
ABSTRACT
Purpose
To test physicochemical and biological properties of PEG-poly(aspartate) [PEG-p(Asp)] block copolymer micelles entrapping doxorubicin hydrochloride (DOX) through ionic interaction.
Methods
PEG-p(Asp) was synthesized from 5 kDa PEG and 20 Asp units. Carboxyl groups of p(Asp) were present as benzyl ester [PEG-p(Asp/Bz)], sodium salt [PEG-p(Asp/Na)] or free acid [PEG-p(Asp/H)]. Block copolymers and DOX were mixed at various ratios to prepare polymer micelles, which were subsequently characterized to determine particle size, drug loading and release patterns, and cytotoxicity against prostate (PC3 and DU145) and lung (A549) cancer cell lines.
Results
PEG-p(Asp/Bz), Na- and H-micelles entrapped 1.1, 56.8 and 40.6 wt.% of DOX, respectively. Na- and H-micelles (<100 nm) showed time-dependent DOX release at pH 7.4, which was accelerated at pH 5.0. Na-micelles were most stable at pH 7.4, retaining 31.8% of initial DOX for 48 h. Cytotoxicity of Na-micelles was 23.2% (A549), 28.5% (PC3) and 45.9% (DU145) more effective than free DOX.
Conclusion
Ionic interaction appeared to entrap DOX efficiently in polymer micelles from PEG-p(Asp) block copolymers. Polymer micelles possessing counter ions (Na) of DOX in the core were the most stable, releasing drugs for prolonged time in a pH-dependent manner, and suppressing cancer cells effectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Potent anticancer drugs are often small molecules that are poorly soluble in water and highly toxic (1). Small molecules (<1,000 atoms) diffuse into normal tissues readily and get removed from the body quickly (2). Non-specific tissue distribution of anticancer drugs leads to systemic toxicity while rapid clearance of drugs from the body makes it difficult to keep the dose levels for effective cancer treatment (3). Organic solvents and solubilizers are frequently used to dissolve drugs in aqueous solutions at therapeutic dose levels (4). Ionization of drug molecules is a technique that is also used to improve drug solubility in water (5). However, these traditional formulations fail to reduce toxicity and improve therapeutic efficacy of anticancer drugs simultaneously (6).
Large molecules are known to circulate in the blood stream longer than small molecules, and accumulate preferentially in tumor tissues where leaky blood vessels and immature lymphatic drainage are present (7). This phenomenon is referred to as the enhanced permeation and retention (EPR) effect (8). Nanoscaled (<200 nm) particles have demonstrated the ability to take advantage of the EPR effect and accumulate in tumors (9). Nanoparticles entrapping various therapeutic agents act as drug carriers that can circulate in the blood longer, increase drug concentrations in tumors, and thus improve the chemotherapeutic efficacy (10). Polymer micelles, liposomes, solid-lipid nanoparticles, and dendrimers are examples of nanoparticulate drug carriers that have been successfully used in preclinical and clinical applications (11–14). These nanoparticles have provided promising drug carrier platforms that can increase drug solubility, achieving controlled delivery and release of anticancer drugs in targeted tumor tissues (15). Particle size, surface properties, and physicochemical stability are important factors for nanoparticles to maximize tumor-preferential drug delivery (16). Nanoparticulate drug carriers are often modified with small molecules that can strongly interact with proteins that over-express on the cancer cell surface, and thus deliver drugs more effectively to cancer cells (17). Therapeutic outcomes of drug carriers can be enhanced by optimizing these drug delivery techniques (18).
Despite such findings, it is still unclear how drug release patterns of drug carriers affect the cellular response and intracellular drug uptake of cancer cells (19). The general perception is that drug carriers that release drugs quickly in a disease lesion (either inside or outside cells) would be more effective than the drug carriers that release drugs slowly as long as the drug concentration remains the same. Accumulated data, however, constantly suggest that drug carriers have the potential to not only enhance the therapeutic efficacy of their drug payloads, but also behave as novel drugs through a different mode of action (20–24).
We have been developing multifunctional drug carriers using poly(ethylene glycol)-poly(amino acids) block copolymer micelles that can achieve controlled drug release and tumor-targeted drug delivery (25,26). Polymer micelles are spherical nanoparticles prepared from self-assembling block copolymers (27,28). The micelles have a hydrophobic core enveloped by a hydrophilic shell. The core-shell structure is unique to entrap drug molecules inside the micelles and protect the therapeutic agents from precipitation and protein adsorption in the body. The core and shell of the micelles can be modified separately or concurrently to introduce functional groups, drug-binding linkers, cross-linkers, and cell-targeting molecules. Our previous studies demonstrated that polymer micelles possessing drug-binding linkers, which can degrade in response to in vivo stimuli, deliver anticancer drugs more efficiently to tumor tissues with reduced systemic toxicity (29,30). We were able to fine-tune the drug release patterns by modifying the drug-binding linkers with hydrazone, ester and spacers in combination (31–33). One of the polymer micelles was prepared from poly(ethylene glycol)-poly(aspartate hydrazide) block copolymers to which doxorubicin (DOX), an anthracycline anticancer drug, was conjugated through a pH-sensitive hydrazone bond (34). Our observations indicated that drug release from polymer micelles can be controlled successfully by modulating the hydrolysis rates of the drug-binding linkers, yet at the same time, the interaction between the drug molecules and polymers appeared to be crucially important to maintain the particle stability of polymer micelles. An ionizable moiety on a drug molecule also plays an important role in stabilizing the micelles and controlling the drug release rates favorably. These results, therefore, suggest that drug release can be controlled by hydrophobic and ionic interactions between drug molecules and micelle-forming block copolymers, providing the simplest experimental model to elucidate the influence of drug release patterns on cellular response more directly.
Micelle formation is an entropy-driven process, and particle stability is determined primarily by the core environment and drug molecules entrapped (35,36). Hydrophobic and lipophilic drugs are known to stabilize the micelle core through hydrophobic interactions between drug molecules and polymer chains (37–39). Stable micelles improve drug entrapment, circulate in the body for longer time, and eventually enhance drug delivery to tumors (40–42). In comparison to the hydrophobic interaction, the ionic interaction is more convenient to prepare polymer micelles because all materials involved in the micelle formation can readily dissolve in aqueous solutions, making them favorable for biomedical applications (43–45). Ion complexes between charged molecules are unstable in the presence of counter ions. Ionizable moieties on drug molecules often hamper effective drug entrapment in polymer micelles. Cross-linking and salt-precipitation are methods often used to stabilize these ion complexes. DOX contains a tetrahydro-naphthacenedione ring conjugated with a daunosamine amino sugar through a glycosidic bond, which contribute to π-π stacking and ionization of the drug molecule, respectively. The hydrochloric acid salt form of DOX (DOX-HCl) is highly soluble in water. As the counter ions of DOX-HCl, Na ions in the micelle core were expected to affect entrapment and release of DOX.
