
Abstract
Antibody drug conjugates (ADCs) are a class of therapeutics that combine the target specificity of an antibody with the potency of a chemotherapeutic. This therapeutic strategy can significantly expand the therapeutic index of a chemotherapeutic by minimizing the systemic exposure and associated toxicity of the chemotherapeutic agent, while simultaneously maximizing the delivery of the chemotherapeutic to the target. The abundance of antibody targets, coupled with advances in antibody engineering, conjugation chemistry, and examples of early clinical success, have stimulated interest in developing ADCs. However, developing and optimizing the highly complex components of ADCs remain challenging. Understanding the pharmacokinetics (PK) and consequently the pharmacokinetic-pharmacodynamic (PKPD) properties of ADCs is critical for their successful development. This review discusses the PK properties of ADCs, with a focus on ADC-specific characteristics, including molecular heterogeneity, in vivo processing, and the implications of multiple analytes. The disposition of ADCs and the utility of PKPD modeling are discussed in the context of providing guidance to assist in the successful development of these complex molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
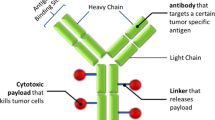
In recent years, a number of novel targeted therapies have been developed to treat cancer. These therapies combine the promise of improved anti-tumor activity with a more tolerable safety profile. Among them, antibody drug conjugates (ADCs) are particularly promising for their high specificity to tumor and high cytotoxicity to cancer cells. ADCs are monoclonal antibodies (mAb) bearing cytotoxic drugs covalently bound via a chemical linker (1,2) (Fig. 1). Often referred to as “targeted chemotherapy”, ADCs are designed to be superior to either antibody therapeutics or chemotherapy alone by overcoming their limitations while preserving the merits from both. In contrast to antibody therapeutics, the cytotoxic agent carried by an ADC could significantly enhance the potency of antibodies beyond their conventional mechanism of action, such as disrupting signal transduction, ADCC (Antibody-Dependent Cellular Cytotoxicity) or CDC (Complement-Dependent Cytotoxicity) (3,4). For instance, Trastuzumab-DM1 has shown efficacy in the trastuzumab refractory patient population and, with its dual mechanism of action, has the potential to be first line therapy for HER2 positive patients (5). The targeted delivery of the cytotoxic drug; meanwhile, can widen their typically narrow or non-existing therapeutic index and bypass drug resistance mechanism(s) that often limit chemotherapeutic effectiveness (6,7). One of the examples is maytasine, which failed in the development as chemotherapeutic in 1980s (8) for lack of therapeutic window, and has been successfully utilized as a conjugating drug in trastuzumab-DM1 (9). For chemotherapeutics that are subject to resistance mechanisms, Kovtun et al. showed that intracellular delivery of maytansinoid DM1 through ADC could bypass the multi-drug resistance (10). In addition, since the utility of the antibody in the context of ADC is predominately for delivery, the ADC strategy enables the expansion of potential targets to any selectively enriched cell surface antigens regardless of their function.

Schematic of ADC stucture. ADCs are monoclonal antibodies bearing cytotoxic drugs covalently bound via a chemical linker.
Another class of antibody conjugates, which uses mAbs conjugated with a particle emitting radioisotope to deliver radiation directly to the tumor, has been successfully developed in treatment of lymphoreticular malignancies (11,12). This approach, known as radioimmunotherapy (RIT), requires a distinct set of optimization and exhibits unique pharmacokinetic and distribution profiles from the small molecule drug conjugates discussed in this review, and interested readers may benefit from several excellent reviews on this topic (4,13-17).
Advances in antibody engineering, conjugation chemistry, and clinical successes of ADCs have heightened interest in their development. More than 20 ADCs are currently in various stages of development and one of them, brentuximab vedotin (ADCETRISTM), was recently approved for the treatment of certain Hodgkin lymphomas and anaplastic large cell lymphomas(18). However, the first approved ADC, gemtuzumab ozagomicin (Mylotarg®) was withdrawn from the market after a recent study showed that postremission treatment with gemtuzumab ozagomicin in older AML patients did not provide benefits regarding any clinical end points (19). This serves as a poignant reminder of the challenges in optimizing and developing safe and effective ADCs.
Optimization of the design and application of these complex molecules is often conducted empirically, with a lack of quantitative and mechanistic understanding of their behavior. An integrated understanding of the pharmacokinetic and pharmacodynamic principles and their applications to target selection, antibody design, linker/drug selection, and drug-to-antibody ratio (DAR) optimization help guide the rational development of ADCs with the best safety and efficacy profiles. In addition, PKPD facilitates ADC development by informing translation of drug effect from in vitro to in vivo, and from preclinical to clinical. In this review, we discuss the PK properties of ADCs, with a focus on ADC-specific characteristics, including molecular heterogeneity, in vivo processing, and the implications of multiple ADC analytes. The disposition of ADCs and the utility of PKPD are discussed in the context of providing guidance to assist the successful development of these complex molecules.
Factors Affecting ADC PK
Structurally, the antibody component of the ADC accounts for the majority of the therapeutic agent (approximately 98% of total ADC by molecular weight). Biologically, the PK of ADCs is strongly influenced by the underlying antibody backbone conferring properties such as target specific binding, neonatal Fc receptor (FcRn)-dependent recycling, and Fc (Fragment, Crystallizable) effector functions. Similarly, the absorption, distribution, metabolism, and elimination (ADME) properties of ADCs possess positive attributes associated with unconjugated antibodies, including slow clearance, long half-life, low volume of distribution, and proteolysis-mediated catabolism. However they also retain less desirable characteristics, including poor oral bioavailability, incomplete absorption following intramuscular or subcutaneous administration, immunogenicity, and nonlinear distribution and elimination (20).
Beyond these similarities, many characteristics of ADCs are distinct from those of an unconjugated antibody, which need to be considered during ADC development. Most notably, ADCs are heterogeneous mixtures of molecular entities or drug species, specifically, antibodies with multiple molecules conjugated at different locations: characteristics that require consideration when evaluating their pharmacology as well as bioanalytical and PK properties. In addition, ADCs consist of two pharmacologically distinct components, the antibody and the cytotoxic small molecule drug (hereafter referred to as drug); this distinction necessitates the understanding of the behavior and fate of both components in vivo. In the following sections, we’ll address those characteristics and their impact on the PK of ADCs.
