
Abstract
Purpose
To identify the aggregation mechanism and the stability characteristics of three different monoclonal antibodies under acidic conditions.
Methods
The aggregation kinetics is analyzed by a combination of light scattering, size exclusion chromatography and fluorescence techniques and the aggregation data are correlated to protein structure, hydrophobicity, charge and antibody subclass.
Results
In the investigated conditions, the antibody aggregation follows a mechanism consisting of two-steps: reversible monomer oligomerization followed by irreversible cluster-cluster aggregation. The kinetics of the two steps is differently affected by the operating conditions: mild destabilizing conditions induce formation of oligomers which are stable within weeks, while stronger denaturing conditions promote aggregation of oligomers to larger aggregates which eventually precipitate. For different antibodies significant differences in both oligomerization and growth rates are found, even for antibodies belonging to the same subclass. For all antibodies the aggregate formation is accompanied by a structure re-organization with an increase in the ordered β-sheet structures. At low pH the aggregation propensity of the investigated antibodies does not correlate with antibody subclass, surface net charge and hydrophobicity of the non-native state.
Conclusions
The aggregation mechanism of three antibodies in acidic conditions as well as differences and analogies in their stability behavior has been characterized.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the last years, many therapeutic proteins have been developed versus cancer and several immunological disorders (1,2). The successful application of this important class of drugs requires avoiding any form of degradation. Protein aggregation is the major form of instability occurring during production, shipping and storage (3,4). Aggregation during production must be avoided because it leads to loss of valuable product, although aggregates could be removed by chromatographic steps. Moreover, protein aggregates formed during shipping or storage may potentially induce immunological reactions in patients (5).
A potential source of protein aggregation is represented by the several types of stresses (thermal, acidic, mechanical, interaction with surfaces) which protein may face during the various production steps: expression, purification and formulation (6). In all of them proteins are exposed to changes of operating parameters such as pH, temperature, salt concentration, buffer composition, shear rate and surfaces. An example is represented by the purification process, which is the main part of the downstream portion of recombinant protein production: during this step the conditions of pH and buffer composition are changed in order to favor chromatographic adsorption or desorption. These parameters may affect protein structure stability and protein-protein interactions, inducing aggregation (7,8).
Depending on the operating conditions, several aggregation mechanisms and types of aggregates have been observed (9,10). The monomeric protein can aggregate either in the native state or in a non-native conformation and aggregation can be either reversible or irreversible. Many experimental studies investigated independently the effect of one or few variables on protein unfolding and aggregation, with large attention given to pH and ionic strength (11–14). Buffer composition (15), particularly anions (16), affects significantly aggregation propensity. In addition, temperature (17), protein concentration (18,19) and interactions with surfaces (20,21) are other relevant parameters for protein aggregation behavior.
The aggregation propensity of a given protein depends not only on the environment but also on the intrinsic primary sequence and structure of the protein. Antibodies (mAbs) are multi-domain proteins consisting of a constant domain (Fc) and two identical domains containing the variant region (Fab). IgG is one class of immunoglobulins exhibiting several subclasses in humans (IgG1, IgG2, IgG3 and IgG4) (22). mAbs within each subclass share the constant Fc domain and differ in the variable Fab domain. In order to correlate aggregation propensity and structure properties, several recent works compare different IgG subclasses (23–25). Particular attention is given in elucidating the role of the single domains (Fc and Fab) in protein self-assembling. In parallel with the experimental approach, in silico studies try to identify aggregating-prone regions or “hot” spots in order to optimize protein sequence engineering against aggregation (26,27). Protein glycosylation was also found to have a potential role in modulating aggregation (28–30).
The large number of intrinsic and extrinsic factors involved in protein stability complicates the rationalization of the problem. On one hand, different proteins share common features in aggregation behavior and the different observed aggregation mechanisms have been classified in a limited number of classes (31,32). On the other hand, it is still not possible to predict a priori the stability of a given protein, and the optimization of formulations and processing conditions requires ultimately specific studies. Investigation of the aggregation mechanism and of analogies among different proteins under different operating conditions may allow us reaching general conclusions about the environmental or structure protein characteristics which determine its aggregation.
In this work, we investigate the aggregation mechanism and the stability characteristics of an IgG2 and two IgG1 proteins in acidic conditions. Low pH values are required by purification steps commonly applied in industrial processing, such as Protein A Chromatography and virus inactivation. These conditions favor the formation of aggregates which have to be removed, thus leading to significant loss in the process yield (8). We characterize the aggregation kinetics in a sufficiently large range of operating conditions by Dynamic Light Scattering (DLS), Size Exclusion Chromatography (SEC) and fluorescence techniques. In the first part of the work, the stability behavior of the IgG2 protein is described in detail, while in the second part the aggregation data of the different antibodies are compared and correlated to proteins structure, thermal stability, hydrophobicity and surface charge.
Materials and Methods
Materials
IgG2 (pI between 7.35 and 8.15), IgG1.1 (pI about 8.5), and IgG1.2 (pI between 8.5 and 9) were stored as 45 g/L solutions in 25 mM sodium phosphate at pH 6.0 with 100 mM NaCl at 4°C.
The solutions for aggregation studies were prepared by manually diluting the mother solution to 1 g/L by selected buffer solutions. For selected conditions the aggregation experiments were repeated preparing the protein solution with an alternative procedure: the mother solution was extensively dialyzed in acidic buffers at 4°C before inducing aggregation by salt addition and incubation at proper temperature. The same results were obtained for the solutions prepared according to the two methods, thus indicating that the dilution applied in the first method is sufficient to neglect the contribution of the buffer composition of the mother solution on the aggregation behavior at low pH.
For each condition three repetitions were performed and average values were considered. All buffers for aggregation studies were filtered using 0.20 μm cut-off sterile syringe filters Pall® Acrodisc® 32 mm (PALL Life Sciences, NY, USA). 0.5 g/L of sodium azide was added to all solutions to prevent formation and proliferation of bacterial growth. All chemicals were supplied by Sigma Aldrich (Buchs, CH).