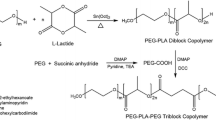
For these reasons, in this study, we prepared polymer micelles with three different core environments using poly(ethylene glycol)-poly(aspartate) (PEG-P(ASP)) block copolymers and doxorubicin hydrochloride (DOX-HCl) as shown in Fig. 1. The carboxyl groups of PEG-P(ASP) were protected by benzyl esters (Bz), ionized by sodium salt (Na), or remained as free acids (H) to entrap DOX through hydrophobic or ionic interactions in the presence and absence of counter ions. These micelles are denoted as Bz-, Na-, and H-micelles, respectively. Entrapment, release and cytotoxicity of DOX were assessed to understand the effect of controlling drug release patterns on cellular response to DOX-loaded polymer micelles. Drug entrapment yields were determined before and after purification of polymer micelles to which DOX was entrapped at various feeding ratios (25~800%) with respect to the number of carboxyl groups of PEG-P(ASP). Drug release patterns were observed at pHs 7.4 and 5.0 (corresponding to the physiological and lysosomal conditions, respectively) for 48 h, which we confirmed previously as the time period the micelles can be present in tumor tissues. Cytotoxicity of Bz/Na/H micelles was determined with human lung (A549) and prostate (PC3 and DU145) cancer cell lines. Lastly, time-dependent changes in intracellular drug uptake were monitored by quantifying DOX accumulated inside cells following the incubation of cells with Bz/Na/H micelles. Therefore, this study is expected to provide experimental evidence and better understanding on drug release from polymer micelles and subsequent cellular response.

Preparation of drug-loaded block copolymer micelles with different core properties. The micelles entrapping doxorubicin (DOX) were prepared from PEG-poly(aspartate) block copolymers that have hydrophobic benzyl groups (Bz), sodium ions (Na), and free carboxylic acid (H) on the side chain, denoted as Bz, Na and H micelles, respectively. Ionic interaction neutralizes the charge between block copolymers (-COO-Na+ or -COO-H+) and DOX (-NH2/HCl). The anthracycline ring of DOX is responsible for hydrophobic interactions between drug molecules in the micelles.
MATERIALS AND METHODS
Materials
α-Methoxy-ω-amino poly(ethylene glycol) (PEG-NH2, 5 kDa) was purchased from NOF Corporation (Japan). L-aspartic acid β-benzyl ester, triphosgene, doxorubicin hydrochloride (DOX), benzene, anhydrous N,N-dimethylformamide (DMF), dimethylsulfoxide-d6 (DMSO-d6), deuterium oxide (D2O), anhydrous ethyl ether, anhydrous hexane, anhydrous tetrahydrofuran (THF) were purchased from Sigma-Aldrich (USA). Regenerated cellulose dialysis bags with molecular weight cut off (MWCO) 6–8 kDa, Slide-A-Lyzer G2 dialysis cassettes with MWCO 10 kDa, phosphate buffer solution (pH 7.4, 10 mM), acetate buffer solution (pH 5.0, 10 mM) and acetonitrile (ACN) were purchased from Fisher Scientific (USA). Amicon-Ultra centrifugal ultrafiltration kits with MWCO 30 kDa were from Millipore (USA).
Cell Lines
DU-145 (human prostate cancer cells), PC3 (human prostate cancer cells), A549 (human lung cancer cells), and F12K cell culture medium were obtained from American Type Culture Collection (USA). Fetal bovine serum (FBS) and RPMI 1640 medium were purchased from Atlanta Biologicals (USA). Hyclone phosphate buffer saline (PBS), trypsin-EDTA (0.25% trypsin and 2.21 mM EDTA) and other cell culture supplies (e.g. 96-well culture plates, pipettes and flasks) were from Fisher Scientific (USA). Cells were cultured in either RPMI1640 or F12K media containing 10% FBS in a humidified atmosphere with 5% CO2 at 37°C.
PEG-PBLA Block Copolymer Synthesis
Our synthesis protocol is shown in Fig. 1. β-Benzyl-L-aspartate N-carboxy anhydride (BLA-NCA) was prepared using the Fuchs-Farthing method as described previously (46). Triphosgene (1.3 eq) and β-benzyl-L-aspartate mixed in dry THF at 50 mg/ml. The reaction was conducted in N2 at 45°C until the solution became clear. Anhydrous hexane was slowly added to the reaction solution, followed by recrystallization of BLA-NCA at −20°C overnight. Purified BLA-NCA was polymerized in anhydrous DMSO at 45°C for 2 days by using amino-terminated PEG as a macroinitiator. The amount of BLA-NCA was adjusted with respect to PEG to prepare poly(ethylene glycol)-poly(β-benzyl L-aspartate) (PEG-PBLA) block copolymers with 20 Asp units. The reaction solution was precipitated in anhydrous ethyl ether, followed by freeze drying of PEG-PBLA. 500 MHz 1H-NMR was used for characterization.
PEG-p(Asp/Bz/Na/H) Block Copolymer Synthesis
PEG-PBLA was used as PEG-p(Asp/Bz) in which all carboxyl groups of p(Asp) segment were protected by benzyl ester. PEG-PBLA was deprotected by 0.1N sodium hydroxide (NaOH) to prepare PEG-p(Asp/Na) block copolymers that have sodium carboxylates on the side chain. The deprotection reaction was conducted until PEG-PBLA solutions turned completely clear. NaOH and benzyl alcohol byproducts were removed by dialysis of the deprotected polymers against deionized water. PEG-p(Asp/H) was prepared by ionic exchange of PEG-p(Asp/Na) in 1N HCl solution, which was dialyzed against deionized water to remove NaCl byproducts completely. All purified block copolymers were collected by freeze-drying. Products were characterized by 1H-NMR (500 MHz) and GPC (Shimadzu LC20, PEG standard, 100 mM PBS mobile phase at 40°C).
Preparation of Polymer Micelles
We fixed the polymer concentration (10 mg/mL) for preparation of all micelles and added DOX in solutions at pre-determined mixing ratios by mass. Polymer micelles entrapping drugs through hydrophobic interaction (Bz-micelles) were prepared by mixing PEG-p(Asp/Bz) and DOX in acetonitrile (ACN) at 10 mg/ml. The mixing ratios between DOX and PEG-p(Asp/Bz) were 0.25, 0.5, 1, 2 and 8 (DOX/carboxyl groups). The organic solution was dialyzed against deionized water (MWCO 6–8 kDa) and purified further by centrifugal ultrafiltration (MWCO 100 kDa) until no further DOX was observed in the filtrates. Prepared Bz-micelles were filtered through 0.45 μm filters, and freeze-dried. Polymer micelles entrapping drugs through combinational hydrophobic and ionic interactions were prepared by mixing DOX with either PEG-p(Asp/Na) or PEG-p(Asp/H) in deionized water. The mixing ratios of DOX and carboxyl groups on the block copolymers were also 0.25, 0.5, 1, 2 and 8 equivalents. Centrifugal ultrafiltration (MWCO 100 kDa) was conducted to remove any unbound DOX from the micelles. Complete removal of free DOX from the micelles was confirmed by measuring DOX in the filtrates at 485 nm (DOX has strong absorbance at 485 nm). The aqueous solutions were dialyzed against deionized water (MWCO 6–8 kDa), filtered through 0.45 μm filters, and freeze-dried.