Heterogeneity of an ADC
An ADC is commonly comprised of a mixture of species, each with distinct structural identity. One source of ADC heterogeneity arises from its manufacturing process. The manufacturing of an ADC involves conjugation of the drug to the antibody, a process that is accomplished by controlled chemical reactions involving specific amino acid residues on the antibody. Cysteine and lysine residues on the monoclonal antibody (mAbs) are often chosen for conjugation due to their amenability to chemical modification (21). Conjugation results in a mixture of ADC species differing not only in the number of drugs attached to the antibody, i.e. drug antibody ratio (DAR) but also in the sites of drug linkage (21,22) (Fig. 2). The amount of drug attached to the antibody is generally reported as an average DAR, which describes a heterogeneous population of ADCs with differing numbers and locations of cytotoxic drugs. For instance, ADCs conjugated by cysteines are mixtures of conjugated antibodies with a DAR ranging from 0 to 8, representing drugs conjugated to some or all of the cysteines that in unconjugated antibodies form the interchain disulfide bonds (21). ADCs conjugated via lysine residues are also mixtures that, with the large number of possible lysine conjugation sites, have the potential for even greater variability in the number of conjugated drugs and their locations (22,23). A second source of ADC heterogeneity results from biological or chemical processes following in vivo administration. These processes can result in the loss of drug from the ADC in systemic circulation, commonly referred to as deconjugation. The mechanisms involved in deconjugation depend on the linker used in the ADC and the site of conjugation, and can include both enzymatic and chemical processes (24). The result of these biological or chemical processes is the loss of cytotoxic drugs, or drug-related species, from the ADC, lowering the average DAR and, potentially, changing the behavior and efficacy of the ADC.

Schematic of ADC heterogeneity. ADCs are a heterogeneous mixture of different DAR species, with individual molecules exhibiting a range of DARs. Adapted with permission from Kaur et al., Mass Spectrometry of Antibody-Drug Conjugates in Plasma and Tissue in Drug Development, in “Characterization of Protein Therapeutics Using Mass Spectrometry,” Guodong Chen, Ed., Springer Press, New York, in press, 2012.
The dynamically changing heterogeneity of ADCs can pose challenges for ADC quantitation, characterization, and optimization. For example, the potency or plasma clearance of an ADC, properties critical to its pharmacologic activity, may depend on the number of conjugated drugs. To overcome this, several approaches have been explored to minimize the production-related heterogeneity of an ADC, particularly cysteine-based conjugates. McDonagh et al. engineered out the cysteines associated with the interchain disulfides resulting in an antibody with fewer cysteines for conjugation and potentially less heterogeneity (25). Alternatively, Junutula et al. reported a novel class of THIOMAB-drug conjugates (TDC) with site-specifically engineered cysteines, which provided a more defined DAR with minimized heterogeneity and increased therapeutic index (26,27). While these technologies allow better control of ADC heterogeneity, the in vivo processes causing heterogeneity in drug load still exist. Currently, the effects of DAR or site of drug conjugation on ADC PK, efficacy, and tolerability (28) are not well understood and difficult to discern. However, the ability to characterize these distinct analytes is critical for designing and optimizing ADCs.
ADC Analytes and Their Pharmacokinetic Significance
The bioanalytical strategy for most drugs is designed to provide quantitative measurement of the analytes necessary for understanding the behavior of the drug in relation to a patient population or disease state. Both, the antibody and small molecule components of an ADC, are critical to its activity, requiring assays suited to measuring these disparate components. These assays may include, but are not limited to, the following: total antibody (conjugated and unconjugated antibody), conjugated antibody, conjugated drug, unconjugated antibody, and unconjugated (free) drug. Each analyte provides unique information regarding ADC behavior in vivo and, singly or in combination, facilitates understanding of ADC PK.
The total antibody (Tab) concentration includes both the conjugated and unconjugated forms of an ADC and is usually determined using an enzyme-linked immunosorbent assay (ELISA)-based format (Fig. 3). The Tab PK profile describes the antibody-related PK behavior of the ADC and provides the best assessment of the in vivo stability and integrity of the antibody over time. The Tab PK profile of ADC serves a key role in ADC optimization, particularly in evaluating the impact of conjugation and selecting a drug load.

Typical ELISA formats for ADC analytes. (a) Total antibody assay: capture of ADC antibody using antigen or target extracellular domain (ECD), with detection using labeled antibody to ADC antibody. (b) Conjugated antibody assay: capture of ADC using anti-cytotoxic drug antibody, with detection using labeled antigen or extracellular domain. Adapted with permission from Kaur et al., Mass Spectrometry of Antibody-Drug Conjugates in Plasma and Tissue in Drug Development, in “Characterization of Protein Therapeutics Using Mass Spectrometry,” Guodong Chen, Ed., Springer Press, New York, in press, 2012.
Conjugated antibody concentrations in systemic circulation are usually determined using an ELISA assay format that measures a mixture of ADC species bearing at least one conjugated cytotoxic drug (Fig. 3). Since the detection in this assay requires the presence of both intact antibody and cytotoxic drug components of the ADC, conjugated antibody concentration is commonly used as an estimate of the active ADC concentration, and is the basis for most ADC PK analyses. However, the interpretation of this concentration is complicated. Two simultaneous processes drive changes in the circulating conjugated antibody concentrations: elimination of intact ADC from circulation and the complete loss of cytotoxic drug (complete deconjugation to DAR 0) from the antibody resulting in unconjugated antibody no longer detected by the assay. In addition, the conjugated antibody assays may have different sensitivity to changes in drug load since it does not differentiate between ADCs with varying numbers of conjugated cytotoxic drugs (DARs). Therefore, because ADC species with different DARs may have different potencies, the measured concentrations may not accurately reflect the associated pharmacologic activity (29). This can be a problem, for example, when comparing the pharmacokinetics of ADCs using assays with different sensitivities to the amount of drug conjugated to the antibody. Even if the PK properties of ADC species are similar, differences in the composition of the circulating ADC mixture (DAR distribution) could lead to different pharmacologic activities and make it difficult to link concentration to physiological effect.
Approaches to measuring conjugated drug vary, but the typical approach involves cleavage of the cytotoxic drug from the antibody followed by cytotoxic drug quantification; providing a measure of the total amount of cytotoxic drug covalently bound to the antibody (30). The interpretation of conjugated drug assay has some similarities with the conjugated antibody assay in the sense that conjugated drug concentration determined by the former assay also describes a mixture of ADC species bearing different amounts of drug; and, changes in conjugated drug concentration could reflect both elimination of ADC from systemic circulation and loss of cytotoxic drug from the antibody. However, in contrast to the conjugated antibody assay, the conjugated drug assay provides limited information about the concentration of the antibody to which the drug is bound, meaning that low concentrations of high DAR species may appear equivalent to high concentrations of low DAR species (30). For example, the conjugated drug concentration of 25 nM of antibody with an average DAR of 4 would be equivalent to 100 nM of antibody with an average DAR of 1. For this scenario, it is conceivable that despite the similarity in conjugated drug concentration the pharmacologic effect may be different.