Light Scattering
Dynamic light scattering (DLS) of aggregating protein solutions was measured in situ at a fixed angle of θ = 173° using a Zetasizer Nano (Malvern, UK) with laser beam of wavelength λ 0 = 633 nm. The so-called Z-average size is obtained by fitting the autocorrelation function with the method of cumulants. From the first cumulant the average particle diffusion coefficient, D, is evaluated and the Z-average hydrodynamic radius, R h , of the solution is calculated by applying the Stokes-Einstein equation:
Where k is the Boltzmann constant, T is the temperature and η is the solution viscosity.Before incubation, all solutions were filtered with a 0.02 μm cut-off, Anotop 10 syringe filter (Whatman, Kent, UK). For selected conditions, DLS measurements were repeated by filtering the solutions before incubation with a 0.20 μm cut-off, Millex syringe filter (Millipore, Billerica, MA, USA). The same results were obtained for samples prepared with the two different cut-off filters, indicating that the applied filtration before incubation does not affect the aggregation kinetics.
For each investigated condition, at least three independent samples were prepared and analyzed, and average values are reported. This procedure applies also to the other techniques used in this work. Evidences of the reproducibility of the data obtained with DLS as well as with the other techniques are reported in the Supplementary Material.
Static light scattering (SLS) of fractionated samples was measured by coupling an on-line static light scattering detector (Wyatt, Dernbach, DE) to the size exclusion column. The molecular weight of the fractionated species (MW) has been evaluated by the Wyatt Astra software® using the Zimm plot equation at low protein concentration:
where c is the protein concentration, R is the excess Rayleigh scattering ratio at the angle θ, P is the form factor and K is an optical constant defined as \( K = 4{\pi^2}{\left( {{{{dn}} \left/ {{dc}} \right.}} \right)^2}{n^2}N_A^{{ - 1}}\lambda_0^{{ - 4}} \), where n is the refractive index of the solvent, dn/dc is the refractive index increment, N A is the Avogadro number and λ 0 is the wavelength of the incident light. For dn/dc the value of 0.185 mL/g, typical for protein aqueous solutions, has been assumed.
Size Exclusion Chromatography (SEC)
Size exclusion chromatography (SEC) was performed with a Superdex 200 10/300 GL, 10 mm×300 mm size-exclusion column (GE Healthcare, Uppsala, SE) mounted on a Agilent 1100 series HPLC unit (Santa Clara, CA, USA) consisting of a quaternary pump with degasser, an autosampler, a column oven, and a UV detector. Protein solutions for SEC analysis were incubated in glass vials at appropriate temperatures. At selected time points along the aggregation process aliquots of 30 μL taken directly from the vials were injected into the SEC column by an automatic injector and eluted for 60 min at a constant flow rate of 0.5 mL/min using as mobile phase a solution of 100 mM sodium sulfate and 25 mM Na2HPO4 at pH 7.0. The UV absorbance chromatograms were detected at 280 nm. Samples were not centrifuged before the injection. Residual monomer amount was evaluated by calculating the area under the monomer peak in the non-normalized SEC chromatograms with the Agilent ChemStation software (Santa Clara, CA, USA). When the baseline between the peaks of monomer and dimer was not resolved, the monomer peak was defined by dropping a perpendicular in the valley between the monomer and the dimer peak at a fixed elution time.
Thioflavin-T (ThT) and 8-Anilinonaphthalene-1-sulfonate (ANS) binding
ThT and ANS fluorescence were measured using an EnSpire 2300 Multilabel Plate Reader (Perkin Elmer, Waltham, MA, USA) with a standard 96 wells black plate with a transparent bottom (ProxiPlate-96, Perkin Helmer, USA). 25 μM ThT or ANS were added to 1 g/L protein solutions, corresponding to a dye-to-protein molar ratio of 3.7. ThT emission at 485 nm was recorded after excitation at a wavelength of 450 nm, while ANS emission at 490 nm was recorded after excitation at a wavelength of 403 nm. To avoid heterogeneity in the measurements, fluorescence values were averaged over 9 different points inside each well (grid 3 × 3). Three repetitions were performed for each sample. To investigate the hydrophobicity of the monomeric proteins before aggregation, ANS spectra of 0.3 g/L protein solutions in proper buffers were acquired by recording ANS emission from 420 to 600 nm.
Circular dichroism (CD)
Circular dichroism (CD) spectra of 0.3 g/L protein solutions in proper buffers were acquired on a Jasco-815 CD spectrophotometer (Jasco, Easton, MD, USA). Far-UV CD spectra were recorded from 260 to 190 nm with the temperature of the cell holder controlled at 20°C. A quartz cuvette with 0.1 cm path length was used. Spectra obtained after buffer subtraction were corrected for protein concentration, smoothed using the Savitsky-Golay function and normalized with respect to the number of aminoacids to obtain mean residue ellipticities (MRE).
Thermal stability of 0.3 g/L protein solutions in 25 mM citric acid buffer at pH 3.0 without salt was evaluated by recording the spectra at several temperatures heating from 15°C to 90°C with a step of 1°C. The protein unfolding was monitored at 210 nm.
Zeta Potential
Zeta potential values of proteins (z) have been evaluated by a Zetasizer Nano (Malvern, Worcestershire, UK) measuring the electrophoretic mobility (μ) via laser Doppler effect. From the electrophoretic mobility, μ, the zeta potential, z, is calculated according to Henry equation:
where ε and η are the dielectric constant and the viscosity of the medium, respectively.
0.3 g/L protein solutions in 25 mM citric acid buffer in the pH range from 3.0 to 6.0 and in 25 mM phosphate buffer in the pH range from 7.0 to 9.0 have been measured at 25°C. Five repetitions of three independent samples were recorded for each condition and average values have been considered.
