a significant decrease (p < 0.05) in droplet size of emulsion was observed upon increasing the concentration of Labrafil 1944 CS until 15% w/w. Upon further increasing the concentration of Labrafil 1944 CS to 30% w/w, a significant increase (p < 0.05) in the droplet size was observed. On the other hand, a continuous improvement in PDI of the resultant nanoemulsion was noted upon increasing the concentration of Labrafil 1944 CS from 0 to 30% w/w (See Supplementary Material Fig. S1).

Construction of ternary phase diagram and identification of extreme vertices design field in the three-component system of Capmul MCM EP-Cremophor RH 40-Labrafil 1944 CS (a) explored self-emulsifying region (b) obtained self-emulsifying region (c) extreme vertices design field with the boundary limitations and explored experimental points.
Formulation Optimization of Tmx-QT-SNEDDS
Figure 2c shows the compositions predicted by the extreme vertices design of 3-component system. The responses of sixteen Tmx-QT-SNEDDS batches at a fixed Tmx loading (10 mg/g) and their statistical analysis are shown in Table II and Table III, respectively. All the responses were separately fitted into various polynomial models and the model with the highest R2 value was considered as, fitting model (Table II).
Effect of the Formulation Components on Dependent Variables
Equations 1, 2 and 3 represents the polynomial equations for analysis of QT loading, droplet size and PDI of resultant emulsion after dilution of Tmx-QT-SNEDDS, respectively.
Where A, B and C are the concentration of Cremophor RH 40, Labrafil 1944 CS and Capmul MCM EP, respectively. ANOVA results confirmed the adequacy of the linear model (Model Prob > F is less than 0.05) for QT loading in Tmx-QT-SNEDDS, whereas quadratic and cubic model was found to be satisfactory for prediction of droplet size and PDI of emulsion resulted after the dilution of Tmx-QT-SNEDDS. Countour plots for the analysis of QT loading, droplet size and PDI of Tmx-QT-SNEDDS are shown in Fig. 3. All the independent variables were found to have the significant (p < 0.05) effect on QT loading in Tmx-QT-SNEDDS, where contribution of Capmul MCM EP was found to be maximum followed by Cremophor RH 40 and Labrafil 1944CS. A proportionate increase in QT loading was observed upon increasing the concentration of Capmul MCM EP, while the former was decreased upon decreasing the concentration of Cremophor RH 40 at a fixed concentration of Capmul MCM EP (Fig. 3a). As evident from Eq. 2, all the independent variables, individually have the significant effect (p < 0.05) whereas their combination showed the insignificant effect (p > 0.1) on droplet size of emulsion resulted after dilution of Tmx-QT-SNEDDS. A significant increase (p < 0.05) in the droplet size was observed upon increasing the concentration of Capmul MCM EP, whereas the same was also increased upon decreasing the concentration of surfactant at a fixed concentration of Capmul MCM EP (Fig. 3b). As evident from Eq. 3, the PDI of emulsion resulted after dilution of Tmx-QT-SNEDDS depends upon the complex multiple factors. At a fixed concentration of Capmul MCM EP, a significant decrease in PDI was observed, upon increasing the concentration of Labrafil 1944 CS, whereas it was increased upon increasing the concentration of the Cremophor RH 40 (Fig. 3c). Validation of polynomial equations was also performed by selecting the 5 arbitrary ratios of Capmul MCM EP, Cremophor RH 40 and Labrafil 1944 CS, from the constrained region (Fig. 2c). Low magnitudes of error as well as the significant values of R2 indicated that the applied model is predictable is nature and is valid (Data not shown).

Formulation optimization of liquid SNEDDS of Tmx and QT; contour plots revealing the effect of independent variables on (a) QT loading, (b) droplet size and (c) PDI; (d) desirability plot (e) overlay plot for numerical and graphical optimization. All the experiments were conducted in presence of Tmx (10 mg/g of formulation).
Desirability and Overlay Plot
Various criterias were considered to geneate numerical and graphical conditions for the optmization of Tmx-QT-SNEDDS compostion (See Supplementary Material Table S II). Fig. 3d shows the desirability graph, which was plotted for the first solution found via the numerical optmization and desirabilty value corresponded to be 0.663. Based on the desirabilty value, Tmx-QT-SNEDDS containing Capmul MCM EP, Cremophor RH 40 and Labrafil 1944 CS in the ratio of 40:30:30 was selected as the optmized compostion. Figure 3e shows the overlay plot, generated after selecting the graphical ranges of various response. Polynomial equations were employed to predict the data of these responses and various responses were found to be 79.80 nm, 0.0781 and 19.44 mg/g for droplet size, PDI and QT loading, respectively.
Preparation of s-Tmx-QT-SNEDDS
Selection of Suitable Solid Carrier
Table IV shows the characteristics of lyophilized SNEDDS and emulsions resulted after reconstitution of lyophilized SNEDDS, which were freeze-dried in the presence of various solid carriers. A crushed or collapsed cake, which was difficult to reconstitute (>5 min), was produced in case of water-soluble sugars such as sucrose, mannitol and trehalose (See Supplementary Material Fig. S2). Further, a significant increase (p < 0.05) in droplet size and PDI of emulsion was observed after reconstitution of solid SNEDDS. On the other hand, free flowing s-SNEDDS, which was able to reconstitute within 5 min and maintained all the quality attributes after reconstitution, were obtained after lyophilization of Tmx-QT-SNEDDS in the presence of water insoluble carriers (Aerosil 200 and Avicel). Among all the solid carriers screened, Aerosil 200 resulted in free flowing powder, which was reconstituted by mere shaking (< 2 min) and resulted in emulsion with better droplet size and PDI, thus selected for further screening.
Optimization of solid carrier concentration
The responses of nine batches of solidified SNEDDS, which were lyophilized in the presence of varying total solid content and SNEDDS: Aerosil 200 ratio, have been summarized in Table V. Statistical parameters of different responses are summarized in Table VI. All the responses were separately fitted into the different polynomial model and the model with the highest R2 was considered as the fitting model (Table VI).