Cytotoxic drug released from the ADC is a concern and may be associated with loss of efficacy or increased toxicity. Cytotoxic drug loss from an ADC can occur by multiple chemical and enzymatic processes resulting in different structural products. Assays for drug containing products usually employ LC-MS or ELISA methods (23,31). LC-MS methods are highly specific for the measured analyte, while ELISA methods may be less specific and able to quantitate multiple analytes of similar structure. In most cases, unconjugated cytotoxic drug concentrations are used to infer the systemic exposure to the cytotoxic drug released from the ADC. However, a critical aspect of assay selection is ensuring that the analyte(s) selected for measurement are relevant. To do this, prior knowledge of the identity, including metabolism of the cytotoxic drug, pharmacologic activity, and prevalence of these products, is necessary. Unfortunately, the measurements of unconjugated drug analytes and drug related products are rarely reported with the exception of two recent studies (32,33), which makes it difficult to determine whether the measured concentrations are meaningful. While unconjugated cytotoxic drug concentrations can be very low, this information is usually not accompanied by the appropriate context (analyte potency, % dosed, etc.), which makes interpretation of measured concentrations difficult (5,34-36). As our understanding of ADC products expands, clearly articulated strategies for measuring the relevant cytotoxic drug -related products are critical for understanding the pharmacologic significance of these products, as well as potential drug-drug interaction (DDI) risk associated with the cytotoxic drug.
Evolving analytical technologies provide more specific and detailed analysis of the circulating ADC species, offering greater insight into their PK behavior. Among those technologies, affinity capture LC-MS is a powerful method by which the ADC is specifically extracted from plasma, then analyzed using LC-MS/MS (37). The power of this method lies in its ability to provide direct measurement of average concentrations of drug associated with antibody. Xu et al. used affinity capture LC-MS to assess the site-specific loss of cytotoxic drug from a TDC providing valuable insights into the impact of conjugation site on linker stability, a critical factor in ADC safety and efficacy (37). This technology was also applied to determine T-DM1 (38) DAR distribution, allowing a better understanding of in vivo changes in DAR. These examples illustrate the value of evolving analytical technologies in exploring the behavior of ADCs, which in turn can lead to improvements in ADC design and development.
Interpretation of ADC Analytes and PK
As described above, several assays are available for characterizing the PK of ADCs. However, no single assay is able to capture all aspects of the in vivo behavior of these complex molecules, such as the rate of drug loss from an ADC (i.e. linker stability), the effect of conjugation on ADC clearance, and ultimately the exposure-response relationship. Thus, integrating information from multiple assays is critical for the interpretation of ADC pharmacologic effects and ADC optimization. Some important parameters in ADC optimization from integrated PK data are described here.
-
1.
Effect of drug conjugation on antibody exposure and clearance. The conjugation of cytotoxic drugs to an antibody has the potential to affect the pharmacokinetic behavior of the antibody. Comparison of Tab PK of the ADC with the Tab PK of unconjugated antibody (administered as unconjugated antibody) provides information regarding the effect of conjugation on antibody clearance (Fig. 4a) (31,39). For reasons not well understood, conjugation can cause an increase in the clearance of the antibody, which has been observed for several ADCs (22,40-42). In addition, conjugation with higher drug antibody ratio (DAR) tends to have faster clearance than conjugation with lower DAR. For example, cAC10-vc MMAE ADCs with high DARs were observed to have a faster Tab clearance than lower DAR ADCs (22). While the impact of the increase in clearance is not known, it may result in more rapid delivery of cytotoxic drug-bearing ADC to the normal organs or tissues with potentially toxic consequences (22).
-
2.
Rate of drug loss from ADC. Loss of drug from the ADC can result in decreased efficacy and changes in the toxicity associated with ADC administration. To understand this phenomenon, comparative assessment of Tab PK with either conjugated antibody PK or conjugated drug PK can provide qualitative guidance on the rate of drug loss from the ADC. Theoretical plots of this comparison are shown in Fig. 4b.
-
a.
When comparing Tab PK with conjugated antibody PK, it is typically observed that conjugated antibody concentrations decline more rapidly than Tab concentrations, for reasons explained earlier (ADC concentration changes result mostly from two simultaneous processes vs. one process for Tab). The degree of divergence of the curves is indicative of the rate of complete drug loss from the ADC (i.e. DARn-to-DAR zero transition). This is due to the nature of the conjugated drug assay, which measures all ADC bearing one or more drugs. A greater divergence of the conjugated antibody PK from the Tab PK infers a more rapid loss of drug from the ADC. The Tab and conjugated antibody concentrations have been reported for numerous ADCs, with little interpretation of the significance of the difference between these concentrations (5,36,43-46). The comparison of different linkers or conjugation sites, by means of comparing relative linker stability, which can be profoundly affected by site and type of conjugation was recently discussed (27,47,48). Illustrative cases for differences in linker stability between disulfide and thioether linkers, and the effect of conjugation site on thiomab-ADC linker stability were also recently reported (47,48).
-
b.
Comparison of Tab and conjugated drug concentrations are best done with concentrations in molar units (Fig. 4b). This allows for clearer visual assessment of the concentration-time profiles of analytes with widely differing molecular weights. Interpretation of the relationship between Tab and conjugated drug concentrations is, perhaps, less intuitive than for conjugated antibody. Conjugated drug concentrations decline more rapidly than Tab concentrations because two processes drive the decrease in conjugated drug concentrations: loss of drug from the ADC and elimination of ADC, while Tab concentrations changes are driven solely by elimination of ADC and unconjugated antibody. As such, the difference in the concentration decrease can be used to infer the rate of drug loss from the ADC (30,31). At the time of dosing, the difference in molar concentrations reflects the starting average DAR, and at some time after dosing the two concentrations (Total antibody and conjugated drug) may intersect when the average DAR equals 1.
-
a.
-
3.
Accumulation of unconjugated antibody. Unconjugated antibody concentrations (ADC antibody that bears no cytotoxic drug), either as part of the drug product or as a result of complete loss of the cytotoxic drug from the ADC in vivo) is rarely measured directly (43), but can be inferred from concentrations of total antibody and conjugated antibody (44,45). However, given that many of the antibodies used for ADCs have little or no biological activity, the concentration of unconjugated antibody is infrequently calculated and is of limited utility.

Typical concentration time profiles of unconjugated antibody and ADC analytes following an intravenous bolus dose. (a) Comparison of plasma concentration profile of Total antibody (following unconjugated antibody administration) with Total antibody (following ADC administration). Faster decrease in Tab concentrations suggests that pharmacokinetics of the antibody are affected by conjugation. Arrow indicates impact of conjugation on Total antibody clearance. (b) Comparison of plasma concentration profiles following ADC administration. Total antibody (Tab, blue) has multi-exponential profile typical of antibody. Conjugated antibody (gray) shows more rapid decrease in concentration as a result of antibody elimination and cytotoxic drug deconjugation. Conjugated drug (orange) starts at higher concentration than Tab, reflecting its DAR, then decreases more rapidly than Tab due to antibody elimination and cytotoxic drug deconjugation. Arrows indicate effect of deconjugation on clearance. Free drug (green) concentrations are much lower, increase with time to reflect delay in deconjugation from ADC, and decline over time.