Results
Effect of operating conditions on IgG2 stability
The stability of the IgG2 antibody at room temperature was described in a previous work (7): the protein is stable above a critical pH value of 4.5 for several days, while for lower pH values the antibody reversibly aggregates to dimers and trimers within few hours (7). Here we investigate the aggregation behavior in the temperature range from 4°C to 37°C and pH range from 3.0 to 4.5 in the presence and in the absence of different salts (25 mM citric acid buffer with and without 150 mM NaCl or Na2SO4). A similar qualitative behavior was observed in the presence of NaCl and Na2SO4, with faster aggregation occurring in the presence of Na2SO4. For sake of simplicity we report in the following only the results obtained in the presence of sodium sulfate. Results and discussion on the effect of the salt are reported in the Supplementary Material. The aggregation kinetics is followed in situ by dynamic light scattering (DLS) and samples taken at different incubation times are analyzed by size exclusion chromatography (SEC). To describe the aggregation state of the IgG2 under different conditions, we show in Fig. 1 the normalized size exclusion chromatograms of 1 g/L protein solutions after a characteristic incubation time of 6 h in 25 mM citric acid buffer with 0.15 M Na2SO4 for several pH and temperature values. The peak at about 25 min elution time corresponds to monomeric antibody, while the peaks at shorter elution times represent aggregates. Aggregates larger than about six monomeric units are excluded from the column volume and elute all simultaneously in the void peak at 18 min (Fig. 1i). At the considered salt concentration, three types of behavior can be identified in the pH-temperature plane investigated in Fig. 1 after 6 h of incubation time: 1) stable solutions (Fig. 1a, b, d) which exhibit the same SEC chromatogram of the non-stressed sample; 2) formation of soluble oligomers (Fig. 1c, e, f, g, h), and 3) formation of submicron aggregates, which will be defined in the following as high molecular weight (HMW) aggregates (Fig. 1i). Aggregation is promoted either by decreasing pH or by increasing temperature. In particular, by increasing temperature from 4°C to 25°C and from 25°C to 37°C the critical pH value for oligomer formation shifts from 3.0 to 4.0 and from 4.0 to 4.5, respectively. Moreover, at pH 3.0 and 37°C the formed oligomers are not stable and grow to larger aggregates within hours, while in the conditions of Fig. 1c, e, f, g, h the formed oligomers are rather stable within weeks. It is worth noticing that in this work we refer to the kinetic stability of the protein solutions. Therefore, we specify the time scale corresponding to the observed stability/aggregation of a given solution. Oligomers solutions which are thermodynamically unstable and aggregate within weeks can be considered rather stable from a kinetic point of view within hours of incubation.

Normalized SEC chromatograms of 1 g/L protein solution after 6 h of incubation in 25 mM citric acid buffer with 0.15 M Na2SO4 at several pH and temperature values
At pH 3.0 and 37°C (conditions of Fig. 1i) mass lost is observed during the SEC analysis of samples taken after 3 h of incubation, indicating formation of large aggregates. On the other hand, full mass recovery is obtained for all the soluble oligomer distributions measured under the conditions of Fig. 1c, e, f, g, h, for which the area under the chromatograms is constant during time. For these samples, the molecular weight (MW) of the soluble oligomers has been measured by static light scattering. The measurements indicate the presence of mainly dimer and trimer, as shown for a representative SEC chromatogram with the corresponding MW in the Supplementary Material. In the following, the soluble oligomers will be treated as a single lumped species corresponding to all the oligomers eluting between 16 and 24 min in the SEC chromatograms, without distinguishing between dimers, trimers and larger oligomers.
It is well known that the off-line SEC analysis suffers from limitations due to interactions between the aggregates and the stationary phase of the column or possible formation of monomer from reversible aggregates upon dilution with the mobile phase (33). Such non-ideal effects are strongly affected by the buffer composition of the mobile phase (34). It is therefore important to check the reliability of the SEC chromatograms by performing comparison with orthogonal techniques (33). For the soluble oligomer chromatograms, where full mass recovery is observed, the reliability of the SEC chromatograms has been verified by coupling an on-line DLS detector (Wyatt, Dernbach, DE) with the SEC column, so as to estimate the average hydrodynamic radius of the oligomer distribution determined from the SEC technique. This value was found to be reasonably in agreement with the hydrodynamic radius of the non-fractionated sample evaluated in situ by DLS, therefore indicating that the SEC chromatograms of the soluble oligomers can be considered reliable (7). Moreover, measurements of selected samples were repeated using a TSK gel G3000SWxl column (Tosoh Bioscience LLC, Montgomeryville, PA) with 100 mM sodium sulfate and 25 mM Na2HPO4 at pH 6.0 as mobile phase. The results obtained with the G3000SWxl column and mobile phase at pH 6.0 were rather similar to the results obtained with the Superdex 200 column and mobile phase at pH 7.0, thus confirming the reliability of the results.
The oligomers observed in Fig. 1 can be either intermediate species in the system evolution to the larger aggregates or the product of an alternative aggregation mechanism. In order to better investigate these two cases, we followed the oligomer stability for longer times and in different conditions.
In Figure 2a the three situations which appeared less stable for the incubation time of 6 h (Fig. 1i, f, h) are now monitored for 15 h at the corresponding temperature in Fig. 1 by showing the Z-average hydrodynamic radius, <Rh>, measured by DLS as a function of time. It is seen that the oligomers in Fig. 1i are not stable and undergo massive aggregation after about 12 h, while the other two remains substantially stable in the 15 h. The DLS intensity signal for the oligomers in Fig. 1i increases in the first 12 h due to aggregation, and later decreases, indicating precipitation of the aggregates. At the end of the aggregation process the aggregates are visually insoluble, and precipitate on the bottom of the cuvette (data reported in the Supplementary Material).

Time evolution of the Z-average hydrodynamic radius, <R h >, measured by dynamic light scattering for a 1 g/L protein solution in 25 mM citric acid buffer with 0.15 M Na2SO4. (a) at 37°C and pH 3.0 (1I) (○), at 25°C and pH 3.0 (1 H) (▲), and at 37°C and pH 4.0 (1 F) (□); (b) at 25°C and pH 3.0 (1 H) (▲), and at 37°C and pH 4.0 (1 F) (□).
It is worth noticing that the DLS analysis applied in this work provides only an average value of the aggregate distribution without giving reliable information about the shape of the distribution. Moreover this technique cannot detect properly the aggregation kinetics of large size aggregates, when aggregates precipitation occurs. In this work we detect the initial aggregation of monomer and oligomers by monitoring changes of the average hydrodynamic radii in the size range from few to 150 nm, corresponding to non-sedimentating conditions. We do not address aggregation kinetics of larger aggregates and change of size distribution, for which other techniques such as field-flow fractionation and microscopy techniques are required (35).