Effect of lyophilization mixture on various responses of s-Tmx-QT-SNEDDS
Statistical parameters for various responses of solidified SNEDDS are presented in Table VI. ANOVA results of droplet size indicated the suitability of two factor interaction (2FI) model wheres linear model was found to be suitable for PDI, TTmx75, TQT75 (Model Prob. > F is less than 0.05). Analysis of various responses of s-SNEDDS are presented in the form of polynomial equations (Eq. 4–7).
where A and B are the total solid content (TOS) (%) and SNEDDS: Aerosil, respectively. As clearly predicted by polynomial equations, both the indepenent variables (total solid content and liquid SNEDDS: Aerosil 200 ratio) have significant effect on the all quality attributes of s-SNEDDS (TTmx75, TQT75, droplet size and PDI). Response surface plots of various responses as the function of total solid content and liquid SNEDDS: Aerosil 200 ratio are presented in Fig. 4. All the batches of s-SNEDDS revealed significantly higher release of QT in comparison with Tmx. At a constant total solid content, a proportionate increase in TTmx75 and TQT75 was observed upon increasing the proportion of the liquid SNEDDS in the lyophilization mixture. On the other hand, release of both the drugs was increased upon increasing the total solid content in the lyophilization mixture, while keeping the liquid SNEDDS: Aerosil 200 ratio constant (Fig. 4a and c). Furthermore, droplet size of nanoemulsion resulted upon dilution of solidified SNEDDS, was also increased proportionately with increase in liquid SNEDDS: Aerosil 200 ratio (Fig. 4b). Similar findings were also observed for PDI, leading to proportionate increase with total solid content and liquid SNEDDS: Aerosil 200 ratio (Fig. 4d). Validation of polynomial equations was also performed by selecting the 5 arbitrary total solid content and liquid SNEDDS: Aerosil 200 ratio in the lyophilization mixture. Significant values of R2 and low magnitudes of error indicated that the applied model is predictable in nature and is valid (Data not shown).

Formulation optimization of solid SNEDDS of Tmx and QT; contour plots for the analysis of (a) TTmx75 (b) droplet size (c) TQT75 (D) PDI; (e) desirability (f) overlay plot for the numerical and graphical optimization of concentration of aerosil 200 for lyophilization.
Desirability and Overlay Plots
Various criterias were applied to genrate the optimum numerical and graphical conditions for the optmization of concentration of solid carrier in the lyophillization mixture (See Supplementary Material Table S III). Figure 4e and f shows the desirability and overlay plots, plotted for the first solution found via the numerical and graphical optmization for the concentration of Aerosil 200 in lyophillization, respectively. Numerical optmization indicated a desirability value of 0.338 that corresponded to about 5.24% w/w and 1.61 of total solid conetent and liquid SNEDDS: Aerosil 200 ratio in the lyophillization mixture, respectively. Polynomial equations were employed to predict the data of various responces, which were found to be 85.00 nm, 0.161, 44.43 min and 40.99 min for droplet size, PDI, TTmx75, TQT75, respectively.
Characterization of s-Tmx-QT-SNEDDS
Reconstitution of s-Tmx-QT-SNEDDS
Table VII shows the drugs content (%) of s-Tmx-QT-SNEDDS and characteristics of nanoemulsion resulted after dilution with SGF (pH 1.2). s-Tmx-QT-SNEDDS revealed higher (>95%) content of both the drugs and reconstituted in shorter period of time (100 ± 2 s) with the minimal agitation to form nanoemulsion with smaller droplet size (<100 nm) and narrow PDI (< 0.2).
Surface Morphology
Figure 5 shows the SEM photograph of pure Tmx base, pure QT, Aerosil 200 and s-Tmx-QT-SNEDDS. Discrete crystalline particles was evident in the SEM photograph of pure Tmx and QT (Fig. 5a and b) whereas of Aerosil 200 exhibited amorphous particles with highly porous surface (Fig. 5c). Interestingly, s-Tmx-QT-SNEDDS revealed relatively smoother surface with pores filled with SNEDDS particles. (Figure 5d).

SEM photograph of (a) pure Tmx (b) pure QT (c) Aerosil 200 (d) s-Tmx-QT-SNEDDS. (Inset images in Fig.11c (5,000 X) and 11 D (5000X) represents the SEM photograph of individual particles).
Solid-State Characterization of s-Tmx-QT-SNEDDS
DSC Analysis
An overlay of DSC curves of pure Tmx, pure QT, Aerosil 200, physical mixtures and s-Tmx-QT-SNEDDS is presented in Fig. 6a. Pure Tmx and pure QT showed a sharp endothermic peak at about 100°C and 315°C, respectively, whereas no peak was present in DSC thermogram of Aerosil 200 over the entire range of tested temperature. Interestingly, no endothermic peaks of either drug were appeared in DSC thermogram of s-Tmx-QT-SNEDDS, whereas both the peaks, although with the reduced intensity were present in the DSC thermogram of physical mixture.

Solid state characterization (a) Overlay of the DSC pattern (b) Overlay of XRD pattern of free Tmx, free QT, physical mixture, Aerosil 200, s-Tmx-QT-SNEDDS.
XRD Analysis
Figure 6b depicts an overlay of XRD patterns of free drugs, Aerosil 200, physical mixture and s-Tmx-QT-SNEDDS. XRD pattern of free Tmx and QT exhibited the characteristics sharp peaks at multiple diffraction angles, whereas no peak was observed in XRD pattern of Aerosil 200. Notably, no peak characteristic of either Tmx or QT was recorded in the XRD pattern of s-Tmx-QT-SNEDDS whereas physical mixture showed characteristic crystalline peaks for both the drugs.
Stability Studies
Stability in SGF and SIF
Table VIII shows characteristics of nanoemulsions resulted after reconstitution of s-Tmx-QT-SNEDDS with simulated GI fluids (SGF pH 1.2; SIF pH 6.8). In both the condition reconstitution was achieved in <2 min with mere shaking and resulted into nanoemulsions with transmittance > 90%. Further, no sign of phase separation or drug precipitation or turbidity was observed in SGF as well as in SIF after the stipulated time.
Stability After Dilution to Different Folds
Table IX shows characteristics of nanoemulsions resulted after reconstitution of s-Tmx-QT-SNEDDS with SGF (pH 1.2) in varying folds of dilution (200, 400, 600 and 800 folds). Nanoemulsions resulted from reconstitution of s-Tmx-QT-SNEDDS were found to be robust to all dilution and did not show any sign of phase separation and drug precipitation.