While the above-described approaches provide useful guidance in assessing ADC PK, more complex integration of different analytical methods are essential in providing mechanistic understanding of the disposition of the ADC. Leipold et al. (38) described a quantitative model using T-DM1 Tab, conjugated antibody, and ADC catabolites PK assessment linking the various analytes, formation and elimination, thus providing mechanistic information about their behavior. Such models can be useful in designing new ADCs or predicting analyte concentrations in humans.
From these integrated assessments of ADC analyte concentrations, it is possible to make inter-molecule and inter-species comparison of ADCs. However, it is important to note that the uncertainty and variability inherent in the assay design used to determine the various analyte concentrations may be compounded when the different assay data are integrated into quantitative analyses, leading to substantial uncertainty in the interpretation. Careful review of the different assay formats is essential. In addition, the analytical method employing different reagents and the dynamic evolution of analytes can also be obstacles for comparison of PK and PK/PD relationship across different molecules and studies. Stephan et al. showed that signal intensity and analyte recovery largely depend on ELISA assay formats, binding avidity and DAR distribution (29,30). For these reasons, caution is advised in making direct comparison of ADC PK and PKPD without careful consideration of the differences in analytical methods, assay formats, and materials.
Despite these challenges, assessment and integration of ADC PK can be valuable not only in understanding a single ADC, but also in evaluating multiple ADCs with different structural and pharmacologic characteristics; allowing improved design and development of these complex molecules.
ADC Disposition
The systemic PK profiles of an ADC provide only partial narratives describing its disposition. The full absorption, distribution, metabolism, and elimination (ADME) properties of ADCs are also critical for the interpretation of the PK and PK/PD relationship and in turn influencing the selection and development of the successful clinical candidate molecules. Since ADCs, by virtue of their structure, have characteristics of both large and small molecule drugs, characterization of their ADME properties may require a hybrid approach (40,49). Aspects of the importance of understanding of ADC ADME properties, as they apply to PKPD relationship, will be discussed in the following sections. Since ADCs have been largely administered via intravenous or intraperitoneal (mainly in preclinical studies) route and their absorption properties are similar to those of unconjugated antibodies, their absorption characteristics will not be discussed here.
Distribution
The structure of ADCs is dominated by the antibody backbone, and consequently, ADC distribution behavior is usually similar to unconjugated antibodies. Initial distribution is typically limited to the vascular space, with a central compartment volume of distribution similar to plasma volume (~50 mL/kg) (50,51). With time, distribution extends to the interstitial space, with a steady state volume of distribution of approximately 150–200 ml/kg. Similar to unconjugated antibodies, diffusion of ADCs across vascular endothelial cells is very slow, and convection is believed to be the primary mechanism responsible for the transport of antibody from plasma to interstitial fluid. Similar to unconjugated antibodies, ADC distribution can also be affected by target antigen expression and internalization (40).
The presence of a conjugated cytotoxic drug increases the importance of understanding the distribution of the ADC and, in particular, of the cytotoxic drug. Distribution of unconjugated antibodies to non-target tissues, via antigen non-specific or specific processes, may have little pharmacologic effect; while distribution and accumulation of an ADC to the same tissues may have profound pharmacologic/toxic effects as a result of uptake of ADC and subsequent release of the cytotoxic drug or other cytotoxic drug-related catabolites.
With this in mind, there are several situations where attention to ADC distribution can be critical in understanding the pharmacologic and toxic effects. For some ADC targets, antigen is shed by the tumor or normal tissues resulting in appreciable quantities of shed antigen in systemic circulation (46,52). As a result of this, ADC can bind to the circulating antigen affecting changes of ADC distribution and, ultimately, the elimination of ADC. Binding of ADC to soluble antigen can result in clearance of this complex by the liver (52,53) with the potential for liver toxicity related to the delivery of the high amount of cytotoxic drug to that organ.
Even in the absence of circulating antigen, conjugation of cytotoxic drug to an antibody may affect tissue distribution relative to that of unconjugated antibodies. For example, conjugation of antibodies with MMAE (monomethyl auristatin E) has been shown to affect tissue distribution in rodent studies, with increased uptake in the liver when compared to the unconjugated antibody (40). This phenomenon is not unique to rodents or auristatins, as evidenced in the case of antibody hu3S193 and its corresponding ADC CMD-193 where conjugation with calicheamicin had a dramatic impact on the ADC distribution to normal tissues and tumors in human patients; resulting in lower uptake into tumor and greater distribution to liver (41,42). In these examples the distribution of the antibody component of the ADC was followed by labeled antibody analogue (i.e. label on antibody). However, it is also of great importance to understand the tissue distribution of the cytotoxic drug, whether it is bound to the antibody or unbound. Alley et al. have done studies tracking both the antibody and cytotoxic drug components of the ADC with dual labeling radioisotopes on antibody and cytotoxic drug. They found that the normal tissue distribution of the cytotoxic drug, an auristatin (MMAE), was similar to that of the antibody in all tissues except for the liver, where concentrations of cytotoxic drug were higher than antibody (54). The authors suggested that this difference could be attributed to the clearance function of the liver. These examples illustrate that ADC and cytotoxic drug distribution could differ between normal tissues and tumor and highlight the importance of understanding tissue distribution of ADCs, which in turn affects their pharmacologic activity or toxicity. In some cases, low level of target antigen expression in normal tissues and their subsequent uptake of ADC may lead to decreased ADC delivery to tumor and/or increased delivery of cytotoxic drug to normal tissues; a phenomenon that could affect the therapeutic index of the ADC (Boswell et al. submitted).
Metabolism/Catabolism and Elimination
Therapeutic antibodies are thought to be eliminated from the body predominantly via target-mediated and non-specific uptake into cells followed by proteolytic degradation mechanism (20,55). However, ADCs, bearing cytotoxic drugs, may retain activity (or become active) following release from, or degradation of their associated antibodies. Unlike unconjugated antibodies, which catabolic products are typically inert small peptides and amino acids, these degradation products may retain high cytotoxic potency. The safety concern or drug-drug interaction may necessitate the investigation of their identity and pharmacologic properties. In this review, we adopt a convention in terminology that denotes catabolism as the degradation of an ADC to its antibody or small molecule component parts (e.g. lysosomal degradation or linker cleavage); and metabolism, as the processes by which the low molecular weight cytotoxic drug (or cytotoxic drug-containing products) is chemically modified (e.g. oxidative metabolism by cytochrome P450 enzymes) in the body.
The formation of cytotoxic drug-containing products from ADCs may occur by two concurrent processes: deconjugation and catabolism. The deconjugation process includes release of cytotoxic drug-containing products from the ADC via enzymatic or chemical processes, with preservation of the antibody backbone. The catabolism process includes proteolytic catabolism of the conjugated antibody and formation of cytotoxic drug-containing catabolites (free drug or drug-amino acid conjugates) (Fig. 5). These processes can occur simultaneously and their relative contribution to the production of cytotoxic drug-containing catabolic products depend on factors such as linker stability, site of conjugation, and total drug load. For ADCs with linkers susceptible to enzymatic or chemical cleavage, such as disulfide and hydrazone bonds, release of cytotoxic drug by deconjugation process may be predominant. An additional mechanism for the loss of the cytotoxic drug from an ADC via transfer of linker-drug (MC-MMAF) to albumin in plasma is also described for mAb-MC-MMAF (54). It is possible that the same exchange interaction could occur between ADCs with similar linker structures and other thiol-containing constituents (e.g. glutathione, cysteine) present in serum/plasma. This example also points out that in addition to the potential safety implications of these cytotoxic drug-bearing plasma constituents, thorough investigation of the catabolism of ADCs is critical as the nature of the cytotoxic drug-containing products may be difficult to predict a priori.