Characterization of Polymer Micelles
Particle sizes of polymer micelles were determined at 2 mg/ml by dynamic light scattering measurement (Zetasizer Nano-ZS, Malvern, UK). Drug-loading was determined by UV/VIS spectrometry at 485 nm using calibration curves of free DOX in deionized water. The drug loading for each polymer micelle was determined by the polymer mass (mg DOX/mg polymer), or also described as weight% (wt%). Long-term stability of polymer micelles was tested in frozen solutions or as a powder form by reconfirming drug-loading and particle sizes for 6 months. All measurements were made in triplicate.
Drug Release Experiments
Drug release patterns of polymer micelles were determined at different pHs, which simulate the physiological condition in the blood (pH 7.4) and acidic environment in intracellular lysosomes (pH 5.0). Polymer micelles (5 mg/ml) were added to dialysis cassettes (MWCO 10 kDa, Slide-A-Lyzer G2, n = 3) floating in >1,000 fold buffer solutions by volume. The micelles were dialyzed at 37°C for 48 h. Drugs remaining in each dialysis cassette were quantified by UV/VIS spectrometry (485 nm) at 0, 0.5, 1, 3, 6, 24 and 48 h. Drug release patterns were analyzed by determining both% released and calculating the area under the concentration curves (AUC). The AUC values were determined between 0–6 h (AUC0-6h) and 6–48 h (AUC6-48h), defined to the drug released in an early and late stages during the overall period of drug release.
Cytotoxicity Assays
Biological activity of DOX-loaded Bz/Na/H micelles was evaluated by using three human cancer cell lines, which include DU145 (human prostate cancer in early disease stage), PC3 (human prostate cancer in late disease stage), and A549 (human non-small cell lung cancer). Growing cells were seeded on 96-well plates (4–5,000 cells/well) and exposed with polymer micelles at various concentrations for 72 h. The concentrations were adjusted based on either DOX.. Cell viability was determined by a Resazurin metabolism assay measuring fluorescence (excitation 560 nm and emission 590 nm). Three independent assays were repeated (n = 8). Data were shown average cell viability ± standard deviation (mean ± SD). The half maximal inhibitory concentration (IC50) values were determined by GraphPad Prism (version 5.02 for Windows, GraphPad Software). One-way analysis of variance (ANOVA) was used to determine statistical significance (p < 0.01).
Cytotoxic Assay (8 × 8 Combination)
We examined potential combinational effects of polymers on cytotoxicity of DOX to PC3 cancer cells. Cells were seeded on a 96-well plate (5,000 cells/well) and allowed to attach overnight. Cells were then exposed to different mixing ratios of DOX and polymers for 72 h starting with the lowest concentration of each and ending with both of the highest concentrations, mixing the concentrations per well.
Drug Stability Confirmation
Chemical and biological stability of DOX from polymer micelles were tested by HPLC analysis and in vitro cytotoxicity assays, respectively. HPLC analysis was conducted by using Shimadzu LC20 equipped with a SPD-M20A photodiode array detector (400–700 nm) and an Eclipse XDB-C18 column (4.6 mm × 150 mm, 5 μm, Agilent Technologies) column at 40°C. The mobile phase (45% ACN/55% H2O) was run at a flow rate of 1 ml/min. Biological stability was determined by incubating samples for 0, 24, 48 and 72 h at 37°C prior to cytotoxicity assays following the aforementioned protocol.
Immunoblot Analysis
Exponentially growing cells (PC3) were treated with free DOX or Bz/Na/H micelles at the IC50s for 72 h. Nuclear and cytosolic fractions were isolated from these cells as previously reported with slight modification (47). Non-treated and DOX-treated PC3 cells (2 × 10−6) were trypsinized and washed with ice-cold PBS. Cell pellets were washed twice with ice-cold nucleus buffer (150 mM NaCl, 1 mM KH2PO4, 5 mM MgCl2, and 1 mM EGTA). The cells were then incubated in nucleus buffer containing 0.03% Triton in wet ice for 10 min and centrifuged, collecting the cytosolic fraction in the supernatant. To collect nuclear extract, cell pellets were washed twice with ice-cold nucleus buffer, resuspended in 0.35 M NaCl and gently mixed for 30 min at 4°C. All procedures were performed in the presence of protease inhibitors (Complete Mini, Roche Diagnostics, Germany). Cell products were stored at −20°C. Whole cell lysates were prepared from the cells following the procedure previously outlined (48). The cells were incubated for 20 min at 4°C with lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 0.5% SDS, 1% sodium deoxycholate and 1% Nonidet P-40) supplemented with protease inhibitors, sonicated and centrifuged to collect the whole cell lysate. Protein content in the nuclear and cytosolic extracts and the whole cell lysates were determined using a colorimetric assay (BCA protein assay) following the manufacturer’s protocol (Thermo Scientific, USA).
The lysates were then loaded onto a NuPAGE Novex 4–12% Bis Tris gel (Invitrogen, USA) and electrophoresed. Proteins were transferred onto a nitrocellulose membrane (Invitrogen, USA) and blocked with 5% non-fat milk in PBS with 0.05% Tween-20 (PBS-T) unless recommended differently by the antibody supplier. Membranes were incubated with anti-Bcl-2 (Epitomics), anti-Bax (P19) (Santa Cruz Biotechnology), anti-NF-κB p65 (Cell Signaling Technology), anti-Topoisomerase IIα (Cell Signaling Technology), anti-Phospho-Histone H2Ax Ser139 (Cell Signaling Technology), anti-Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Abcam, Cambridge) and anti-Acetyl-Histone H3 (Upstate-Millipore) overnight at 4°C. After secondary antibody incubation, proteins were visualized using ECL (GE Healthcare) on the Image Station 2000 MM (Eastman Kodak) equipped with the Molecular Imaging Software (version 4.04, Eastman, Kodak) used for densitometry analysis. Band intensities in whole lysate and cytosolic fraction samples were normalized with respect to GAPDH and band intensities in nuclear fraction samples were normalized with respect to Acetyl-Histone H3. Prism Software was employed for the graphical representation of the results.