Accelerated Stability Studies
Table X shows the characteristics of nanoemulsion resulted upon reconstitution of s-Tmx-QT-SNEDDS, stored at accelerated storage condition (40°C/75% RH) for 6 months. All the formulation attributes of s-Tmx-QT-SNEDDS and nanoemulsion resulted upon its reconstitution was retained after storage at the accelerated condition (40°C/75% RH) for stipulated time.
In Vitro Release of s-Tmx-QT-SNEDDS
Figure 7 shows the in vitro release profile of Tmx (Fig. 7a) and QT (Fig. 7b) from the s-Tmx-QT-SNEDDS in simulated GI fluids. An insignificant difference (p > 0.05) in the cumulative release of free drug mixture from both the dialysis membrane (MWCO 1 kD and 12 kD) was observed at all the time points, while the same showed an significant difference (p < 0.001) in case of s-Tmx-QT-SNEDDS. After 8 h, > 95% of Tmx and QT was released from s-Tmx-QT-SNEDDS through the dialysis membrane of MWCO 12 kD whereas only 13.89 ± 2.34% of Tmx and 15.14 ± 3.23% of QT were released from s-Tmx-QT-SNEDDS through the dialysis bag of MWCO 1 kD, respectively.

In vitro release profile of s-Tmx-QT-SNEDDS and free drug mixture of Tmx and QT (1:2 w/w) from different dialysis membrane (a) release profile of Tmx (b) release profile of QT (each data point is represented as mean ± SD, n = 6).
Caco-2 Cell Culture Experiments
Qualitative Cell Uptake
Figure 8 represents CLSM images of Caco-2 cells incubated with free C-6 and C6-Tmx-QT-SNEDDS (1μg/ml, 1 h). As evident, significantly higher fluorescence was visible inside the cells following 1 h incubation with C6-Tmx-QT-SNEDDS, indicative of proficient uptake of SNEDDS by Caco-2 cells.

Uptake of C6-Tmx-QT-SNEDDS and free C-6 and by Caco-2 cells (a) and (c) represents the green fluorescence of C-6 while (b) and (d) corresponding DIC images. Scale bar 20 μm.
Quantitative Cell Uptake
Confluency of the Caco-2 monolayer was monitored as a function of TEER, which was found to be in the range of 900–1,100 Ω /cm2. The concentration and time dependent apical uptake was observed in case of both free drugs and s-Tmx-QT-SNEDDS; however, the magnitude of uptake was significantly higher in later case (Fig. 9). Significant increase in the apical uptake was observed upon increasing the concentration upto 1 μg/ml in both the cases beyond which a plateau phase was observed (Fig 9a). Time dependent studies further revealed maximum uptake till 1 h in both the cases (Fig. 9b) with significant decrease (p < 0.05) in the uptake efficiency upon further increasing the incubation time till 2 h. Of note, incubation of free drug combination (free Tmx + free QT, 1:2 w/w) with Caco-2 monolayer revealed ~2-fold higher uptake of Tmx as compared to free Tmx, while that of QT remain unaffected. Interestingly, s-Tmx-QT-SNEDDS revealed 9.63- and 8.44-fold higher uptake of Tmx and QT in comparison with free Tmx and free QT, respectively.

Cellular uptake of free drugs, their combination and SNEDDS in Caco-2 cells monolayer (a) concentration dependent uptake (b) time dependent uptake. Each data point represented as mean ± SD (n = 4). Free QT was added in double concentration as compared to free Tmx.
Caco-2 Cell Cytotoxicity
The obvious toxicity concern associated with increased uptake of SNEDDS formulation was addressed by evaluating the effect of formulations on viability and integrity of Caco-2 monolayers. The viability of Caco-2 cells in monolayer was found to be >90% in all the cases and no significant (p > 0.05) difference was observed among free drugs, their combination and s-Tmx-QT-SNEDDS (Fig. 10a). Further, insignificant changes (p > 0.05) in the integrity of Caco-2 monolayers was observed when measured as a function of alterations in TEER values (Fig. 10b). Notably, about ~55% reduction in TEER values was observed in case of positive control (SDS 0.1% w/v). The results of TEER values were further corroborated by actin visualization. Continuous band architecture around the periphery of cells, referred as actin rings, was observed in case of control, free drug combination and SNEDDS in contrast to disruption of actin rings with cell detachment in case of positive control (Fig.10c).

Effect of free drugs and s-Tmx-QT-SNEDDS on Caco-2 cells (a) % cell viability of Caco-2 cells (b) alterations in TEER values (b) actin visualization.

Schematic representation for method of preparation of s-Tmx-QT-SNEDDS.
DISCUSSION
The present investigation discloses the statistical design, development and characterization of dual drug loaded solid SNEDDS. Subsequent to oral administration, the said formulation is spontaneously reconstituted upon contact with GI fluids and forms nanoemulsion with droplet size < 100 nm. The formulation strategy included the solubilization of both the drugs (Tmx and QT) in appropriate oil followed by the addition of appropriate surfactant and co-surfactant. The resultant mixture was heated at 40–45°C for 30 s to form an isotropic liquid Tmx-QT-SNEDDS, which was finally lyophilized in the presence of solid carrier (Aerosil 200) (Fig. 11).