By prolonging the experiment duration to 45 days we see that also oligomers in Fig. 1h, f aggregate but in a longer time scale involving tens of days. Oligomers in Fig. 1h undergo massive aggregation after about 30 days, while precipitation of micron aggregates formed by the oligomers in Fig. 1f was observed after more than two months.
It is worth noting that the profile of the time evolution of the average size is similar in all the investigated conditions, as seen by comparing the slower kinetics in Fig. 2b to the faster kinetics at 37°C and pH 3.0 in Fig. 2a. A slow, almost linear increase of the average size during the initial times is followed by a fast acceleration leading to the formation of insoluble aggregates in the micron range which, eventually, precipitate.
From the data shown in Fig. 2 we can conclude that the oligomers observed in Fig. 1 are intermediates in the aggregation process leading to larger micron precipitates. The kinetics of oligomer-oligomer aggregation and, therefore, the oligomers life-span depend on the operating conditions, and particularly on pH and temperature, i.e., on the denaturing conditions.
These results evidence the multi-variable nature of this stability problem, which involves a combined effect of salt, pH and temperature on the aggregation kinetics. The latter is most likely related to unfolding of the native structure, induced with different combinations of operating parameters. On the other hand, the aggregation mechanism seems to be the same in all conditions as discussed in the next section.
IgG2 Aggregation Mechanism
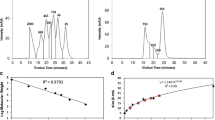
The mechanism of aggregation was investigated in more detail for the following reference conditions: 1 g/L protein solution in 25 mM citric acid buffer with 0.15 M Na2SO4 and 37°C. In Fig. 3 the first 3 h of the time evolution of the normalized SEC chromatograms and of the corresponding average molecular weight of the soluble oligomers in the reference condition and different pH values are compared. In particular, we consider in the sequel three pH values: 3.0, 4.0 and 4.5, which correspond to the operating conditions i, f and c in Fig. 1, and therefore are referred to 1i, 1 f and 1c in the sequel, respectively. It is seen that the time evolution of the chromatograms at pH 4.5 (Fig. 3a) and pH 4.0 (Fig. 3b) is similar, with a kinetics slightly faster at pH 4.0. On the other hand, at pH 3.0 already after 1 h species larger than trimer are observed (Fig. 3c and d).

Time evolution of normalized size exclusion chromatograms of 1 g/L protein solution in 25 mM citric acid buffer with 0.15 M Na2SO4 at 37°C and several pH values: (a) pH 4.5 (1c); (b) pH 4.0 (1 f); (c) pH 3.0 (1i); (d) Time evolution of the average molecular weight of the soluble oligomers eluting between 16 and 24 min in the chromatograms in (a), (b) and (c) normalized with respect to the molecular weight of the monomer (MW 0 ). When not visible, error bars are smaller than the symbols. (*) for the sample at pH 3.0 (1i) after 3 h of incubation time, 15.6 ± 7% of mass lost is observed during SEC analysis. At pH 4.0 and 4.5 only dimers and trimers are visible, while at pH 3.0 aggregation proceeds to larger aggregates.
From the non-normalized chromatograms the residual monomer amount has been evaluated by the ratio between the area under the monomer peak and the area of the control monomeric sample. In Fig. 4a the time evolution of the residual monomer amount at the different pH values is compared. Despite the difference in molecular weight, after 3 h of incubation the residual monomer at all pH values is comparable, being 11.9 ± 0.4% at pH 3.0, 23.4 ± 2.6% at pH 4.0 and 24 ± 2.9% at 4.5.

(a) Residual monomer fraction versus time evaluated from the non-normalized SEC chromatograms of Fig. 3; (b) time evolution of the residual monomer fraction (▲) (right axis) and of the average hydrodynamic radius (○) (left axis) at pH 3.0 (1I).
The difference in aggregate size at the different pH values increases further at 6 h of incubation. The corresponding SEC chromatograms are shown in Fig. 1 (i.e., 1c, 1 f and 1i), while the DLS data are shown in Fig. 2a, indicating an average radius, <R h >, of 30 ± 1 nm at pH 3.0 and of 10 ± 1 nm at pH 4.0. Despite such a difference in size, the difference in residual monomer between the two pH values is only of about 10%, being the residual monomer equal to 6.3 ± 2% at pH 3.0 and 17 ± 1.5% at pH 4.0 (Fig. 3d).
At longer times (about 10–12 h), for the case pH = 3.0 (Fig. 2a) a strong acceleration in aggregation is observed through the strong increase of the Z-average hydrodynamic radius measured by DLS. Since this occurs when the monomer consumption is almost complete, as shown in Fig. 4b, we can conclude that most likely the formation of HMW aggregates is not due to monomer addition but to cluster-cluster aggregation.
We investigated the reversibility of the aggregates formed during the aggregation (i.e., the possibility to dissociate the aggregates into monomers by reverting to the original solution conditions) (10) by taking two samples after a selected time of incubation: one sample was immediately analyzed by SEC, while the second one was diluted twenty-folds in 25 mM citric acid buffer at pH 3.0 without salt, and analyzed after 3 h. As shown in Fig. 5, the dilution of samples taken in the very early stages (after 5 min of incubation) leads to a significant monomer recover from oligomers. This clearly indicates that the initial oligomerization step at 37°C is reversible, as observed earlier for the same oligomerization process at room temperature (7). On the other hand, the dilution of samples taken after 10 h of incubation does not lead to a decrease in the aggregate average size or to monomer formation, indicating that the formed aggregates are mostly irreversible.

Mass percentage of oligomers (black bars) and monomers (grey bars) determined from the SEC technique for (a) a 10 g/L mAb sample taken after 5 min incubation in 25 mM citric acid buffer at pH 3.0 and 37°C; (b) the same sample as (a) diluted 20 folds in 25 mM citric acid buffer at pH 3.0 without salt and incubated for 3 h at 25°C.