Determination of Intracellular DOX Concentration
A549 cells were cultured in F-12K cell culture medium supplemented with 10% fetal bovine serum (FBS) and seeded onto 96-well plates at a cell density of 10,000 cells per well. After 24 h incubation period, cell culture medium was aspirated and the cells were exposed to various concentrations of free DOX (0.025–500 μM) for 0.5, 1.0, 3.0, and 6.0 h at 37°C. Following the incubation periods, cells were washed with PBS three times and replaced with 100 μL PBS. Fluorescence of each cell solution was measured at an excitation wavelength of 485 nm and emission wavelength of 560 nm to determine the concentrations of DOX in the cells. Time-dependent changes in intracellular drug uptake were subsequently monitored. Cells (10,000 cells/well) were seeded on 96-well plates. After a 24 h incubation period, cell culture medium was aspirated and the cells were treated with Bz/Na/H micelles and free DOX for 0.5, 1, 3, 6, 24, 48, and 72 h at 37°C. The drug concentrations were 50 μM or 100 μM. Following the incubation periods, cells were washed with PBS three times and lysed with 100 μl DMSO. Fluorescence of each cell solution was measured at 560 nm with 485 nm excitation.
RESULTS
Block Copolymer Synthesis
1H-NMR and GPC analyses confirmed that PEG-PBLA was successfully synthesized, producing neither unreacted PEG nor p(Asp) homopolymers (data not shown). Peak ratios between PEG (3.5 ppm) and benzyl esters (7.4 ppm) showed that PEG-PBLA consisted of 5 kDa PEG and 20 Asp units. Deprotection of benzyl esters from PEG-PBLA produced PEG-p(Asp/Na), which was fluffy powder and readily soluble in water causing no aggregation. PEG-p(Asp/H) was brittle powder, yet also dissolved easily in water with no aggregation. GPC measurements revealed that PEG-p(Asp/Bz) formed large aggregates in aqueous solutions while both PEG-p(Asp/Na) and PEG-p(Asp/H) present as individual polymer chains. 1H-NMR analysis confirmed the absence of benzyl alcohol, a byproduct during the polymer deprotection. Molecular weight determined by GPC suggested that no degradation of the block copolymer backbone was observed during deprotection and ionic exchange procedures using NaOH and HCl, respectively.
Polymer Micelle Preparation and DOX Entrapment
Drug loading experiments revealed that DOX was entrapped in polymer micelles more effectively through ionic interactions than hydrophobic interaction. Figure 2 shows that approximately 20% of DOX were entrapped by PEG-p(Asp/Bz) as the mixing ratio between the drug and carboxyl groups on the block copolymer increased from 25% to 100%. When the mixing ratio was higher than 100%, most DOX molecules were washed off through ultrafiltration without binding to the Bz-micelles. There was a significant decrease in sample recovery from Bz-micelles across the ultrafiltration membrane when 800% DOX was added, which is presumably because the pores on the membrane were clogged by DOX molecules at high concentrations. Na- and H-micelles entrapped DOX efficiently even at the 200% mixing ratio, suggesting that DOX was bound to block copolymers through ionic interaction (up to 100%) and additionally through hydrophobic interaction (>100%). PEG-p(Asp/Na) and PEG-p(Asp/H) appeared to entrap 1.25 fold DOX than its capacity, which was calculated from 62.6% DOX retention at 200% mixing ratio.

DOX entrapment yields of Bz/Na/H micelles. *Data were not collected due to sample precipitation.
Drug entrapment yields decreased drastically after repetitive purification of all Bz/Na/H micelles by centrifugal ultrafiltration. Final DOX entrapment yields for Bz-, Na-, and H-micelles were 1.1%, 56.8% and 40.6%, respectively. These results indicate that the high drug entrapment observed initially (>100%) was attributed to DOX molecules weakly bound to the polymers. It is still noticeable that Na- and H-micelles entrapped 52 and 37 times more DOX than Bz-micelles, respectively. Bz/Na/H micelles purified by ultrafiltration were subsequently characterized to determine particle size and stability.
Particle Size and Shelf Stability of Polymer Micelles
DLS measurements showed that all micelles entrapping DOX exhibited a particle size below 100 nm (Table I), which is clinically relevant for passive tumor targeting. Bz-micelles were the smallest in particle size with an average of 51 nm. Na- and H-micelles were 93.4 nm and 90.7 nm, respectively. To confirm shelf-stability, polymer micelles were stored as either powder or solutions for 6 months, repeating freezing and thawing randomly. When stored as powder, Bz-micelles were not reconstituted readily irrespective of storage period (hours to months). Bz-micelle solutions remained clear causing no precipitation, when stored as frozen solutions. However, DOX filtrates were always observed after ultrafiltration of Bz-micelles regardless of storage at 4°C or −20°C. On the other hand, Na- and H-micelles showed excellent shelf-stability as either powder or solutions. Freeze-dried powders of Na- and H-micelles were readily reconstituted to prepare micelle solutions at various concentrations, which were tested up to 10 mg/ml based on DOX loading. Thawing frozen solutions did not cause DOX release from Na- and H-micelles even after centrifugal ultrafiltration. These results indicate that ionic interaction is effective to entrap DOX molecules in polymer micelles during storage as powder or solutions. HPLC measurements confirmed that DOX released from the micelles appeared at minute 8, the same time as freshly prepared DOX (data not shown). A single peak of DOX was observed on the HPLC, indicating that DOX in the micelles neither underwent degradation nor dimer formation during storage.
DOX Release from the Micelles
Drug release experiments revealed that Bz-micelles entrapping DOX through hydrophobic interaction released more than 50% DOX in 6 h at both pH 7.4 (64.6%) and 5.0 (64.1%) as shown in Fig. 3. There was no significant difference in drug release patterns of Bz-micelles between pH 7.4 and 5.0. On the other hand, Na- and H-micelles remained stable, entrapping 75.8% and 63.47% DOX at pH 7.4 during the same period (0–6 h). Drug release from Na- and H-micelles was accelerated as pH decreased from 7.4 to 5.0. Interestingly, drug release patterns after 6 h become identical when Na- and H-micelles were incubated at pH 5.0. Figure 4 summarizes the area under the curve (AUC) values of DOX released from the micelles at different pHs shown in Fig. 3. In the early stage (0–6 h), Na- and H-micelles retained 68.8% and 42.8% more DOX than PEG-p(Asp/Bz) at pH 7.4. The difference in drug remaining in the micelles became greater in the late stage (6–48 h). DOX was entrapped 3.30 and 2.59 times more in the Na- and H-micelles over the Bz-micelles. Drug release patterns at pH 5.0 indicated that the micelles entrapping DOX through ionic interaction released 44.2% (Na-micelles) and 37.9% (H-micelles) less DOX than Bz-micelles in the early stage at pH 5.0. In the late stage at pH 5.0, the drug release was accelerated. It is intriguing that Na- and H-micelles released 52.5% less DOX than Bz-micelles. Complete DOX release was not observed from any micelle tested in this study for 48 h.