Selection of suitable oil is critical for the formulation of liquid SNEDDS, as it increases the overall pay load of lipophilic or poorly aqueous soluble drugs and precisely absorbed via the lymphatic pathway in GI tract, thereby prevents GI and first pass metabolism (37,50). As per our knowledge, this is the first report where SNEDDS have been employed for solubilization of dual drug within a single carrier system. The same was achieved by selecting a suitable oil which have propensity to solubilize both the drugs in appreciable amount (36). Tmx, owing to its higher log P (~7) in comparison with QT (~1.8), exhibited significantly higher solubility in all the tested oils in comparison with QT (Fig. 1). Furthermore, recommended oral dose of QT (250–500 mg/day; tid) is significantly higher as compared to that of Tmx (10–20 mg/day; od). Henceforth, a criteria was made to select oil, which solubilizes maximum amount of QT and Tmx in a dose equivalent or more than its therapeutic dose. Capmul® MCM EP contains the mixture of mono-, di- and triglyceride of fatty acid esterified with the glycerol, revealed optimum solubility of QT (31.93 ± 2.34 mg/g) as well as of Tmx (133.32 ± 7.32 mg/g). The specialized property of Capmul® MCM EP to solubilize both the drugs with dissimilar log P could be attributed to interaction of both the drugs with the diglyceride chains of oil. QT, owing to its pentaflavanol structural unit, has a strong propensity to form H-bonds with the glycerol moiety present in the oil. On the other hand, appreciably higher solubility of Tmx in Capmul MCM EP could be attributed to the hydrophobic interaction between the aliphatic chain and aromatic rings of Tmx. Isothermal stress testing (IST) was also employed to determine the compatibility between the solubilized drugs (Tmx and QT) in Capmul MCM EP (51). An insignificant change in the retention time or absence of any new peak in HPLC chromatogram of any of the IST sample suggested the compatibility of both the drugs in Capmul MCM EP (Supplementary Material Fig. S3). Furthermore, quantitative analysis of various IST samples revealed an insignificant change in the recovery of Tmx and QT in comparison with that of control samples, suggesting no chemical interaction or the formation of any byproduct during the stress stability condition (See Supplementary Material Table S IV).
Surfactants are usually added in liquid SNEDDS to impart the spontaneity by virtue of instantaneous emulsification which lead to the formation of nanoemulsion (droplet size < 100 nm) (52). Various non-ionic surfactants were screened as a function of emulsification time and characteristics of nanoemulsion resulted upon dilution (% transmittance, droplet size and PDI). Slower solubilization rate of Pluronic™ (F-127 and F-68) in aqueous phase hampers the uniform deposition on the oil–water interface and lead to a nanoemulsion with relatively higher emulsification time (5–10 min). On the other, all the surfactants of Tween series (Polyoxyethylene sorbitan esters) (Tween 20, Tween 80, Tween 40 and Tween 60) and Gelucire appeared to be poor emulsifier for Capmul MCM EP owing to their reduced affinity to interact and retain at the oil–water interface. Interestingly, superior emulsification ability of Cremophor RH 40 among all the screened surfactants could be ascribed to insertion of long chains of fatty acid and PEG unit into the core of oil droplet, which ultimately lead to efficient stabilization of oily droplets (53). Finally, Cremophor RH 40 was selected as the suitable surfactant for emulsification of Capmul MCM EP. Later on, selection of a specific co-surfactant was carried out based on the improvement in emulsification ability of Cremophor RH 40 and characteristics of resultant nanoemulsion (% transmittance, droplet size and PDI). Similar to surfactants, co-surfactant also lowers the interfacial tension by adsorbing at the oil–water interface along with the surfactant molecules (54,55). An ideal co-surfactant along with the surfactant forms a steady layer at the oil–water interface and minimizes the interfacial tension. All the co-surfactant except Labrafil 1944 CS resulted into nanoemulsion having unacceptable PDI (>0.4). This might be explained based on lack of close assembly of co-surfactants with Cremophor RH 40 at the oil–water interface (53). Interestingly, Labrafil 1944 CS (macrogol-6 glycerides EP) along with Cremophor RH 40 competently stabilizes the oily droplets in aqueous environment, owing to the presence of equivalent length of long chains in both the molecules, which might firmly insert into the core of oily droplet. A further enhancement in the stabilization propensity of both the molecules could be achieved by the possibility of hydrogen bonding between the PEG architecture of surfactant/co-surfactant and fatty acids of Capmul MCM EP (53).
Pseudoternary phase diagrams were constructed to identify the self-emulsification region for a combination of oil, surfactant and co-surfactants, generally on the basis of droplet size and PDI of emulsion resulted upon dilution (56). All the composition of explored region resulted into nanoemulsion with desired droplet size (< 200 nm) and PDI (< 0.2) (Fig. 2b). However, increased probability of water penetration into the oily droplets at the higher Cremophor RH 40 concentration (> 80%) might lead to disruption of the oil–water interface and ejection of oily droplets into the aqueous phase, which ultimately leads to the formation of emulsion with unacceptable droplet size and PDI (35). Subsequent to the identification of self-emulsifying region for Capmul® MCM EP-Cremophor RH 40-Labrafil 1944 CS based placebo system; optimization of liquid SDDS (Tmx-QT-SNEDDS) composition was carried out based on the loading of both the drugs and characteristics of emulsion (droplet size and PDI) resulted after the dilution with deionized water. Further, we anticipated that characteristics of emulsion resulted upon solubilization of Tmx in Capmul® MCM EP-Cremophor RH 40-Labrafil 1944 CS based system and subsequently after dilution should remain unaffected as compared to equivalent placebo systems. Tmx possess a significantly higher solubility in Capmul MCM EP (118.60 ± mg/g) as compared to in Cremophor RH40 (17.80 ± 3.23 mg/g) and Labrafil 1944 CS (15.23 ± 4.21 mg/g), which enable the preferential localization of Tmx in the hydrophobic core of oily droplet rather than at the oil–water interface. To further proof our hypothesis, pseudoternary phase diagram was constructed with varying (50:1, 40:1 and 30:1 w/w) Capmul MCM EP: Tmx ratios. An insignificant change in the area of self-emulsification region was observed, upon increasing the loading of Tmx from 50:1 w/w to 30:1 w/w (See Supplementary Material Fig. S4). At varying Tmx loading, all the liquid SNEDDS compositions resulted into emulsion with droplet size and PDI of <100 nm and 0.2 respectively. Based on the above finding, Tmx equivalent to its clinical dose (10 mg) was considered as the final loading in liquid SNEDDS (10 mg/g) which was further employed for the optimization of QT loading for which extreme vertices mixture design was employed.