Diagram of theoretical ADC catabolism. The formation of cytotoxic drug-containing products from ADCs may occur by two concurrent processes: deconjugation and catabolism. The deconjugation process includes release of cytotoxic drug-containing products from the ADC via enzymatic or chemical processes and unconjugated antibody, with preservation of the antibody backbone. The catabolism process includes proteolytic catabolism of the antibody and formation of cytotoxic drug-containing catabolites (free drug or drug-amino acid conjugates).
Another mechanism for producing cytotoxic drug-containing catabolites is ADC catabolism; a process which, as expected, is similar to that for unconjugated antibodies, and is driven by either receptor-mediated endocytosis or fluid-phase pinocytosis with subsequent trafficking to the lysosome, followed by enzymatic degradation (Fig. 5) (49,54,56-58). Depending on linker type, cytotoxic drug-containing catabolites are produced either by cleavage of the linker or by catabolism of the antibody with, in some cases, further intracellular processing (59). Studies of several linker-drug combinations have shown that formed catabolic products depend on linker-drug structure. Disulfide or protease cleavable ADCs such as mAb-SPP-DM1 or mAb-vc-MMAE undergo primarily linker cleavage with cytotoxic drug release (49,56); and ADCs with noncleavable linkers such as mAb-MCC-DM1 and mAb-MC-MMAF, produce catabolic products that contain the linker-drug conjugated to the amino acid (eg. lys-MCC-DM1 or cys-MC-MMAF) (60). Knowledge of the products of ADC catabolism can be valuable in several ways. Some catabolic products, such as S-methyl-DM4 and MMAE have been shown to contribute to drug efficacy by engaging in “bystander effect”, whereby cell-membrane-permeable ADC catabolites formed in tumor cells are able to diffuse from the target-expressing cell to neighboring cells causing their death (56,57), perhaps providing an advantage in tumors with heterogeneous target expression. However, the pharmacologic activity of ADC catabolites formed in the tumor and normal tissues may not be limited to effects on only tumors, but may diffuse into the plasma and exhibit systemic effects, including toxicity and drug-drug interactions.
As described above, the size and structure of ADC catabolic products are similar to small molecule therapeutics. These catabolites may be subject to the metabolism and elimination processes associated with small molecule drugs, including CYPs and drug transporters. While ADC-specific catabolism guidance has not been developed, a well-established framework has been described for assessing metabolites for small molecule therapeutics (61-65). There is a theoretical potential for ADC catabolites to engage in drug-drug interactions (DDIs) with other small molecule therapeutics, affecting the serum or plasma concentrations of either the ADC catabolite or other co-administered medications. However, it is worth noting that given the concentrations of cytotoxic drug released from ADCs are low; the risk of DDI is presumably low. A discussion of this topic is outside the scope of this review, and interested readers are encouraged to refer to the available literature (66) (in press) and draft guidance from FDA (67).
Investigating the mechanisms and products of ADC catabolism requires a multi-pronged approach using both in vitro and in vivo systems. Appropriate in vitro studies including catabolism studies in target-expressing cell lines and plasma stability studies across species can elucidate disposition mechanisms, identify ADC catabolites, and establish the relevance of preclinical species. For example, a recent report on the effect of conjugation site on the in vivo stability and therapeutic activity of TDCs nicely demonstrated the utility of such an integrated approach. Plasma stability data of TDCs along with data from in vivo studies confirmed that the stability and therapeutic activity of the antibody conjugate were affected positively by succinimide ring hydrolysis and negatively by maleimide exchange with thiol-reactive constituents in plasma (48). Another excellent example is the studies on catabolic fate and pharmacokinetic characterization of T-DM1 in rats and humans (68). Catabolites identified in rats using multiple methodologies were monitored and assessed in humans, which provides valuable information on ADC disposition across species.
Immunogenicity
Similar to other large molecule biotherapeutics, ADCs have the potential to induce immune responses in animals and humans. Both intrinsic (product-related) and extrinsic (patient-related) factors can influence the development of ATA (69), ADCs with product-related variants may lead to increased risk of immunogenicity. There is the theoretical risk that conjugation with linker-drug and tertiary structural distortion of the mAb structure resulting from conjugation could lead to a higher incidence of immunogenicity. To date, substantial immunogenicity has not been reported for the two most advanced ADCs in the clinic (32,33). Recent data from T-DM1 trials showed that the incidence of ATAs is low (4.5%) in patients exposed to repeated doses of T-DM1. The immunogenicity of Herceptin® (trastuzumab) was reported in 1 out of 903 patients (Herceptin® (Trastuzumab) package insert); however, comparisons of ATA incidence between products is difficult, given the qualitative or quasi-quantitative nature of ATA methods. Most importantly, there appears to be no obvious effect of ATA on the PK, safety, or efficacy profiles of T-DM1 (32). Brentuximab vedotin reported similar lack of impact from its immunogenicity (33). As with other mAbs, assessment of the clinical importance of immunogenicity of ADCs requires rigorous monitoring (70-72).
Application of PKPD to ADC Development
The development of ADCs provides unique opportunities and challenges for the use of PKPD principles. As with other therapeutics, PKPD can aid in understanding exposure-response relationships, determining the optimal dose and dose regimen, predicting human PK, facilitating the translation of nonclinical data to clinical outcome, and allowing quantitative understanding of mechanistic pharmacology (73). However, the application of PKPD to ADC development has its own challenges.
Notably, the pharmacologic activity of ADCs may be attributed to more than one mechanism (e.g. target-specific efficacy/toxicity and non-specific toxicity) and more than one active ADC-related product (e.g. heterogeneous mixture of intact ADC species and catabolic products), each with potentially different potencies. As described previously, obtaining the necessary information related to these mechanisms and species can require considerable efforts and time.
Exposure-Response
PK/PD modeling provides a quantitative link between drug dose and pharmacologic effect (response), a key component of most drug development programs. A thorough assessment of exposure-response (E-R) relationships can advise on the dose, dose regimen, and dose adjustments in patients (74), in addition to providing a platform for translating pharmacologic effects between animal species to human. For ADCs, E-R is of particular significance due to their typically narrow therapeutic index (relative to unconjugated antibodies) and therefore the need for dose and regimen optimization. The presence of multiple active species in systemic circulation complicates the generation of an E-R relationship, particularly if variability in the relative concentrations or potencies exists between human and animal species (e.g. in translating response from nonclinical to clinical). If, for instance, the key analyte driving drug effect differs between patients and animal species, the strategy for drug monitoring and for modeling the E-R relationship will need to reflect this difference accordingly. However, PKPD coupled with a thorough understanding of the relevant analytes can help to overcome these challenges.