Drug release patterns of Bz/Na/H micelles at pHs 7.4 and 5.0 (37°C). Free DOX was used as a control to determine the dialysis efficiency and data normalization.

Area under the curve (AUC) of drug release patterns of Bz/Na/H micelles at pHs 7.4 and 5.0 (37°C). The AUC values estimate the degree of drug exposure to cancer cells in the early (0–6 h) and late (6–48 h) incubation stages.
Cell Viability Assays
Bz/Na/H micelles inhibited all cancer cells tested (DU145, PC3 and A549) in concentration-dependent manners (Fig. 5). DU145 was the most sensitive to free DOX, followed by A549 and PC3. In comparison with free DOX, Na- and H-micelles were equally or slightly more effective to all cell lines. Bz-micelles showed variable cytotoxicity following storage for different periods. Surprisingly, Na-micelles appeared more potent than free DOX, the drug payload, in all cell lines tested (p < 0.01). Such enhanced cytotoxicity of Na-micelles was reproducible in multiple cytotoxicity assays conducted in this study. No deterioration was seen in biological activity of Na-micelles irrespective of storage periods. H-micelles also showed cytotoxicity similar to free DOX, indicating that the micelles released DOX in an active form. Enhanced cytotoxicity was not observed in H-micelles. Empty polymer micelles showed no cytotoxicity when tested in all three cell lines. IC50 values were not obtained since there was no cell death (data not shown). The 8 × 8 assay performed revealed that the trend in cytotoxicity did not change when the concentration of empty polymer increased in the case of all three polymer types as seen in Fig. 6. These results confirmed that enhanced cytotoxicity of Na-micelles was not attributed to potential toxicity of empty micelles.

The half maximal inhibitory concentration (IC50) values of free DOX and micelles against various cancer cell lines (PC3, A549, and DU145).

Combination cytotoxicity assays (8 × 8) to investigate potential effects of empty Bz/Na/H micelles on cellular response to DOX.
Chemical and Biological Stability of DOX
Differential cytotoxicity of micelles at the same drug concentration suggested that DOX that was released from the micelles but not immediately taken up by the cells could lose its bioactivity and was no longer toxic to the cancer cells. In addition, since Na and H micelles released DOX slowly for a full 48 h, it was expected that the drug released from the micelles was protected from the cell culture media while inside the micelle and therefore retained its bioactivity when released from the micelle and taken in by the cells. To test these hypotheses, DOX was added to cell culture plates in media containing 10% FBS and allowed to incubate at different time points (0, 24, 28, 72 h incubation). Table II summarizes the results, which indicate that DOX showed consistent IC50 values irrespective of pre-incubation time. Therefore, chemical and biological deterioration of DOX under our experimental conditions appeared negligible.
Western Blotting
Cell survival and apoptotic cell death signaling pathways were investigated by the Western blotting. DOX induces cell apoptosis by inhibiting the molecular function of topoisomerase II (TOP2) which is an enzyme essential in the separation of entangled daughter strands during DNA replication. The expression levels of TOP2 (Fig. 7a) were normalized with respect to H3 (Fig. 7b), indicating that free DOX and micelles did not change TOP2 expression levels in the cell. Two Bcl-2 family members (anti-apoptotic Bcl-2 and pro-apoptotic Bax) were subsequently monitored (Fig. 7c). We observed no significant change in Bcl-2/Bax expression among free DOX and micelle formulations (Fig. 7d). Nevertheless, the expression levels of total and cleaved Poly ADP-ribose polymerase (PARP) confirmed that free DOX and micelles caused considerable PARP cleavage (Fig. 7e). PARP cleavage was particularly evident with Bz and Na micelles. Contrary to TOP2, the expression of γH2Ax, which is the phosphorylated form of the histone H2Ax, increased when the cells were treated with free DOX and micelles (Fig. 7f). However, there was no significant difference in the γH2Ax expression among the cells exposed to different DOX formulations (Fig. 7g). By isolating nuclei from the cytosolic fraction, the migration pattern of the nuclear factor kappa B (NFκB) from the cytosol to the nucleus was determined. Our results show that NFκB expression levels did not change significantly in the cytosol after treating the cells with free DOX and micelles (Fig. 7h). Interestingly, the nuclear NFκB level decreased more than 60 times in the treated cells, while no DOX formulations changed the protein turnover in the nuclear compartment (Fig. 7i).

Western blotting assays to determine the expression levels of Top2, γH2Ax, total/cleaved-PARP, Bcl-2/Bax and NFκB in PC3 cells treated with free DOX and Bz/Na/H micelles.
Time-Dependent Intracellular Drug Uptake
We quantified intracellular DOX concentrations in cancer cells exposed to free DOX and Bz/Na/H-micelles for 72 h. Figure 8 shows that intracellular drug concentration continued to increase in the cells exposed to free DOX, Na- and H-micelles. It is noticeable that Na- and H-micelles showed higher intracellular DOX concentrations than free DOX. However, the intracellular DOX concentrations were less than 10% of drug concentrations in cell culture media. DOX concentrations inside cells were approximately 9 μM when the cells were exposed to 100 μM DOX for 72 h. Experiments were repeated by incubating cancer cells in media at 50 μM DOX, yet the results were reproducible and the intracellular DOX concentration was less than 10% of the drug concentration in the media after 72 h incubation. An unexpected result was that Bz-micelles, which released DOX quickly at both pHs 7.4 and 5.0, showed the lowest intracellular DOX concentration levels. In order to confirm that there were no human errors, we quantified DOX in cell culture media containing free DOX and Bz/Na/H-micelles, and the amount of DOX detected in cell culture media were the same.

Time-dependent quantification of intracellular DOX concentrations in A549 cells treated with free DOX and Bz/Na/H micelles.
DISCUSSION
In this study, we produced three types of polymer micelles from PEG-p(Asp/X) block copolymer, where ‘X’ indicates carboxyl groups protected by benzyl esters (Bz), ionized in a sodium salt form (Na), or desalted as free acid (H). All block copolymers were successfully prepared causing no degradation on the polymer backbone during the deprotection and ion-exchange procedures (Fig. 1).