Linear model navigated by the applied design indicated a direct relationship between the loading of QT and composition of liquid SNEDDS (Table III). Polynomial equation for the loading of QT in Tmx-SNEDDS reveled the contribution in the order of Capmul MCM EP > Cremophor RH 40 > Labrafil 1944 CS, owing to the maximum solubility of QT in Capmul MCM EP followed by Cremophor RH 40 (21.59 ± 2.67 mg/g) and Labrafil 1944 CS (15.11 ± 3.23 mg/g). Droplet size of emulsion resulted upon dilution of liquid SNEDDS determine the rate and extent of drug release vis-à-vis their absorption in GI tract (50). Overall bulk of oily phase as well as hydrodynamic diameter of oily droplet might increase, which ultimately lead to increase in the droplet size upon increasing the relative proportion of oil in Tmx-QT-SNEDDS. Additionally, inability of surfactant at lower concentration to sufficiently reduce the interfacial tension lead to increase in droplet size (36). We further envisaged that a surfactant/co-surfactant balance (SCB) at the oil–water interface could have a crucial role in predicting the PDI of the emulsion. Any disturbance in the SCB could lead to the increased penetration of water into oily droplets and resulted into ejection of oil in the aqueous environment, which overall resulted into the formation of non-uniform droplets and unacceptable PDI (53). An increased propensity of Cremophor RH 40 to form a steady layer along with Labrafil 1944 CS at the oil–water interface might lead to improvement in PDI upon increasing the concentration of Labrafil 1944 CS. Finally, higher model F-value and correlation coefficient close to unity for all the responses of Tmx-QT-SNEDDS indicated that applied model could be used to navigate the design space (Table III). Further, a specific composition of Tmx-QT-SNEDDS was optimized based on the criteria of desirability, which is an arbitrary scale and usually ranked between 0.00 (minimal) to 1.00 (maximal) (57). The key objective of the present study was to prepare the dual drug loaded SNEDDS, where we mainly emphasized to load maximum amount of QT, owing to its limited solubility in Capmul MCM EP. Additionally, the concentration of surfactant in SNEDDS formulation should also be minimized because oral administration of SNEDDS with higher surfactant concentration may lead to the undesirable toxicity and GI irritation. It was also desirable that liquid Tmx-QT-SNEDDS upon dilution revealed minimum droplet size as well as PDI. Considering all these facts, the desirability value was found to be 0.663, which corresponded to relative proportion of Capmul MCM EP, Cremophor RH 40 and Labrafil 1944 CS in the ratio of 40:30:30 w/w.
The developed Tmx-QT-SNEDDS was lyophillized in the presence of a solid carrier by the step wise freeze drying cycle developed and patented by our goup (45). As per our knowledge, this is the first report where step-wise lyophillzation has been employed for the solidification of SNEDDS. Stepwise lyophillzation imparts a controlled freezing/thermal conditions and slow cooling which ultimately lead to the uniform deposition of SNEDDS formulation onto solid carrier. Generally, solid SNEDDS is prepared by manual mixing of oily liquid with solid carrier (58). However, the executed method is usually associated with the non-uniform adsorption of liquid SNEDDS, incomplete desorption after reconstitution and irregular release profile of the drugs (42). Herein, we envisage the formulation of s-SNEDDS by stepwise lyophillization of diluted SNEDDS in the presence of solid carrier rather than by manual mixing of isotropic liquid with the intert solid carrier. Selection of a suitable solid carrier for the lyophillzation of the liquid SNEDDS, is a critical step because it overall affects the release behavior, drug loading capacity and stability of the formulation. The poor reconstitution behavier of solid SNEDDS, lyophillized in the presence of water soluble sugars, could be attributed to partial solubilization of water soluble sugars in the hydrophilic component of liquid SNEDDS (59). Water insoluble adsorbent type of solid carriers (Aerosil 200 and Avicel) resulted into free flowing solid SDEDDS which retained all the critical formulation attributes after reconstitution. Adsorbent type of solid carriers imparts higher surface area for the adsorption of liquid SNEDDS. Considering all above facts, Aerosil 200 was selected as the suitable solid carrier for the lyophillization of Tmx-QT-SNEDDS, which possess a significantly higher surface area (200 m2/g) as compared to Avicel (20 m2/g).
Subsequently, 32 full factorial design was implemented to optimize the concentration of Aerosil 200 for lyophilization of liquid Tmx-QT-SNEDDS. Rapid release of both the drugs upon contact with GI fluids was sought from s-SNEDDS; hence criteria of TTmx75 and TQT75 were taken into consideartion. Significantly higher release of QT in comparison with Tmx from all s-SNEDDS could be attributed to higher solubility of QT in release media. Slower release of both the drugs at higher solid content and increased proportion of liquid SNEDDS in lyophilization mixture, could be attributed to increased hindrance and incomplete desorption of SNEDDS components from s-SNEDDS. Additionally, hindered functionality of surfactant and minimal space available for efficient emulsification at higher solid content could also increase the droplet size upon reconstitution of s-Tmx-QT-SNEDDS. On the other hand, saturated adsorption capacity of Aerosil 200 at higher proportion of liquid Tmx-QT-SNEDDS could increase the coalescence of the oil droplets thereby resulting in to increase in the droplet size. Similar findings were also observed for PDI, leading to proportionate increase with total solid content and ratio of Aerosil to liquid SNEDDS (Fig. 4d). Finally, an optimum concentration of solid carrier in the lyophilization mixture was determined based on the desirability value, which was found to be 0.338. Based on desirabaility factor, the total solid content and liquid SNEDDS: Aerosil 200 ratio in lyophilization mixture was estimated to be 5.24% w/w and 1.61, respectively.
Reconstitution behavior of s-Tmx-QT-SNEDDS was further evaluated to ensure that the formulation is able to form nanoemulsion upon contact with GI fluids within minimal period. s-Tmx-QT-SNEDDS were reconstituted to form nanoemulsion in < 2 min while maintaining all the quality attributes (droplet size < 100 nm and PDI < 0.2). Surface morphology of s-Tmx-QT-SNEDDS was evaluated by SEM analysis, which revealed the non-porous surface where all the pores of Aerosil 200 were completely filled by liquid SNEDDS. Solid state of both the drugs in s-Tmx-QT-SNEDDS was further endorsed by DSC and XRD analysis, which revealed amorphous nature of both drugs in s-Tmx-QT-SNEDDS. Additionally, DSC and XRD analysis collectively indicated the solubilization of both the drugs within ternary component of SNEDDS and the same not precipitate during the entire course of lyophilization.