From the experimental evidences collected so far we can envision the pH-induced aggregation process as a two-step process. In the first one, monomeric antibodies form soluble oligomers consisting mainly of dimers-trimers. Dilution of the oligomer mixture in a salt-free solution leads to monomer recover from the oligomers, indicating that this first step can be considered reversible. In the second step, an auto-accelerating cluster-cluster aggregation takes place. The transition between the two steps, and therefore the oligomers life-span, can occur at different times and it is indeed accelerated at pH 3.0 with respect to pH 4.0 and 4.5.
Changes in protein secondary structure during aggregation were assessed by Thioflavin-T (ThT) and 8-Anilinonaphthalene-1-sulfonate (ANS) binding studies (36), which give information about the β-sheet content (37) and the hydrophobicity of the protein (38), respectively. In Figure 6a the ThT fluorescence values versus time are shown for the three pH values investigated, corresponding to conditions 1 f, 1c and 1i. It is seen that the fluorescence signal increases with an extent proportional to aggregation, indicating that the latter is accompanied by β-sheet formation: at pH 3.0 a continuous increase of fluorescence is observed, while at pH 4.0 and 4.5 only an initial increase related to oligomer formation is detected. A similar behavior was observed for ANS binding.

(a) ThT fluorescence values for 1 g/L protein solution in 25 mM citric acid buffer with 0.15 M Na2SO4 at 37°C at pH 3.0 (1i) (□), pH 4.0 (1 f) (■) and pH 4.5 (1c) (○); (b) ThT fluorescence values () and Z-average hydrodynamic radius (●) versus monomer conversion for the reference reaction at pH 3.0 (1i); (c) ThT (○) and ANS (■) fluorescence values versus Z-average hydrodynamic radius for the reference reaction at pH 3.0 (1i).
In Figure 6b the correlation between aggregation and structure changes is evidenced for the more unstable condition, 1I, by plotting the ThT fluorescence values and < R h > versus monomer conversion: both quantities increase non-linearly with respect to monomer conversion and follow a similar profile, thus proving the strong correlation between the two quantities. This is confirmed by plotting ThT and ANS values versus < R h >, as reported in Fig. 6c: the fluorescence values grow rapidly with the average size indicating the structure re-arrangement of the formed oligomers. With increasing average size, the change in fluorescence values reduces until reaching a plateau: this corresponds to the fast acceleration in aggregation kinetics observed in Fig. 2a. During this step, the aggregates grow too fast to allow internal re-arrangements.
Re-organization of microdomains inside the clusters and formation of distorted intermolecular β-sheet during aggregation have already been observed for the antibody under investigation (7) as well as for others (12,14,39). However, the features of such re-structuring are still unclear. We compared the increase of ThT fluorescence during mAb aggregation observed above with the increase found during aggregation of an equivalent amount of insulin in similar conditions of pH and ionic strength. Under such conditions insulin forms fibrillar aggregates with a large amount of amyloid β-sheet structures. After 15 h incubation, in the case of insulin the ThT fluorescence value increases from 470 ± 50 a.u. to 470000 ± 4000 a.u, while in the case of mAb its value increases from 1400 ± 500 a.u. to 9400 ± 400 a.u. The value of ThT fluorescence of mAb aggregates is therefore about 2% of the value for insulin fibrils, indicating that the β-sheet formation in antibody aggregates is quite limited with respect to the extremely regular architecture of amyloid fibrils.
Based on the experimental evidences collected so far we can propose the aggregation mechanism schematically depicted in Fig. 7. The aggregation is initiated by a reversible oligomerization involving mainly monomers, dimers and trimers. In suitable conditions, for example of pH and temperature, such oligomers are not stable and grow via irreversible cluster-cluster aggregation within hours. The formed large aggregates become progressively less soluble and, eventually, precipitate. In all the investigated conditions the aggregation follows a unique mechanism whose kinetics depends on the operating condition. The aggregation is associated to a change of secondary structure inside the cluster, as detected by ThT and ANS binding.

Scheme of the developed aggregation mechanism for the antibody under investigation.
Correlation Between Aggregation Stability and IgG Properties
The previous analysis was extended to other two model immunoglobulins belonging to the IgG1 subclass, referred to in the following as IgG1.1 and IgG1.2. The stability of the three antibodies is compared in Table I, where the stability character observed by SEC and DLS after a characteristic time of 6 h at different pH values and temperatures is reported. It can be seen how the aggregation propensity is different for the different antibodies. At pH 4.5 IgG2 is more unstable than the two IgG1s, while at lower pH the situation is more complex: at pH 4.0 and 37°C, IgG1.1 is stable, while IgG1.2 and IgG2 form unstable oligomers which aggregate during time. Precipitation is observed after some weeks for IgG1.2 and after months for IgG2. At pH 3.0 and 25°C, IgG1.2 is the less stable and significant precipitation is observed within 6 h of incubation. Finally, at pH 3.0 and 37°C all antibodies precipitate within hours.
The aggregation kinetics at 37°C and pH 3.0 was followed by DLS, ThT and ANS binding, and monomer conversion was measured by SEC. The DLS data in Fig. 8a show that the aggregation kinetics in the first 3 h follows the order: IgG1.2 > IgG2 > IgG1.1. It is worth noticing the different behavior between IgG2 and IgG1.1: the latter is characterized by an oligomerization step similar to IgG2, but significantly slower, as indicated by the smaller size of the aggregates in this period of time in Fig. 8a and the lower monomer conversion values in Fig. 8b. Once a critical concentration of IgG1.1 oligomers has been reached (at about 5 h), aggregation to larger aggregates occurs with a much faster kinetics with respect to IgG2 (Fig. 8a). On the other hand, IgG1.2 shows in Fig. 8a to exhibit the fastest aggregation kinetics and the fastest oligomerization rate.

Time evolution of the Z-average hydrodynamic radius, <R h >, (a), and of residual monomer (b) for a 1 g/L protein solution in 25 mM citric acid buffer with 0.15 M Na2SO4 at 37°C and pH 3.0; (c) ThT fluorescence values versus Z-average hydrodynamic radius, <R h >, at the same conditions as a): IgG2 (○), IgG1.1 (■) and IgG1.2 (). Continuous lines are a guide to the eyes.