DOX was effectively entrapped in polymer micelles from block copolymers in which carboxyl groups present as Na salt or free acid (Fig. 2). DOX possesses an ionizable amino group on the sugar ring, which is in HCl salt form. Drug entrapment yields obtained in this study suggest that counter ions and ionizable carboxyl groups are both essential for PEG-p(Asp) block copolymers to entrap DOX molecules. DOX can bind to PEG-p(Asp/Na) block copolymers through ionic interaction as Na ions on the block copolymers form a salt bridge producing NaCl as a byproduct. PEG-p(Asp/H) also possessed ionizable carboxyl groups, yet showed less drug entrapment than PEG-p(Asp/Na) with counter ions of DOX. PEG-p(Asp/Bz) in which every carboxyl group was protected showed low DOX entrapment. Drug-loaded polymer micelles showed different particle sizes, depending on the core environment (Table I). Bz-micelles were the smallest (51 nm) while Na- and H-micelles were close to 100 nm. The increased particle sizes would be attributed to fact that more DOX molecules were entrapped in the Na- and H-micelles. It is intriguing that Na-micelles entrapped more drugs (56.8% DOX) than H-micelles (40.6% DOX), yet the particle size increased only 2.98% (93.4 vs 90.7 nm). These results indicate that the cores of Na- and H-micelles might not be condensed as tightly as Bz-micelles although more hydrophobic DOX molecules were entrapped. We postulate that ionic interaction neutralizes the charge between block copolymers (COO-) and DOX (NH3+) first, and then the hydrophobic portion (anthracycline ring) of DOX induces micelle formation. Bz micelles had low drug entrapment likely because DOX remained ionized due to the absence of its counter ion. Shelf-stability revealed that polymer micelles entrapping DOX through ionic interaction were more stable than Bz-micelles as both powder and frozen solutions.
Drug release patterns demonstrated impressive stability of Na- and H-micelles (Fig. 3). These results are surprising because ionic interactions are often considered unfavorable to prepare stable micelles in the presence of counter ions (e.g. buffer solutions or cell-culture media). Both Na- and H-micelles remained stable as they released DOX. No burst release was observed during the 48 h incubation under sink conditions at 37°C. In comparison to Bz-micelles that released DOX quickly at both pH 7.4 and 5.0, Na- and H-micelles clearly showed pH-dependent drug release patterns. Drug release was accelerated as pH decreased from 7.4 and 5.0. In both pHs, the drug release patterns followed the first order kinetics. It must be noted that there was an induction period of DOX release in Na-micelles at pH 5.0 between 0–6 h (Fig. 4). After this induction period, DOX release profiles became the same for Na- and H-micelles. Such characteristic patterns were not observed at pH 7.4. These results suggest that sodium ions play an important role in suppressing DOX release from the micelles. Drug release from the micelles might take place as ionic exchange proceeds between DOX and buffering agents in the micelle cores. Sodium ions seemed to effectively slow down the ionic exchange of DOX in the micelle core. The pKa of the β-carboxyl group of Asp in a polypeptide is known between 4 and 5. Compared to pH 7.4 at which carboxyl groups are fully ionized and thus sodium salts can be trapped in the micelle cores, Na-micelles would have expelled Na salt at pH 5.0 during the induction time between 0–6 h as carboxyl groups became deionized in acidic conditions. At the same time, DOX release from Na- and H-micelles still remained slower than Bz-micelles. These results indicate that the ionizable functional groups in the micelle core can be employed as kinetic barrier to control drug release from polymer micelles.
All polymer micelles entrapping DOX induced cell death in different cancer cell lines (DU145, PC3 and A549) as shown in Fig. 5. HPLC analyses confirmed that DOX released from the micelles appeared at the same elution time of freshly prepared free DOX. These results indicate that polymer micelles can retain chemical and biological properties of DOX for prolonged time, which was monitored for 6 months in this study. One of the interesting observations was that Na-micelles were identified more potent than free DOX (p < 0.01). There are two factors that brought our attention. Firstly, Na-micelles released DOX at the slowest rate among three micelles tested in this study. It is very unusual that drug carriers that release drugs the slowest became the most potent, even compared to the drug payloads. If cytotoxicity is solely dependent on DOX concentrations, then free DOX and Bz-micelles, which released DOX at the fastest rate, should have shown the similar activity. However, our observation showed the opposite results. The results were reproducible. Secondly, there was no drug degradation observed in samples during cell culture. Our initial hypothesis was that DOX in cell culture media might have deactivated during the cell culture. This hypothesis turned out to be incorrect because DOX preincubated for 0, 24, 48 and 72 h in cell culture media under cell incubation conditions (humidified, 5% CO2 and 37°C) did not lose its bioactivity. Figure 6 shows that empty polymer micelles caused no toxicity alone or adverse effects on cytotoxicity of DOX in combination. These results also indicate that DOX was the only active pharmaceutical ingredient (API). Cell toxicity is often involved with metabolites of APIs, yet we can avoid that possibility in our case. Another potential mechanism is that polymer micelles would have circumvented P-glycoprotein (an efflux pump responsible for exporting drug molecules from the cell interior) and delivered more DOX inside cells. This hypothesis is also negligible because all cancer cells used in this study were sensitive to free DOX. Therefore, it is surmised that Na-micelles suppressed cancer cell growth effectively because drugs were supplied to the cancer cells continuously, damaging sensitive cancer cells in an early stage and less sensitive cancer cells in a later stage for prolonged time. Further investigation is necessary to fully elucidate the mechanism on the enhanced DOX activity because drug release kinetics alone cannot explain our unexpected findings in this study.
We attempted to find a molecular determinant that caused different cytotoxicity of cancer cells against the micelles at the same drug concentrations (Fig. 7). Unfortunately, we observed no significant change in Top2 expression in the cells treated by free DOX and micelles (Fig. 7a and b). Top 2 down regulation has been suggested as a mechanism of cellular resistance to Top2 inhibitors partially due to the activation of the proteasome degradation machinery. However, previous studies suggest that Top2 inhibitors induce both cleaved Top2 complexes and mutated Top2 to accumulate in the cell, proposing multiple models to elucidate the interaction between DOX and DNA responsible for cell death. DOX has been shown to induce cytotoxic effects either by preventing Top2 to bind DNA at >10 μM or by inhibiting the final DNA religation step at <1 μM (49). Therefore, these contradictory results might be attributed to that cells were treated with DOX concentrations lower or equal to 1 μM in this study.
We subsequently observed the expression levels of Bcl-2 and Bax (two Bcl-2 family members playing a pivotal role in apoptosis). Bcl-2 is an anti-apoptotic protein isolated from the mitochondrial membrane. Bcl-2 forms heterodimers with a pro-apoptotic protein Bax and counteracts the Bax activity. Bcl-2 often overexpresses as prostate cancer progresses to an aggressive and hormone-independent disease stage. Although Bcl-2 is one of the signature signal pathways during the treatment of DOX, we failed to report any significant change in the expression levels of the aforementioned members of the Bcl-2 family (Fig. 7c and d). Interestingly, we observed a decrease in the pro-apoptotic 23-kDa Bcl-2 form which was not consistent with cell death reported in the cell viability assays. However, these results were consistent with previous studies that showed no change on the Bax and Bcl-2 protein levels after exposure of PC3 to 1 μg/mL of DOX for up to 24 h.