Dose and Dose Regimen Determination
A narrow therapeutic index for ADCs also drives the need to carefully optimize the dose and dose regimen of ADCs. There are considerable clinical precedents for improved tolerability and clinical outcome with oncology agents as a result of modification of dosing regimens (75,76). PKPD methodologies are invaluable tools for quantitative analysis of E-R relationships and simulation of possible outcomes resulting from dose and dose regimen changes. Optimization of dose and dose regimen can be investigated in nonclinical studies, as described by Jumbe et al. (77), where a novel dose-regimen-finding strategy in conjunction with PKPD modeling was used to provide guidance on the optimal clinical dose and dose regimen for T-DM1. This model provided valuable information employed in dose and dose regimen selection in clinical studies during the development of this promising ADC. In the clinic, dose regimen exploration for ADCs has also been conducted but the methods or strategies used to determine the optimal dose have been scarcely discussed. (5,35,46,78). Given the potential for ADC dose and dose regimen optimization to improve clinical outcome; greater effort in this area, and particularly the use of PKPD methodologies, appears warranted.
Human PK Prediction
Another important aspect of translational PK studies is human PK prediction. Predictions of the human pharmacokinetics of ADCs and their cytotoxin-containing catabolic and deconjugation products can be useful in estimating clinical dose and dose regimen, and assessing potential safety risks associated with clinical use of the ADC. Human PK prediction for ADCs has not been extensively reported, which may reflect the limited clinical experience with new generations of linker drug combinations. As discussed earlier, ADC PK measured by its total antibody or conjugated antibody is driven by its mAb moiety rather than its small molecule component. Non-human primates, such as cynomolgus monkeys, share high target antigen homology and similar cross reactivity to antibody with human. Consequently, both target mediated and non-target mediated mechanisms impacting on antibody clearance are comparable between two species. These attributes often make cynomolgus monkey an appropriate species in predicting human PK behavior. Recommendations for mAb human PK projection can be applied to ADCs, i.e., estimating human CL using cynomolgus monkey PK data and an allometric scaling exponent of 0.85 (79,80). However, this approach does not account for the changes to the mAb as a result of conjugation that could impact ADC clearance, such as increased hydrophobicity and structural distortion. These changes could potentially alter ADC distribution and susceptibility to nonspecific proteolytic catabolism (40), and skew the clearance from the predicted value.
Although this strategy, in principle, may work well in predicting human PK for antibody related PK parameters, it is often more challenging in predicting PK behavior for the small molecule drug-related analytes, such as released free cytotoxic drug or cytotoxic drug-containing catabolic products. PK prediction of these small molecule analytes is more complex and subject to not only species differences in their formation, but also in the disposition of the these products. Early experience from ADCETRIS® (brentuximab vedotin) found higher plasma free MMAE levels in patients that were not predicted based on cynomolgus monkey studies (33). Further study of this topic, particularly examination of the mechanistic differences between species in ADC catabolism and disposition and the PK of the resulting products can help not only in the development of current clinical candidates but in the design of new ADCs.
Mechanistic Pharmacology
The area where PKPD can specifically influence ADC development is improving the understanding of the mechanistic processes associated with ADC behavior. The complex interplay of the ADC species, its catabolites, and biologic processes, makes it difficult to make intuitive predictions of the effects of changes in ADC structure (e.g. DAR distribution, linker type, site of conjugation) on pharmacologic response. PKPD modeling, in conjunction with thorough experimentation, can provide essential quantitative mechanistic information for ADC design. Some thorough studies of the ADC in vitro catabolism, in vivo tumor and normal tissue uptake and catabolism in tissues, as well as the relationship of DAR distribution to efficacy and toxicity of ADCs have generated a rich data set (49,54,56,58). This information, coupled with the appropriate PKPD model could:
-
Provide better understanding of the mechanism of action of ADCs
-
Provide critical guidance regarding the optimization of dose and dose regimen
-
Guide extrapolations of ADC behavior between animal species
-
Allow predictions of tumor response in animal and clinical studies.
Indeed, a step in this direction was the work of Leipold et al. using PK modeling in conjunction with quantification of individual deconjugation products by affinity LC-MS, to quantitatively evaluate the linker stability and the catabolite formation rates for T-DM1 (38,81). This work provided insights into the mechanistic behavior of T-DM1, and developed a framework for future work such as prediction of the concentrations of potentially toxic catabolites, and investigation of differences in ADC PK and catabolism in animal species.
Conclusions
The simple concept of “antibody-linker-drug” carries great promise in oncology applications; it is, however, complicated by the need to optimize three different moieties, and synchronize them to generate the most desirable pharmacologic effects. As a hybrid between antibody therapeutics and small molecule cytotoxic drugs, ADCs exhibit unique pharmacological and PK properties. Among them, the heterogeneity from both production and in vivo processing, the necessity to monitor multiple active ADC analytes, and less understood catabolic and metabolic species, demanding for further development of meaningful PKPD relationships. PK and PKPD modeling are powerful tools to integrate these complex interactions with the benefits of better understanding of ADC behavior, and molecule selection, linker drug optimization, dose and regimen selection, and clinical monitoring. The growth of the interest in ADCs, the evolvement of powerful analytical tools, and generation of crucial mechanistic data indicate a promising future for ADC PKPD.
References
Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14:154–69.
Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev. 2006;5:147–59.
Waldmann TA. Immunotherapy: past, present and future. Nat Med. 2003;9:269–77.
Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1:118–29.
Burris 3rd HA, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, Tan-Chiu E, Krop IE, Michaelson RA, Girish S, Amler L, Zheng M, Chu YW, Klencke B, O’Shaughnessy JA. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398–405.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43.
Kustova Y, Espey MG, Sung EG, Morse D, Sei Y, Basile AS. Evidence of neuronal degeneration in C57B1/6 mice infected with the LP-BM5 leukemia retrovirus mixture. Mol Chem Neuropathol. 1998;35:39–59.
Reider PJ, Roland DM. The alkaloids. In: Brossi A, editor. Vol. 23, New York: Academic; 1984.
LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17:6437–47.
Kovtun YV, Audette CA, Mayo MF, Jones GE, Doherty H, Maloney EK, Erickson HK, Sun X, Wilhelm S, Ab O, Lai KC, Widdison WC, Kellogg B, Johnson H, Pinkas J, Lutz RJ, Singh R, Goldmacher VS, Chari RV. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010;70:2528–37.
Chamarthy MR, Williams SC, Moadel RM. Radioimmunotherapy of non-Hodgkin’s lymphoma: from the ‘magic bullets’ to ‘radioactive magic bullets’. Yale J Biol Med. 2011;84:391–407.