During the entire course of transit in GI tract, s-Tmx-QT-SNEDDS are supposed to encounter with variable dilution of GI fluids and constitute to form a nanoemulsion. Therefore, it was necessary to investigate their stability in simulated GI fluids and substantiate the solubilized state of both the drugs after dilution with GI fluids. s-Tmx-QT-SNEDDS were found to retain all the quality attributes and stable in all the dilutions of simulated GI fluids without any sign of drug precipitation or phase separation.
Long-term stability of s-Tmx-QT-SNEDDS was predicted by assessing their stability under the accelerated condition (40°C/RH 75%) for 6 months according to ICH guidelines. s-Tmx-QT-SNEDDS was found to retain all the physical and performance quality attributes (reconstitution time, droplet size, PDI and drug content) after 6 months of storage at accelerated condition. Solid state of both the drugs in s-Tmx-QT-SNEDDS might also change during the course of storage at the accelerated condition owing to crystallization of drugs. The molecular intactness of both the drugs in s-Tmx-QT-SNEDDS was confirmed by the DSC and XRD analysis, which collectively confirmed the solubilized state of both the drugs while being stored at the accelerated conditions (See Supplementary Material Fig. S5).
The in vitro drug release profile of s-Tmx-QT-SNEDDS was evaluated in SGF (pH 1.2) for 2 h and in SIF (pH 6.8) for 6 h using dialysis membrane of different molecular weight cut off. In our recent report, we envisaged the use of two dissimilar dialysis membranes for predicting the in vitro drug release from liquid self-emulsifying formulation (39). Similar approach was also implemented for the developed s-Tmx-QT-SNEDDS. As discussed in the previous section, s-Tmx-QT-SNEDDS formulation was able to emulsify within minimal time (< 2 min) and form drug loaded oily droplets and micelles upon dilution with aqueous medium. Desorption of the SNEDDS components (oil, surfactant and co-surfactants) from the inert solid carrier results into release of solubilized drugs in GI environment. However, an incomplete desorption of liquid excipients from the inert solid carrier impairs their efficient release in the GI tract (42). Herein, we have evaluated the in vitro release of solidified SNEDDS, where we observed that 75% of both the drugs were released within 30–45 min when carried out from dialysis membrane of 12 kD MWCO. Our findings are also meets the compendial requirement of dissolution. However, an obvious difference between the cumulative drug releases of both the drugs was observed when release was performed with dialysis membrane of 12 kD and 1 kD. We propose that dialysis membrane of 1 kD allows the passage of free drugs only whereas it retain all the drug loaded micelles or oily droplets. On the other hand, all the components of SNEDDS including free drugs could be analyzed in the release medium. On the basis of above finding, we hypothesize that solubilization of both the drugs were maintained following to their dilution with different GI fluids.
Finally, in vitro Caco-2 cells were employed to endorse the feasibility of transport through the GI tract and establish the oral delivery potential of s-Tmx-QT-SNEDDS. Caco-2 cells monolayer based model is well-established in vitro model to study the uptake pattern, uptake mechanism, pre-systemic metabolism and cyto-toxicological evaluation of SNEDDS. Recently, in our group we have also established a correlation between the in vitro Caco-2 cell uptake and in vivo pharmacokinetics of quercetin loaded SNEDDS (39). Confocal images of Caco-2 cells clearly indicated the higher uptake of both the drugs from SNEDDS within 1 h incubation (Fig. 8). These results are in line with our recent report, where we revealed maximum uptake of SNEDDS following 1 h of incubation with Caco-2 cells (39). Further, quantitative uptake of s-Tmx-QT-SNEDDS in Caco-2 monolayer was carried out to endorse the results of CLSM analysis. S-Tmx-QT-SNEDDS revealed 9.63- and 8.44-fold higher uptake of Tmx and QT, respectively when compared to free drug counterparts, which could be attributed to rapid transcellular uptake of SNEDDS (22). Nevertheless, about 2-fold appreciation in uptake of Tmx was observed upon incubation of Caco-2 monolayer with combination of Tmx with free QT (1:2 w/w). The previous finding of Shin and co-worker, which revealed the enhanced oral bioavailability of Tmx upon co-administration with free QT, supports our results (60). Notably, no visible sign of Caco-2 cell cytotoxicity was observed at all tested concentrations in spite of remarkable increase in the uptake, indicative of the safety profile of developed formulation (Fig. 10). The contributing factors include employment of biocompatible GRAS listed excipients (below the IIG limits), careful selection of the excipients (to have negative zeta potential of the formulation) and exhaustive statistical optimization (to minimize the excess surfactant levels).
Monitoring of TEER values and actin visualization of Caco-2 monolayer has been widely recommended for predicting the integrity of intercellular tight junction, transepithelial ion transport and paracellular transport of various drugs or nanoparticles (49). Interestingly, the results of TEER values and actin visualization ruled out the probability of any paracellular uptake mechanism classical to conventional nanocarriers. Hence, the developed SNEDDS is anticipated to be preferentially uptaken by transcellular route via fluid phase pinocytosis (22). Subsequent to uptake, the formulation is subjected to intraluminal processing leading to sequesterization and formation of chylomicrons (61). These secondary vehicles would then be taken up by lymphatic system bypassing the physiological barriers such as solubility, permeability and first pass hepatic metabolism (62).
CONCLUSION
The present study reports a novel solid self-emulsifying formulation containing dual drug of therapeutic significance for improved therapeutic efficacy after oral administration. Exhaustive statistical optimization was employed to load the significantly higher proportion of antioxidant (QT) and the clinical dose of Tmx, while maintaining all the quality attributes of liquid SNEDDS. A novel stepwise lyophilization was employed to solidify the dual drug loaded liquid SNEDDS in the presence of inert solid carrier; the same was also carried out by employing statistical optimization. The developed formulation exhibit advantages of smoother industrial scalability and adaptability (owing to simple manufacturing process), cost effectiveness (owing to absence of any high-energy equipment), and high drug payload (increased solubilization potential). The oral delivery potential of the developed formulation was obvious by significant appreciation in uptake of both the drugs in Caco-2 cell culture model, as compared to free drug counterparts. The developed formulation would be further assessed for in vivo pharmacokinetic, in vitro/in vivo anticancer efficacy and toxicity to completely realize the therapeutic potential of dual drug loaded in SNEDDS.