The ratio of Bax to Bcl-2 is often used to identify cell apoptosis. However, the Bax/Bcl-2 ratio for the DOX-treated PC3 cells was not significantly different from that of the non-treated cells. The limited information obtained by studying the Bcl-2 family can be attributed to variable turnover rates of the Bcl-2 and Bax depending on the concentration and exposure time of DOX (50). PARP is an apoptosis-related protein that plays a role in DNA repair (51). PARP synthesis is generally stimulated by DNA fragmentation in apoptotic cells. After the initial increase in protein levels, PARP is cleaved to an 89 kDa and a 24 kDa fragment by a number of proteolytic enzymes implicated in apoptosis. In Fig. 7e, we observed that the cleaved PARP increased when PC3 were exposed to DOX, which was more obvious with Bz- and Na-micelles. These results are interesting because Bz-micelle showed the most limited cell death. When cells were treated with free DOX, the ratio of the cleaved PARP to total PARP returned to basal levels, suggesting that the cell death was not attributed solely to apoptosis but also to necrosis.
To quantify the extent of DNA damage, the expression levels of phosphorylated H2Ax (γH2Ax) were evaluated in non-treated and DOX-treated PC3 cells (Fig. 7f). H2Ax is a protein that becomes phosphorylated shortly after DNA damage (52), indicating DOX is hitting its therapeutic target (51). Approximately, a 2–3 fold increase in γH2Ax expression was observed after treating the cells with free DOX and micelles (Fig. 7g). These results indicate a characteristic pattern of anthracycline-induced DNA damage.
To obtain an insight on potentially activated survival signaling pathways, we observed NF-κB expression levels in the cytosol and nuclear compartments (Fig. 7h and i). NF-κB is a protein complex involved in immune response, proliferation, differentiation and survival of cells (53). We observed that the NF-κB expression level in the cytosol remained unchanged, yet it decreased in the nucleus corresponding well with the cell viability and the limited Bcl-2 expression levels. NF-κB was previously shown to induce Bcl-2 expression. NF-κB is highly activated in the nucleus of hormone-irresponsive PC3 cells. Therefore, the NF-κB expression restricted in the cytosol of the cells treated with micelles is noteworthy because NF-κB is associated with various malignant phenotypes of cancer cells such as angiogenesis, invasion and metastasis.
Time-dependent changes in intracellular DOX concentrations were lastly investigated (Fig. 8). We selected A549 cells that showed the most significant difference in cytotoxicity between micelles. There results revealed that Na- and H-micelles delivered more drug molecules inside cells even compared to free DOX while Bz-micelles showed almost no drug accumulation in the cells. We measured the drug concentrations in the cell culture media at the same time to confirm the same amount of drugs were present, and there was no difference in DOX levels in the media. Intracellular drug uptake yields also remained less than 10% with respect to drugs added to the media. These results clearly indicate that cellular response to drug-loaded polymer micelles is not dependent solely on the total drug concentrations at which cancer cells are exposed. Therefore, our observations suggest that, if drug carriers continue to be developed by simply focusing on delivering more drugs to tumors and releasing drugs quickly in tumor tissues, drug molecules may not be used efficiently to maximize the therapeutic efficacy, and that slow drug release at an effective dose level for prolonged time would be more promising to enhance cellular response to chemotherapy.
CONCLUSION
Polymer micelles entrapping DOX in three different core environments were prepared and characterized in this study. We investigated the relationship between the drug release patterns of the micelles and cellular response of cancer cells. Drug entrapment yields reveal that counter ions for drug molecules contribute to stabilize drug-loaded polymer micelles and to achieve sustained drug release in buffered aqueous solutions or serum-containing media. Polymer micelles entrapping DOX through ionic interactions (Na- and H-micelles) remained stable as powder or solutions at least 6 months as opposed to the micelles entrapping drugs through hydrophobic interaction (Bz-micelles). DOX release patterns were greatly affected by ionizable groups of DOX as well as presence of counter ions in the micelles. Cytotoxicity of cancer cells was not consistent with drug release patterns of polymer micelles while Na-micelles that released DOX at the slowest rate appeared to be the most potent. Western blotting assays suggest that Bz/Na/H-micelles likely induce cell death through different molecular mechanisms as opposed to the drug payloads. Na- and H-micelles delivered more drugs inside cells than Bz-micelles and free DOX even at the same drug concentrations. In conclusion, our findings suggest that optimizing drug release patterns of drug carriers in tumors would be as crucially as or more important than enhancing tumor accumulation of drugs to reduce toxicity and enhance efficacy of cancer chemotherapy.
REFERENCES
Lvov YM, Pattekari P, Zhang X, Torchilin V. Converting poorly soluble materials into stable aqueous nanocolloids. Langmuir. 2011;27:1212–7.
Cukierman E, Khan DR. The benefits and challenges associated with the use of drug delivery systems in cancer therapy. Biochem Pharmacol. 2010;80:762–70.
Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 1997;26:71–90.
Liu J, Xiao Y, Allen C. Polymer-drug compatibility: a guide to the development of delivery systems for the anticancer agent, ellipticine. J Pharm Sci. 2004;93:132–43.
Oberoi HS, Laquer FC, Marky LA, Kabanov AV, Bronich TK. Core cross-linked block ionomer micelles as ph-responsive carriers for cis-diamminedichloroplatinum(ii). J Control Release. 2011;153:64–72.
Ruenraroengsak P, Cook JM, Florence AT. Nanosystem drug targeting: facing up to complex realities. J Control Release. 2010;141:265–76.
Maruyama K. Intracellular targeting delivery of liposomal drugs to solid tumors based on epr effects. Adv Drug Deliv Rev. 2011;63:161–9.
Maeda H. Tumor-selective delivery of macromolecular drugs via the epr effect: background and future prospects. Bioconjugate Chem. 2010;21:797–802.
Torchilin V. Tumor delivery of macromolecular drugs based on the epr effect. Adv Drug Deliv Rev. 2011;63:131–5.
Blanco E, Hsiao A, Mann Aman P, Landry Matthew G, Meric-Bernstam F, Ferrari M. Nanomedicine in cancer therapy: innovative trends and prospects. Cancer Sci. 2011;102:1247–52.
Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100:572–9.
Musacchio T, Torchilin Vladimir P. Recent developments in lipid-based pharmaceutical nanocarriers. Front Biosci. 2011;16:1388–412.
Seidlits S, Peppas NA. Star polymers and dendrimers in nanotechnology and drug delivery. Nanotechnol Ther. 2007;317–48.
Wong HL, Li Y, Bendayan R, Rauth MA, Wu XY. Solid lipid nanoparticles for anti-tumor drug delivery. Nanotechnol Cancer Ther. 2007;741–76.
Park K. All nanocarriers are created equal. J Control Release. 2008;130:139.
Chan Juliana M, Valencia Pedro M, Zhang L, Langer R, Farokhzad Omid C. Polymeric nanoparticles for drug delivery. Methods Mol Biol. 2010;624:163–75.