By looking at the ThT values in Fig. 8c, it appears that the three antibodies exhibit different initial values, indicating a different structure of the non-native monomers. In particular we see that the aggregation of all antibodies is accompanied by structure changes, particularly during oligomer formation, and the fluorescence values of the final aggregates correlate with that of the initial monomers and not with aggregation rate. The slower IgG1.1 oligomer formation is in fact accompanied by a larger increase of order β-sheet structure with respect to the faster aggregating IgG2. On the other hand, IgG1.2 shows the lowest ThT binding, although its aggregation kinetics is the fastest of the three antibodies. Possibly, because of the fast aggregation kinetics, the internal domains do not have time to re-organize.
In order to elucidate which property of the antibody is crucial for the aggregation propensity at low pH, the monomeric antibodies before aggregation were characterized in terms of thermal stability, secondary structure, hydrophobicity and surface charge. On this purpose, experiments were performed in the absence of salt and at low protein concentration (0.3 g/L) to avoid aggregation. This concentration is different from the concentration considered for the aggregation studies (1 g/L). However, the results of all the applied techniques are based on the relative comparison of the three antibodies and are therefore independent of the protein concentration.
The thermal stability at pH 3.0 was investigated by circular dichroism (CD) in absence of salt and at low protein concentration to reduce aggregation during the measurements. In Figure 9a the normalized CD mean residue ellipticity (MRE) at 210 nm as a function of temperature is reported for the different antibodies. Given the multi-domain nature of the protein, a two-state unfolding model would likely not apply for the considered antibodies (40). A simple sigmoidal function was used to fit the data. The thermal stability follows the order: IgG1.1 (most stable) > IgG2 > IgG1.2 (less stable), which is the same order observed for the aggregation stability in the first 3 h of incubation at pH 3.0 (Fig. 8a).

(a) Thermal stability of the three investigated antibodies: normalized CD mean residue ellipticity (MRE) at 210 nm versus temperature for a 0.3 g/L protein solution in 25 mM citric acid buffer at pH 3.0: IgG2 (○), IgG1.1 (□) and IgG1.2 (△). Lines represent sigmoidal fitting to experimental data. (b) Far-UV CD spectra at 20°C for 0.3 g/L solutions of the three investigated antibodies in 25 mM citric acid buffer at pH 3.0: IgG2 (―), IgG1.1 (− −) and IgG1.2 (― •).
The far-UV CD spectra at 20°C and pH 3.0 without salt are shown in Fig. 9b: the three antibodies show different secondary structures, although all the spectra have a minimum at about 216 nm, characteristic of the large β-sheet content of immunoglobulins. In the IgG1.1 spectrum the minimum is shifted from 216 to 218 nm, but the β-sheet content is comparable to IgG2. The IgG1.2, characterized by a large propensity to aggregate, has a significant lower secondary structure amount compared to the other IgGs, in agreement with the low ThT binding reported in Fig. 8c.
The global hydrophobicity of the three antibodies was assessed by ANS measurements (38). The spectra were recorded at 20°C for 0.3 g/L protein solutions in 25 mM citric acid buffer with 25 μM ANS. In Figure 10a the maximum fluorescence values of the ANS emission spectra for the three antibodies solutions at different pH values are compared. According to ANS analysis at all pH values the protein hydrophobicity follows the order: IgG2 > IgG1.1 > IgG1.2. The IgG1.2 is the less hydrophobic, despite the largest reactivity at pH 3.0. In addition, at pH 4.0 IgG1.1 is more hydrophobic and more stable than IgG1.2. The ANS fluorescence values increase by decreasing the pH value, confirming denaturation and exposure of hydrophobic patches at lower pH.

(a) ANS fluorescence emission values at 485 nm for 0.3 g/L solutions of IgG2 (green bars), IgG1.1 (red bars) and IgG1.2 (blue bars) at 20°C in 25 mM citric acid buffer with 25 μM ANS at different pH values. (b) Zeta potential values as a function of pH for 0.3 g/L solutions of the three investigated antibodies in 25 mM citric acid buffer in the pH range from pH 3.0 to 4.5 and in 25 mM phosphate buffer in the pH range from 6.0 to 9.5: IgG2 (○), IgG1.1 (□) and IgG1.2 (△).
Finally, the zeta potential values of the three antibodies were measured as a function of pH (Fig. 10b): at pH 3.0 the zeta potential follows the order IgG1.1 > IgG2 > IgG1.2, which is in agreement with the ranking of aggregation propensity in the first three hours (Fig. 8a). On the other hand, at pH 4.0 IgG1.1 has the lowest zeta potential, and therefore the lowest surface charge, although it is the most stable of the three antibodies. Moreover, at pH 4.5 the zeta potential of IgG1.1 and IgG2 is similar, despite their different aggregation tendency.
Discussion
For all the three considered antibodies the aggregation propensity increases with decreasing pH at a fixed temperature or with increasing temperature at a fixed pH. In both cases, the change of conditions (pH or temperature) promotes unfolding of the native protein. The formation of soluble oligomers within the first hours of incubation at low pH is consistent with the behavior observed with other IgG1s at pH 3.5 (12,14). In this work, addition of salt induces aggregation of the soluble oligomers to larger aggregates at pH 3.0, while at pH 4.0 and 4.5 in the temperature range from 25 to 37°C the soluble oligomers are stable for several days, in contrast with the formation of HMW aggregates observed with other IgG1s at pH 4.5 and larger temperatures (12,14).
At pH 3.0 the order in thermal stability (Fig. 9a) correlates with the order in aggregation stability in the first 3 h (Fig. 8a), confirming the important role of structure stability in determining aggregation propensity in acidic conditions. In general, antibody thermal stability is significantly reduced at low pH, particularly due to the sensitivity of the CH2 domain in the Fc fragment to acidic conditions (16,24,40). IgG2 antibodies are more prone to aggregate than IgG1 antibodies in destabilizing conditions, i.e. physiological pH and elevated temperature (39), or acidic conditions at room temperature (24). This behavior is generally related to the larger stability of the CH2 domain in IgG1 antibodies with respect to IgG2 antibodies (24,30). However, a general conclusion on IgG subclasses cannot be achieved. In this work, the investigated IgG2 is in fact more unstable than the considered IgG1s at pH 4.5, while IgG1.2 (but not IgG1.1) is more unstable than IgG2 in the pH range from 3.0 to 4.0. These results would indicate that also the Fab fragments contribute to the structure and aggregation stability, in agreement with previous studies reported in the literature (12,14,41).