REFERENCES
Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205–13.
Middleton E, Kandaswami C, Theoharides TC. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev. 2000;52(4):673–751.
Boots AW, Haenen G, Bast A. Health effects of quercetin: from antioxidant to nutraceutical. Eur J Pharmacol. 2008;585(2–3):325–37.
Piantelli M, Maggiano N, Ricci R, Larocca LM, Capelli A, Scambia G, et al. Tamoxifen and quercetin interact with type II estrogen binding sites and inhibit the growth of human melanoma cells. J Invest Dermatol. 1995;105(2):248–53.
Caltagirone S, Rossi C, Poggi A, Ranelletti FO, Natali PG, Brunetti M, et al. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int J Cancer. 2000;87(4):595–600.
Sartippour MR, Pietras R, Marquez-Garban DC, Chen HW, Heber D, Henning SM, et al. The combination of green tea and tamoxifen is effective against breast cancer. Carcinogenesis. 2006;27(12):2424–33.
Ma ZS, Thanh HOAH, Chee Pang NG, Phuc Tien DO, Thanh HN, Hung H. Reduction of CWR22 prostate tumor xenograft growth by combined tamoxifen-quercetin treatment is associated with inhibition of angiogenesis and cellular proliferation. Int J Oncol. 2004;24(5):1297–304.
Brookes PS, Digerness SB, Parks DA, Darley-Usmar V. Mitochondrial function in response to cardiac ischemia-reperfusion after oral treatment with quercetin. Free Radic Biol Med. 2002;32(11):1220–8.
Liu S, Hou W, Yao P, Li N, Zhang B, Hao L, et al. Heme oxygenase-1 mediates the protective role of quercetin against ethanol-induced rat hepatocytes oxidative damage. Toxicol In Vitro. 2012;26(1):74–80.
Gupta C, Vikram A, Tripathi DN, Ramarao P, Jena GB. Antioxidant and antimutagenic effect of quercetin against DEN induced hepatotoxicity in rat. Phytother Res. 2010;24(1):119–28.
Abo-Salem OM, Abd-Ellah MF, Ghonaim MM. Hepatoprotective activity of quercetin against acrylonitrile induced hepatotoxicity in rats. J Biochem Mol Toxicol. 2011;25(6):386–92.
Tabassum H, Parvez S, Rehman H, Banerjee BD, Raisuddin S. Catechin as an antioxidant in liver mitochondrial toxicity: inhibition of tamoxifen induced protein oxidation and lipid peroxidation. J Biochem Mol Toxicol. 2007;21(3):110–7.
Granado-Serrano AB, Martin MA, Bravo L, Goya L, Ramos S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J Nutr. 2006;136(11):2715–21.
Caltagirone S, Ranelletti FO, Rinelli A, Maggiano N, Colasante A, Musiani P, et al. Interaction with type II estrogen binding sites and antiproliferative activity of tamoxifen and quercetin in human non-small-cell lung cancer. Am J Respir Cell Mol Biol. 1997;17(1):51–9.
Lama G, Angelucci C, Bruzzese N, Nori SL, D’Atri S, Turriziani M, et al. Sensitivity of human melanoma cells to Oestrogens, tamoxifen and quercetin: is there any relationship with type I and II oestrogen binding site expression? Melanoma Res. 1998;8(4):313–22.
McVie JG, Simonetti GP, Stevenson D, Briggs RJ, Guelen PJ, de Vos D. The bioavailability of tamoplex (tamoxifen). part 1. A pilot study. Methods Find Exp Clin Pharmacol. 1986;8(8):505–12.
Tukker JJ, Blankenstein MA, Nortier JW. Comparison of bioavailability in man of tamoxifen after oral and rectal administration. J Pharm Pharmacol. 1986;38(12):888–92.
Martin EA, Rich KJ, White INH, Woods KL, Powles TJ, Smith LL. 32P-Postlabelled DNA adducts in liver obtained from women treated with tamoxifen. Carcinogenesis. 1995;16(7):1651–4.
Lettéron P, Labbe G, Degott C, Berson A, Fromenty B, Delaforge M, et al. Mechanism for the protective effects of silymarin against carbon tetrachloride-induced lipid peroxidation and hepatotoxicity in mice: evidence that silymarin acts both as an inhibitor of metabolic activation and as a chain-breaking antioxidant. Biochem Pharmacol. 1990;39(12):2027–34.
Spencer JPE, Chowrimootoo G, Choudhury R, Debnam ES, Srai SK, Rice-Evans C. The small intestine can both absorb and glucuronidate luminal flavonoids. FEBS Lett. 1999;458(2):224–30.
van Leeuwen R, Brundel D, Neef C, van Gelder T, Mathijssen R, Burger D, et al. Prevalence of potential drug–drug interactions in cancer patients treated with oral anticancer drugs. Br J Cancer. 2013;108(5):1071–8.
Thanki K, Gangawal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Rel. 2013;170(1):15–40.
Hu C-MJ, Zhang L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem Pharmacol. 2012;83(8):1104–11.
Zhigaltsev IV, Maurer N, Akhong Q-F, Leone R, Leng E, Wang J, et al. Liposome-encapsulated vincristine, vinblastine and vinorelbine: a comparative study of drug loading and retention. J Control Rel. 2005;104(1):103–11.
Bayne WF, Mayer LD, Swenson CE. Pharmacokinetics of CPX–351 (cytarabine/daunorubicin HCl) liposome injection in the mouse. J Pharm Sci. 2009;98(7):2540–8.
Misra R, Sahoo SK. Coformulation of doxorubicin and curcumin in poly (D, L-lactide-co-glycolide) nanoparticles suppresses the development of multidrug resistance in K562 cells. Mol Pharmaceutics. 2011;8(3):852–66.
Acharya S, Sahoo SK. Sustained targeting of Bcr–Abl + leukemia cells by synergistic action of dual drug loaded nanoparticles and its implication for leukemia therapy. Biomaterials. 2011;32(24):5643–62.
Zhang L, Radovic–Moreno AF, Alexis F, Gu FX, Basto PA, Bagalkot V, et al. Co–Delivery of Hydrophobic and Hydrophilic Drugs from Nanoparticle–Aptamer Bioconjugates. Chemmedchem. 2007;2(9):1268–71.