Levy-Nissenbaum E, Radovic-Moreno Aleksandar F, Wang Andrew Z, Langer R, Farokhzad Omid C. Nanotechnology and aptamers: applications in drug delivery. Trends Biotechnol. 2008;26:442–9.
Alexis F, Pridgen Eric M, Langer R, Farokhzad Omid C. Nanoparticle technologies for cancer therapy. Handb Exp Pharmacol. 2010;55–86.
Pirollo KF, Chang EH. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008;26:552–8.
Grattoni A, Shen H, Fine D, Ziemys A, Gill JS, Hudson L, et al. Nanochannel technology for constant delivery of chemotherapeutics: beyond metronomic administration. Pharm Res. 2011;28:292–300.
Dang TT, Bratlie KM, Bogatyrev SR, Chen XY, Langer R, Anderson DG. Spatiotemporal effects of a controlled-release anti-inflammatory drug on the cellular dynamics of host response. Biomaterials. 2011;32:4464–70.
Greco F, Vicent MJ, Gee S, Jones AT, Gee J, Nicholson RI, et al. Investigating the mechanism of enhanced cytotoxicity of hpma copolymer-dox-agm in breast cancer cells. J Control Release. 2007;117:28–39.
Murakami M, Cabral H, Matsumoto Y, Wu S, Kano MR, Yamori T, et al. Improving drug potency and efficacy by nanocarrier-mediated subcellular targeting. Sci Transl Med. 2011;3:64ra2.
van Vlerken Lilian E, Duan Z, Little Steven R, Seiden Michael V, Amiji Mansoor M. Augmentation of therapeutic efficacy in drug-resistant tumor models using ceramide coadministration in temporal-controlled polymer-blend nanoparticle delivery systems. AAPS J. 2010;12:171–80.
Lee HJ, Ponta A, Bae Y. Polymer nanoassemblies for cancer treatment and imaging. Ther Deliv. 2010;1:803–17.
Bae Y, Kataoka K. Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv Drug Deliv Rev. 2009;61:768–84.
Yokoyama M. Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Del. 2010;7:145–58.
Kataoka K, Kwon GS, Yokoyama M, Okano T, Sakurai Y. Block-copolymer micelles as vehicles for drug delivery. J Control Release. 1993;24:119–32.
Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular ph-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjugate Chem. 2005;16:122–30.
Bae Y, Nishiyama N, Kataoka K. In vivo antitumor activity of the folate-conjugated ph-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjugate Chem. 2007;18:1131–9.
Bae Y, Alani AWG, Rockich NC, Lai TSZC, Kwon GS. Mixed ph-sensitive polymeric micelles for combination drug delivery. Pharm Res. 2010;27:2421–32.
Alani AWG, Bae Y, Rao DA, Kwon GS. Polymeric micelles for the ph-dependent controlled, continuous low dose release of paclitaxel. Biomaterials. 2010;31:1765–72.
Howard MD, Ponta A, Eckman A, Jay M, Bae Y. Polymer micelles with hydrazone-ester dual linkers for tunable release of dexamethasone. Pharm Res. 2011;28:2435–46.
Ponta A, Bae Y. Peg-poly(amino acid) block copolymer micelles for tunable drug release. Pharm Res. 2010;27:2330–42.
Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloid Surface B. 1999;16:3–27.
Yokoyama M, Inoue S, Kataoka K, Yui N, Sakurai Y. Preparation of adriamycin-conjugated poly(ethylene glycol)-poly(aspartic acid) block copolymer - a new type of polymeric anticancer agent. Makromol Chem-Rapid. 1987;8:431–5.
Kwon GS, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Enhanced tumor accumulation and prolonged circulation times of micelle-forming poly(ethylene oxide-aspartate) block copolymer-adriamycin conjugates. J Control Release. 1994;28:334–5.
Yokoyama M, Okano T, Sakurai Y, Kataoka K. Improved synthesis of adriamycin-conjugated poly(ethylene oxide) poly(aspartic acid) block-copolymer and formation of unimodal micellar structure with controlled amount of physically entrapped adriamycin. J Control Release. 1994;32:269–77.
Kwon GS, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Physical entrapment of adriamycin in ab block-copolymer micelles. Pharm Res. 1995;12:192–5.
Kataoka K, Matsumoto T, Yokoyama M, Okano T, Sakurai Y, Fukushima S, et al. Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-l-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J Control Release. 2000;64:143–53.
Kwon G, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Block copolymer micelles for drug delivery: loading and release of doxorubicin. J Control Release. 1997;48:195–201.
Nishiyama N, Kataoka K. Polymeric micelle drug carrier systems: peg-pasp(dox) and second generation of micellar drugs. Adv Exp Med Biol. 2003;519:155–77.
Kim JO, Sahay G, Kabanov AV, Bronich TK. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules. 2010;11:919–26.
Bures P, Peppas NA. Ionic nanoparticulate systems for drug delivery. Nanotechnol Ther. 2007;349–60.
Bontha S, Kabanov AV, Bronich TK. Polymer micelles with cross-linked ionic cores for delivery of anticancer drugs. J Control Release. 2006;114:163–74.
Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular ph change. Angew Chem Int Ed. 2003;42:4640–3.
Urasaki Y, Laco GS, Pourquier P, Takebayashi Y, Kohlhagen G, Gioffre C, et al. Characterization of a novel topoisomerase i mutation from a camptothecin-resistant human prostate cancer cell line. Cancer Res. 2001;61:1964–9.
Robinson BW, Behling KC, Gupta M, Zhang AY, Moore JS, Bantly AD, et al. Abundant anti-apoptotic bcl-2 is a molecular target in leukaemias with t(4;11) translocation. Br J Haematol. 2008;141:827–39.
Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–33.
Devlin HL, Mack PC, Burich RA, Gumerlock PH, Kung HJ, Mudryj M. deVere White RW. Impairment of the DNA repair and growth arrest pathways by p53r2 silencing enhances DNA damage-induced apoptosis in a p53-dependent manner in prostate cancer cells. Mol Cancer Res. 2008;6:808–18.
Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, et al. Histone gammah2ax and poly(adp-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res. 2010;16:4532–42.
Shaheen FS, Znojek P, Fisher A, Webster M, Plummer R, Gaughan L, et al. Targeting the DNA double strand break repair machinery in prostate cancer. PLoS One. 2011;6:e20311.
Hayden MS, Ghosh S. Shared principles in nf-kappab signaling. Cell. 2008;132:344–62.
ACKNOWLEDGMENTS & DISCLOSURES
Authors acknowledge financial support provided by the Kentucky Lung Cancer Research Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eckman, A.M., Tsakalozou, E., Kang, N.Y. et al. Drug Release Patterns and Cytotoxicity of PEG-poly(aspartate) Block Copolymer Micelles in Cancer Cells. Pharm Res 29, 1755–1767 (2012). https://doi.org/10.1007/s11095-012-0697-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-012-0697-5