The analysis of the two-step aggregation mechanism gives information about the relationship between the stability of non-native monomers and the stability of aggregates. On one hand, interactions between non-native monomers should reflect interactions between non-native aggregates. On the other hand, aggregation could reduce the number of hydrophobic patches exposed during unfolding, and aggregates could therefore interact more similarly to native proteins. In this work, exposure of the model IgG2 to mildly destabilizing conditions was sufficient to promote reversible-oligomerization: we believe that during the self-assembling the available hydrophobic patches are covered, and oligomers are rather stable within weeks. When exposed to stronger destabilizing conditions (pH 3.0 and larger temperature), the level of unfolding of the monomeric protein increases, exposing a larger number of hydrophobic patches. We think that under these conditions the oligomerization is not enough to cover the aggregating-prone patches, and in the presence of salt the formed oligomers aggregate to larger ones and eventually precipitate. We conclude that, for the IgG2 under investigation, the extent of monomer unfolding is reflected in the stability of the formed aggregates.
On the other hand, the non-native IgG1.1 monomer has a lower tendency to aggregate with respect to IgG2, likely due to the lower unfolding extent. However, the formed oligomers aggregate much faster than the IgG2 aggregates. This behavior could be explained considering the larger ordered structure of IgG1.1 oligomers, as indicate by ThT binding in Fig. 8c: possibly, during the slow oligomerization step oligomers have more time to re-arrange in a more organized structure, which is more prone to aggregate. However, IgG1.2 shows the largest aggregation rates (Fig. 8a) and the lowest amount of ordered secondary structure (Figs. 8c and 9b).
In the case of amyloidogenic proteins, i.e., a class of proteins self-assembling in regular fibrillar structures known as amyloids, the aggregation propensity was found to correlate with general physicochemical properties of the proteins, namely hydrophobicity, surface charge and amount of ordered structure (42). For the three antibodies investigated in this work, the change in zeta potential and in hydrophobicity due to different protein structure or different pH does not correlate with the antibody aggregation propensity. The results suggest that for multi-domain proteins such as antibodies it is difficult to associate the monomer and oligomer reactivity to generic protein physicochemical properties, such as hydrophobicity or charges. Aggregation kinetics in acidic conditions appears therefore related to specific features of the non-native monomer and oligomers, which are still unclear. Aggregation may occur in specific binding sites, as recently shown for antibody dimer formation at low pH (6).
It is worth noticing that the oligomerization and the structure changes occurring in the early stages of aggregation are the less understood steps also in the kinetics of amyloid fibril formation (43). Investigation of IgG aggregation could therefore give mechanistic insights also in the aggregation of amyloidogenic proteins.
Conclusions
We investigate the stability of an IgG2 and two IgG1s at acidic pH.
Two distinct steps are identified for the IgG2 aggregation: 1) reversible oligomerization involving mainly monomers, dimers and trimers, and 2) irreversible cluster-cluster aggregation leading to larger aggregates in the micron range, which, eventually, precipitate.
Temperature, pH and salt concentration determine both oligomerization and growth rate by affecting the protein conformational state. In particular, mild destabilizing conditions promote formation of stable oligomers; on the other hand, stronger denaturing conditions induce oligomer-oligomer aggregation.
In analogy with IgG2, the stability of the two IgG1s decreases with increasing denaturing conditions, i.e. lowering pH and increasing temperature. However, significantly different aggregation rates are observed for the three antibodies in different operating conditions. In particular, the two IgG1s show lower oligomerization rate, but faster cluster-cluster aggregation rate with respect to IgG2. Moreover, IgG2 forms oligomers with long life-spans in a broad range of operative conditions. For all the three antibodies, the oligomerization step is accompanied by conformational changes with an increase in the ordered, β-sheet structures.
The aggregation propensity of the investigated antibodies in acidic conditions does not correlate with antibody subclass, surface net charge and hydrophobicity of the non-native state. The results indicate that the aggregation tendency is dictated by specific features of the non-native structure, which are still to a good extent unclear.
Abbreviations
- ANS:
-
8-Anilinonaphthalene-1-sulfonate
- CD:
-
circular dichroism
- DLS:
-
dynamic light scattering
- HMW:
-
high molecular weight
- Ig:
-
immunoglobulin
- mAb:
-
monoclonal antibody
- MRE:
-
mean residue ellipticity
- MW:
-
molecular weight
- SEC:
-
size exclusion chromatography
- ThT:
-
thioflavin-T
References
Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16.
Aggarwal S. What’s fueling the biotech engine-2007. Nat Biotechnol. 2008;26:1227–33.
Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm. 2005;289:1–30.
Vazquez-Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng. 2011;108:1494–508.
Rosenberg AS. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006;8:E501–7.
Paul R, Graff-Meyer A, Stahlberg H, Lauer ME, Rufer AC, Beck H, Briguet A, Schnaible V, Buckel T, Boeckle S. Structure and function of purified monoclonal antibody dimers induced by different stress conditions. Pharm Res. 2012;29:2047–59.
Arosio P, Barolo G, Muller-Spath T, Wu H, Morbidelli M. Aggregation stability of a monoclonal antibody during downstream processing. Pharm Res. 2011;28:1884–94.
Shukla AA, Gupta P, Han X. Protein aggregation kinetics during protein a chromatography case study for an fc fusion protein. J Chromatogr A. 2007;1171:22–8.
Van Buren N, Rehder D, Gadgil H, Matsumura M, Jacob J. Elucidation of two major aggregation pathways in an IgG2 antibody. J Pharm Sci. 2009;98:3013–30.
Narhi LO, Schmit J, Bechtold-Peters K, Sharma D. Classification of protein aggregates. J Pharm Sci. 2012;101:493–8.
Brummitt RK, Nesta DP, Chang LQ, Chase SF, Laue TM, Roberts CJ. Nonnative aggregation of an IgG1 antibody in acidic conditions: Part 1. Unfolding, colloidal interactions, and formation of high-molecular-weight aggregates. J Pharm Sci. 2011;100:2087–103.