Song XR, Cai Z, Zheng Y, He G, Cui FY, Gong DQ, et al. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharm Sci. 2009;37(3):300–5.
Lammers T, Subr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, et al. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30(20):3466–75.
Krakovičová H, Etrych T, Ulbrich K. HPMA-based polymer conjugates with drug combination. Eur J Pharm Sci. 2009;37(3):405–12.
Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T, et al. Co–delivery of doxorubicin and Bcl–2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug–resistant cancer cells. Small. 2009;5(23):2673–7.
Kaneshiro TL, Lu Z-R. Targeted intracellular codelivery of chemotherapeutics and nucleic acid with a well-defined dendrimer-based nanoglobular carrier. Biomaterials. 2009;30(29):5660–6.
Jain AK, Thanki K, Jain S. Co-encapsulation of tamoxifen and quercetin in polymeric nanoparticles: implications on oral bioavailability, antitumor efficacy and drug induced toxicity. Mol Pharmaceutics. 2013. doi:10.1021/mp400311j.
Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25(1):47–58.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11:S93–8.
Talegaonkar S, Jain S, Bhusinge U. Nano-colloidal carriers: site specific and controlled drug delivery. New Delhi: CBS Publishers and Distributors; 2010.
Jain S, Jain AK, Swarnakar NK, Harde H, Pharmaceutical composition for enhancing anticancer efficacy of tamoxifen. PCT application number PCT/IB2012/001361, filed on 11/7/2012.
Jain S, Jain AK, Pohekar M, Thanki K. Novel self-emulsifying formulation of quercetin for improved in vivo antioxidant potential: implications on drug induced cardiotoxicity and nephrotoxicity. Free Radic Biol Med. 2013;65C:117–30.
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13):606–12.
Kallakunta VR, Bandari S, Jukanti R, Veerareddy PR. Oral self emulsifying powder of lercanidipine hydrochloride: formulation and evaluation. Powder Technol. 2012;221:375–82.
Van Speybroeck M, Williams HD, Nguyen T-H, Anby MU, Porter CJH, Augustijns P. Incomplete desorption of liquid excipients reduces the in vitro and in vivo performance of self-emulsifying drug delivery systems solidified by adsorption onto an inorganic mesoporous carrier. Mol Pharmaceutics. 2012;9(9):2750–60.
Zhou L, Yang L, Tilton S, Wang J. Development of a high throughput equilibrium solubility assay using miniaturized shake flask method in early drug discovery. J Pharm Sci. 2007;96(11):3052–71.
Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1–2):166–72.
Jain S, Chauhan DS, Jain AK, Swarnakar NK, Harde H, Mahajan RR, Kumar D, Valvi PK, Das M, Datir SR, Thanki K. Stabilization of the nanodrug delivery systems by lyophilization using universal step-wise freeze drying cycle. Indian Patent Application No 2559/DEL/2011 filed on September 06, 2011.
ICH Q1A(R2): Stability testing of new drug substances and products Q1A(R2), ICH harmonized tripartite guideline; Step 4 version: February 6, 2003.
Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355(1):269–76.
Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–86.
Lin Y-H, Chung C-K, Chen C-T, Liang H-F, Chen S-C, Sung H-W. Preparation of nanoparticles composed of chitosan/poly-γ-glutamic acid and evaluation of their permeability through caco-2 cells. Biomacromolecules. 2005;6(2):1104–12.
Neslihan Gursoy R, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–82.
Moyano MA, Broussalis AM, Segall AI. Thermal analysis of lipoic acid and evaluation of the compatibility with excipientes. J Thermal Anal Calorimet. 2010;99(2):631–7.
Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212(2):233–46.
Li W, Yi S, Wang Z, Chen S, Xin S, Xie J, et al. Self-nanoemulsifying drug delivery system of persimmon leaf extract: optimization and bioavailability studies. Int J Pharm. 2011;420(1):161–71.
Tenjarla S. Microemulsions: an overview and pharmaceutical applications. Crit Rev Ther Drug Carrier Syst. 1999;16(5):461–521.
Azeem A, Rizwan M, Ahmad FJ, Iqbal Z, Khar RK, Aqil M, et al. Nanoemulsion components screening and selection: a technical note. AAPS Pharm Sci Tech. 2009;10(1):69–76.
Shafiq S, Shakeel F, Talegaonkar S, Ahmad FJ, Khar RK, Ali M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur J Pharm Biopharm. 2007;66(2):227–43.
Ahnazarova SL, Kafarov VVÄe, Mackovskij VM, Rep'ev AP. Experiment optimization in chemistry and chemical engineering. Mir Publishers; 1982, Moscow.
Oh SJ, Kim O, Lee JS, Kim J, Kim MR, Choi HS, et al. Inhibition of angiogenesis by quercetin in tamoxifen-resistant breast cancer cells. Food Chem Toxicol. 2010;48(11):3227–34.
Oh DH, Kang JH, Kim DW, Lee BJ, Kim JO, Yong CS, et al. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int J Pharm. 2011;420(2):412–8.
Shin SC, Choi JS, Li X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm. 2006;313(1):144–9.
Levin MS, Talkad VD, Gordon JI, Stenson WF. Trafficking of exogenous fatty acids within caco-2 cells. J Lipid Res. 1992;33(1):9–19.
Trevaskis NL, Charman WN, Porter CJH. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008;60(6):702–16.
ACKNOWLEDGMENTS AND DISCLOSURES
The authors are thankful to Director, NIPER for providing the necessary infrastructure and facilities and Department of Science & Technology (DST), Government of India, New Delhi, and financial support. A.K.J and K.T. are grateful to Council of Scientific and Industrial Research (CSIR), GOI, New Delhi, for providing Research fellowships.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1153 kb)
Rights and permissions
About this article
Cite this article
Jain, A.K., Thanki, K. & Jain, S. Solidified Self-Nanoemulsifying Formulation for Oral Delivery of Combinatorial Therapeutic Regimen: Part I. Formulation Development, Statistical Optimization, and In Vitro Characterization. Pharm Res 31, 923–945 (2014). https://doi.org/10.1007/s11095-013-1213-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-013-1213-2