Horning SJ. Future directions in radioimmunotherapy for B-cell lymphoma. Semin Oncol. 2003;30:29–34.
Elgqvist J, Andersson H, Back T, Hultborn R, Jensen H, Karlsson B, Lindegren S, Palm S, Warnhammar E, Jacobsson L. Therapeutic efficacy and tumor dose estimations in radioimmunotherapy of intraperitoneally growing OVCAR-3 cells in nude mice with (211)At-labeled monoclonal antibody MX35. J Nucl Med. 2005;46:1907–15.
Sharkey RM, Goldenberg DM. Cancer radioimmunotherapy. Immunotherapy. 2011;3:349–70.
Steiner M, Neri D. Antibody-radionuclide conjugates for cancer therapy: historical considerations and new trends. Clin Cancer Res. 2011;17:6406–16.
Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23:1137–46.
Zhu H, Baxter LT, Jain RK. Potential and limitations of radioimmunodetection and radioimmunotherapy with monoclonal antibodies. J Nucl Med. 1997;38:731–41.
Ingram I. FDA approves brentuximab vedotin for Hodgkin lymphoma and systemic anaplastic large-cell lymphoma. Oncology (Williston Park) 2011;25:904.
Lowenberg B, Beck J, Graux C, van Putten W, Schouten HC, Verdonck LF, Ferrant A, Sonneveld P, Jongen-Lavrencic M, von Lilienfeld-Toal M, Biemond BJ, Vellenga E, Breems D, de Muijnck H, Schaafsma R, Verhoef G, Dohner H, Gratwohl A, Pabst T, Ossenkoppele GJ, Maertens J. Gemtuzumab ozogamicin as postremission treatment in AML at 60 years of age or more: results of a multicenter phase 3 study. Blood. 2010;115:2586–91.
Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–68.
Singh R, Erickson HK. Antibody-cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol. 2009;525:445–67. xiv.
Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL, Francisco JA. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70.
Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005;14:2436–46.
Sun X, Widdison W, Mayo M, Wilhelm S, Leece B, Chari R, Singh R, Erickson H. Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug Chem; 2011.
McDonagh CF, Kim KM, Turcott E, Brown LL, Westendorf L, Feist T, Sussman D, Stone I, Anderson M, Miyamoto J, Lyon R, Alley SC, Gerber HP, Carter PJ. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Ther. 2008;7:2913–23.
Junutula JR, Bhakta S, Raab H, Ervin KE, Eigenbrot C, Vandlen R, Scheller RH, Lowman HB. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J Immunol Methods. 2008;332:41–52.
Dornan D, Bennett F, Chen Y, Dennis M, Eaton D, Elkins K, French D, Go MA, Jack A, Junutula JR, Koeppen H, Lau J, McBride J, Rawstron A, Shi X, Yu N, Yu SF, Yue P, Zheng B, Ebens A, Polson AG. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood. 2009;114:2721–9.
McDonagh CF, Turcott E, Westendorf L, Webster JB, Alley SC, Kim K, Andreyka J, Stone I, Hamblett KJ, Francisco JA, Carter P. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng Des Sel. 2006;19:299–307.
Stephan JP, Chan P, Lee C, Nelson C, Elliott JM, Bechtel C, Raab H, Xie D, Akutagawa J, Baudys J, Saad O, Prabhu S, Wong WL, Vandlen R, Jacobson F, Ebens A. Anti-CD22-MCC-DM1 and MC-MMAF conjugates: impact of assay format on pharmacokinetic parameters determination. Bioconjug Chem. 2008;19:1673–83.
Sanderson RJ, Hering MA, James SF, Sun MM, Doronina SO, Siadak AW, Senter PD, Wahl AF. in vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11:843–52.
Xie H, Audette C, Hoffee M, Lambert JM, Blattler WA. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J Pharmacol Exp Ther. 2004;308:1073–82.
Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, Burris Iii HA, Lorusso PM, Yi JH, Saad O, Tong B, Chu YW, Holden S, Joshi A. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69:1229–40.
Drug Approval Package ADCEREIS (brentuximab vedotin). http://wwwaccessdatafdagov/drugsatfda_docs/nda/2011/125388_adcetris_toccfm; 2011.
Korth-Bradley JM, Dowell JA, King SP, Liu H, Berger MS. Impact of age and gender on the pharmacokinetics of gemtuzumab ozogamicin. Pharmacotherapy. 2001;21:1175–80.
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–21.
Advani A, Coiffier B, Czuczman MS, Dreyling M, Foran J, Gine E, Gisselbrecht C, Ketterer N, Nasta S, Rohatiner A, Schmidt-Wolf IG, Schuler M, Sierra J, Smith MR, Verhoef G, Winter JN, Boni J, Vandendries E, Shapiro M, Fayad L. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J Clin Oncol. 2010;28:2085–93.
Xu K, Liu L, Saad OM, Baudys J, Williams L, Leipold D, Shen B, Raab H, Junutula JR, Kim A, Kaur S. Characterization of intact antibody-drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography-mass spectrometry. Anal Biochem. 2011;412:56–66.
Leipold D, Moore H, Jumbe S, Baudys J, Saad O, Mai E, Wong W, Jay Tibbitts J. Development of a pharmacokinetic model examining trastuzumab-MCC-DM1, MCC-DM1 and DM1 in normal rats. AACR Meeting Abstracts. Oct 2007; 2007.
Sapra P, Stein R, Pickett J, Qu Z, Govindan SV, Cardillo TM, Hansen HJ, Horak ID, Griffiths GL, Goldenberg DM. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clin Cancer Res: Official J Am Assoc Cancer Res. 2005;11:5257–64.
Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, Fourie A, Chuh J, Koppada N, Saad O, Gill H, Shen BQ, Rubinfeld B, Tibbitts J, Kaur S, Theil FP, Fielder PJ, Khawli LA, Lin K. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011;22:1994–2004.
Scott AM, Tebbutt N, Lee FT, Cavicchiolo T, Liu Z, Gill S, Poon AM, Hopkins W, Smyth FE, Murone C, MacGregor D, Papenfuss AT, Chappell B, Saunder TH, Brechbiel MW, Davis ID, Murphy R, Chong G, Hoffman EW, Old LJ. A phase I biodistribution and pharmacokinetic trial of humanized monoclonal antibody Hu3s193 in patients with advanced epithelial cancers that express the Lewis-Y antigen. Clin Cancer Res. 2007;13:3286–92.
Herbertson RA, Tebbutt NC, Lee FT, MacFarlane DJ, Chappell B, Micallef N, Lee ST, Saunder T, Hopkins W, Smyth FE, Wyld DK, Bellen J, Sonnichsen DS, Brechbiel MW, Murone C, Scott AM. Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD-193 in patients with advanced epithelial cancers. Clin Cancer Res. 2009;15:6709–15.
Henry MD, Wen S, Silva MD, Chandra S, Milton M, Worland PJ. A prostate-specific membrane antigen-targeted monoclonal antibody-chemotherapeutic conjugate designed for the treatment of prostate cancer. Cancer Res. 2004;64:7995–8001.