Brummitt RK, Nesta DP, Chang LQ, Kroetsch AM, Roberts CJ. Nonnative aggregation of an IgG1 antibody in acidic conditions, part 2: Nucleation and growth kinetics with competing growth mechanisms. J Pharm Sci. 2011;100:2104–19.
Ejima D, Tsumoto K, Fukada H, Yumioka R, Nagase K, Arakawa T, Philo JS. Effects of acid exposure on the conformation, stability, and aggregation of monoclonal antibodies. Proteins Struct Funct and Bioinformatics. 2007;66:954–62.
Sahin E, Grillo AO, Perkins MD, Roberts CJ. Comparative effects of ph and ionic strength on protein-protein interactions, unfolding, and aggregation for igg1 antibodies. J Pharm Sci. 2010;99:4830–48.
Serno T, Carpenter JF, Randolph TW, Winter G. Inhibition of agitation-induced aggregation of an IgG-antibody by hydroxypropyl-beta-cyclodextrin. J Pharm Sci. 2010;99:1193–206.
Fesinmeyer RM, Hogan S, Saluja A, Brych SR, Kras E, Narhi LO, Brems DN, Gokarn YR. Effect of ions on agitation- and temperature-induced aggregation reactions of antibodies. Pharm Res. 2009;26:903–13.
Perico N, Purtell J, Dillon TM, Ricci MS. Conformational implications of an inversed ph-dependent antibody aggregation. J Pharm Sci. 2009;98:3031–42.
Chari R, Jerath K, Badkar AV, Kalonia DS. Long- and short-range electrostatic interactions affect the rheology of highly concentrated antibody solutions. Pharm Res. 2009;26:2607–18.
Treuheit MJ, Kosky AA, Brems DN. Inverse relationship of protein concentration and aggregation. Pharm Res. 2002;19:511–6.
Thirumangalathu R, Krishnan S, Ricci MS, Brems DN, Randolph TW, Carpenter JF. Silicone oil- and agitation-induced aggregation of a monoclonal antibody in aqueous solution. J Pharm Sci. 2009;98:3167–81.
Tyagi AK, Randolph TW, Dong A, Maloney KM, Hitscherich Jr C, Carpenter JF. Igg particle formation during filling pump operation: A case study of heterogeneous nucleation on stainless steel nanoparticles. J Pharm Sci. 2009;98:94–104.
Duebel S (ed Handbook of therapeutic antibodies. ed. Weinheim, Wiley, 2007.
Ishikawa T, Ito T, Endo R, Nakagawa K, Sawa E, Wakamatsu K. Influence of ph on heat-induced aggregation and degradation of therapeutic monoclonal antibodies. Biol Pharm Bull. 2010;33:1413–7.
Hari SB, Lau H, Razinkov VI, Chen SA, Latypov RF. Acid-induced aggregation of human monoclonal IgG1 and IgG2: Molecular mechanism and the effect of solution composition. Biochemistry. 2010;49:9328–38.
Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 2008;97:1414–26.
Chennamsetty N, Helk B, Voynov V, Kayser V, Trout BL. Aggregation-prone motifs in human immunoglobulin G. J Mol Biol. 2009;391:404–13.
Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A. 2009;106:11937–42.
Kayser V, Chennamsetty N, Voynov V, Forrer K, Helk B, Trout BL. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol J. 2011;6:38–44.
Schaefer JV, Plueckthun A. Engineering aggregation resistance in IgG by two independent mechanisms: Lessons from comparison of pichia pastoris and mammalian cell expression. J Mol Biol. 2012;417:309–35.
Latypov RF, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 fc. J Biol Chem. 2012;287:1381–96.
Roberts CJ. Kinetics of irreversible protein aggregation: Analysis of extended lumry-eyring models and implications for predicting protein shelf life. J Phys Chem B. 2003;107:1194–207.
Roberts CJ. Non-native protein aggregation kinetics. Biotechnol Bioeng. 2007;98:927–38.
Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJA, Middaugh CR, Winter G. Potential inaccurate quantitation and sizing of protein aggregates by size exclusion chromatography: Essential need to use orthogonal methods to assure the quality of therapeutic protein products. J Pharm Sci. 2010;99:2200–8.
Arakawa T, Ejima D, Li T, Phil JS. The critical role of mobile phase composition in size exclusion chromatography of protein pharmaceuticals. J Pharm Sci. 2010;99:1674–92.
Hawe A, Wiggenhorn M, van de Weert M, Garbe JHO, Mahler H-C, Jiskoot W. Forced degradation of therapeutic proteins. J Pharm Sci. 2012;101:895–913.
Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25:1487–99.
Biancalana M, Koide S. Molecular mechanism of thioflavin-t binding to amyloid fibrils. Biochim Biophys Acta Protein Proteomics. 2010;1804:1405–12.
Slavik J. Anilinonaphthalene sulfonate as a probe of membrane-composition and function. Biochim Biophys Acta. 1982;694:1–25.
Franey H, Brych SR, Kolvenbach CG, Rajan RS. Increased aggregation propensity of IgG2 subclass over IgG1: Role of conformational changes and covalent character in isolated aggregates. Protein Sci. 2010;19:1601–15.
Vermeer AWP, Norde W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404.
Chen S, Lau H, Brodsky Y, Kleemann GR, Latypov RF. The use of native cation-exchange chromatography to study aggregation and phase separation of monoclonal antibodies. Protein Sci. 2010;19:1191–204.
Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–8.
Wu C, Shea J-E. Coarse-grained models for protein aggregation. Curr Opin Struct Biol. 2011;21:209–20.
Acknowledgments AND DISCLOSURES
Financial support of the Swiss National Science Foundation (Grant No. 200020-126487/1) is gratefully acknowledged. We thank also Merck Serono (Vevey, Switzerland) for supplying material, Dr. Thomas Müller-Späth for fruitful discussions and Ms. Yang Yang for help with experimental work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 200 kb)
Rights and permissions
About this article
Cite this article
Arosio, P., Rima, S. & Morbidelli, M. Aggregation Mechanism of an IgG2 and two IgG1 Monoclonal Antibodies at low pH: From Oligomers to Larger Aggregates. Pharm Res 30, 641–654 (2013). https://doi.org/10.1007/s11095-012-0885-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-012-0885-3