Rupp U, Schoendorf-Holland E, Eichbaum M, Schuetz F, Lauschner I, Schmidt P, Staab A, Hanft G, Huober J, Sinn HP, Sohn C, Schneeweiss A. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: final results of a phase I study. Anti-Cancer Drugs. 2007;18:477–85.
Tijink BM, Buter J, de Bree R, Giaccone G, Lang MS, Staab A, Leemans CR, van Dongen GA. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res. 2006;12:6064–72.
Tolcher AW, Ochoa L, Hammond LA, Patnaik A, Edwards T, Takimoto C, Smith L, de Bono J, Schwartz G, Mays T, Jonak ZL, Johnson R, DeWitte M, Martino H, Audette C, Maes K, Chari RV, Lambert JM, Rowinsky EK. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the CanAg antigen: a phase I, pharmacokinetic, and biologic correlative study. J Clin Oncol. 2003;21:211–22.
Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–90.
Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30:184–9.
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS, Blattler WA. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66:4426–33.
Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy 2010;24:23–9.
Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11:81–8.
Pastuskovas CV, Mallet W, Clark S, Kenrick M, Majidy M, Schweiger M, Van Hoy M, Tsai SP, Bennett G, Shen BQ, Ross S, Fielder P, Khawli L, Tibbitts J. Effect of immune complex formation on the distribution of a novel antibody to the ovarian tumor antigen CA125. Drug Metab Dispos. 2010;38:2309–19.
Lovdal T, Andersen E, Brech A, Berg T. Fc receptor mediated endocytosis of small soluble immunoglobulin G immune complexes in Kupffer and endothelial cells from rat liver. J Cell Sci. 2000;113(Pt 18):3255–66.
Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, Benjamin DR. The pharmacologic basis for antibody-auristatin conjugate activity. The Journal of pharmacology and experimental therapeutics; 2009.
Braeckman RA, editor. Pharmacokinetics and pharmacodynamics of protein therapeutics. NY: Marcel Dekker; 2000.
Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, Wilhelm SD, Singh R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2010;21:84–92.
Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, Senter PD, Alley SC. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–97.
Sutherland MS, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, Lewis TS, Meyer DL, Zabinski RF, Doronina SO, Senter PD, Law CL, Wahl AF. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J Biol Chem. 2006;281:10540–7.
Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc Natl Acad Sci USA. 2005;102:17987–92.
Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19:759–65.
Baillie TA, Cayen MN, Fouda H, Gerson RJ, Green JD, Grossman SJ, Klunk LJ, LeBlanc B, Perkins DG, Shipley LA. Drug metabolites in safety testing. Toxicol Appl Pharmacol. 2002;182:188–96.
Smithand DA, Obach RS. Metabolites in safety testing (MIST): considerations of mechanisms of toxicity with dose, abundance, and duration of treatment. Chem Res Toxicol. 2009;22:267–79.
Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–32.
FDA. Guidance for industry: in vivo drug metabolism/drug interaction studies—study design, data analysis, and recommendations for dosing and labeling. In: DHHS, editor. Rockville, MD: U.S. Food and Drug Administration; 1999.
FDA. Guidance for industry: drug metabolism/drug interaction studies in the drug development process: studies in vitro. In: DHHS, editor. Rockville, MD:U.S. Food and Drug Administration; 1997.
Lu D, Girish S, Theil F, Joshi A. Pharmacokinetic and pharmacodynamic-based drug interactions for therapeutic proteins. In: Zhouand H, Meibohm B, editors. Drug-Drug Interaction for Therapeutic Biologics; 2012.
Drug Interaction Studies--Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations (Draft Guidance). http://wwwfdagov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm121568htm; 2012.
Shen BQ, Bumbaca D, Saad O, Yue Q, Pastuskovas CV, Khojasteh SC, Tibbitts J, Kaur S, Wang B, Chu YW, Lorusso PM, Girish S. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine T-DM1: an emphasis on preclinical and clinical catabolism. Curr Drug Metab; 2012.
Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, Barrett YC, Devanarayan V, Gorovits B, Gupta S, Parish T, Quarmby V, Moxness M, Swanson SJ, Taniguchi G, Zuckerman LA, Stebbins CC, Mire-Sluis A. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333:1–9.
Putnam WS, Prabhu S, Zheng Y, Subramanyam M, Wang YM. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28:509–16.
Peng K, Siradze K, Quarmby V, Fischer SK. Clinical immunogenicity specificity assessments: a platform evaluation. J Pharm Biomed Anal. 2011;54:629–35.
Guidance on Immunogenicity Assessment of Biotechnology-Derived Therepeutic Proteins Draft. wwwemaeuropaeu/pdfs/human/biosimilar/1432706enpdf; 2007.
Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, Street SD. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug discovery today; 2011.
FDA. International Conference on Harmonisation Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. http://www.fda.gov/RegulatoryInformation/Guidances/ucm129335.htm (accessed 10 Feb 2012.
Zandvliet AS, Schellens JH, Beijnen JH, Huitema AD. Population pharmacokinetics and pharmacodynamics for treatment optimization in clinical oncology. Clin Pharmacokinet. 2008;47:487–513.
Tabernero J, Pfeiffer P, Cervantes A. Administration of cetuximab every 2 weeks in the treatment of metastatic colorectal cancer: an effective, more convenient alternative to weekly administration? Oncologist. 2008;13:113–9.
Jumbe NL, Xin Y, Leipold DD, Crocker L, Dugger D, Mai E, Sliwkowski MX, Fielder PJ, Tibbitts J. Modeling the efficacy of trastuzumab-DM1, an antibody drug conjugate, in mice. J Pharmacokinet Pharmacodyn. 2010;37:221–42.
Naumovski L, Junutula JR. Glembatumumab vedotin, a conjugate of an anti-glycoprotein non-metastatic melanoma protein B mAb and monomethyl auristatin E for the treatment of melanoma and breast cancer. Curr Opin Mol Ther. 2010;12:248–57.
Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? mAbs 2011;3:61–6.
Ling J, Zhou H, Jiao Q, Davis HM. Interspecies scaling of therapeutic monoclonal antibodies: initial look. J Clin Pharmacol. 2009;49:1382–402.
Leipold D, Bender B, Keyang K, Theil F, Tibbitts J. Understanding the de-conjugation of Trastuzumab-MCC-DM1 through application of a multi-compartmental model of individual drug: antibody species in cynomolgus monkey. AACR Meeting Abstracts, Apr 2009; 2009: 2914; 2009.
Acknowledgments AND DISCLOSURES
The authors would like to thank Amita Joshi, Ola Saad, Daniel Maslyar, Kelong Han, and Frank-Peter Theil for their constructive input and careful review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lin, K., Tibbitts, J. Pharmacokinetic Considerations for Antibody Drug Conjugates. Pharm Res 29, 2354–2366 (2012). https://doi.org/10.1007/s11095-012-0800-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-012-0800-y