
ABSTRACT
Purpose
To provide a definitive assessment of prediction of in vivo CL int from human liver in vitro systems for assessment of typical underprediction.
Methods
A database of published predictions of clearance from human hepatocytes and liver microsomes was compiled, including only intravenous CL b. The influence of liver model (well-stirred (WS) or parallel tube (PT)), plasma protein binding and clearance level on the relationship between in vitro and in vivo CL int was examined.
Results
Average prediction bias was about 5- and 4-fold for microsomes and hepatocytes, respectively. Reduced bias using the PT model, in preference to the popular WS model, was only marginal across a wide range of clearance with a consequential minor impact on prediction. Increasing underprediction with decreasing fu b, or increasing CL int, was found only for hepatocytes, suggesting fundamental in vitro artefacts rather than failure to model potentially unequilibrated binding during rapid extraction.
Conclusions
In contrast to microsomes, hepatocytes give a disproportionate prediction with increasing clearance suggesting limitations either at the active site, such as cofactor exhaustion, or with intracellular concentration equilibrium, such as rate-limiting cell permeability. A simple log linear empirical relationship can be used to correct hepatocyte predictions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Human liver-derived in vitro systems provide currently indispensible tools for prediction of drug clearance which are vital to drug discovery and development. Although in vitro–in vivo prediction methodology has become established over the last 20 years, a tendency towards underprediction of unbound intrinsic clearance (CL int) in vivo of a magnitude of several fold, using either human liver microsomes or hepatocytes, remains. This prediction bias is unresolved from imprecision (arising from human variability and experimental uncertainty), and the cumulative unknowns challenge the quantitative capability of these systems (1).
The relative convenience of using human liver microsomes may be outweighed by the incomplete set of clearance pathways offered, compared with hepatocytes (2–4), particularly given the widespread commercial availability of cryopreserved hepatocytes. However, it appears that cytochrome P450 (CYP) metabolising capacity of cryopreserved hepatocytes may differ from human liver microsomes—average CYP maximal activity has been found to be between 2- and 20-fold less than that of microsomes from the same commercial sources (1). This apparent disadvantage of hepatocytes is not reflected in reported comparisons of intrinsic clearance between the systems (5–7), where prediction bias was found to be similar. Inter-individual variability is normally addressed by pooling of in vitro material from numerous liver donors or assay across several individual donors to give a range. However, representation of human variability could still be distorted because of the log-normal distribution of CYP activity between donors combined with the independence of expression between CYPs (1). The relative performance of human liver microsomes and hepatocytes for prediction of clearance remains unclear.
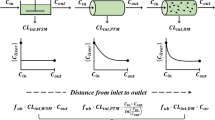
Failure to accurately predict clearance in vivo from either human liver in vitro system has necessitated critical consideration of the wider extrapolation methodology as a potential source of systematic underprediction. Several aspects of the methodology have been focused on in recent years. Scaling of in vitro intrinsic clearance measurements to the whole body has been exhaustively investigated; commonly used scaling factors, for either system, do not appear to be a source of bias or major imprecision (8). Physiological modelling of drug extraction by the liver relies on the simplistic Well-Stirred liver model, and this may be justified, as the accuracy of prediction is not improved upon by the use of either the Parallel Tube or Dispersion liver models, with the exception of only the most highly cleared drugs (9). Liver models incorporating modelling of active uptake processes have yet to be widely adopted, but active uptake may be relatively unimportant for the large number of highly permeable drugs investigated to date (10) and may not have had a major impact on predictions. Recently, however, significantly reduced prediction bias using the Parallel Tube model rather than the more commonly used Well-Stirred model has been reported (7,11), returning attention to drug dispersion in the liver. Another aspect of physiological modelling which has received attention recently is the appropriateness of the steady-state unbound fraction in blood (fu b), which is commonly applied with liver models. It has been suggested that underprediction of clearance may be associated with unequilibrated uptake processes in liver, including binding to plasma protein, and hence the kinetics of metabolism, uptake and binding should be dynamically modelled (12). Possibly related to this is a trend of clearance dependence in prediction, evident in some reported predictions (5,7).
To provide a comprehensive basis for further investigation of the underprediction of clearance, we have compiled a database of predictions from various published studies involving human liver microsomes and/or hepatocytes. The assessment of prediction accuracy and precision was unified with respect to the use of unbound fraction in vitro and apparent intrinsic clearance in vivo (avoiding investigation that used the oral route). The impact of choice of liver model and extent of binding in blood on prediction has been examined, along with dependency on clearance. The main objective was to obtain a clearer profile of prediction bias for both human hepatic microsomes and hepatocytes which, together with potential indications of underlying causes of underprediction, would guide further investigation. The database presented also provides a valuable set of in vivo CL int values for 110 drugs (intravenously dosed) with corresponding in vitro predictions of CL int from cryopreserved hepatocytes (89 drugs) and hepatic microsomes (67 drugs). This compilation should prove to be valuable in the future assessment and comparison of novel in vitro systems that are currently under development, e.g. HepaRG, adenoviral transfected HepG2 cell systems (13,14).
METHODS
In Vitro and In Vivo Data Collation
In vitro intrinsic clearance (CLint) determined either by drug depletion or metabolite formation kinetics reported for human cryopreserved hepatocytes (5–7,15,16) and human liver microsomes (7,16,17) was combined into a database for each system. In vivo plasma clearance (CLp) and unbound fraction in plasma (fup) were collated from the above sources. Drugs were included only if the corresponding in vivo clearance was based on intravenous dose to avoid uncertainty arising from absorption processes. Drugs with in vivo clearance values greater than hepatic blood flow were excluded to avoid additional uncertainty from extra-hepatic effects.
Determination of Unbound In Vitro Intrinsic Clearance
Where in vitro system unbound concentration was available (5,7,15), unbound in vitro CL int (CL int,u) was determined using an estimated unbound fraction in vitro based on the appropriate lipophilicity relationship algorithm for either microsomes (fu mic; (18)) or hepatocytes (fu heps; (19)), below.
where P is microsomal protein concentration, log P/D is the log P value for basic drugs and the log D value for acidic and neutral drugs
where K p is the hepatocyte/medium concentration ratio, K a is the microsomal binding constant (125 for 1 mg of microsomal protein/ml and 1 million cells/ml) and V R is the volume ratio of hepatocytes to medium (0.005 for 1 million cells/ml).
Scaling of In Vitro Intrinsic Clearance to Whole Liver
In vitro intrinsic clearance (CLint) values were scaled to the in vivo equivalent whole liver using average liver and body weight with either a microsomal recovery factor or hepatocellularity value (Eq. 3).
where SF is the either the microsomal average recovery factor of 40 mg microsomal protein/g liver or hepatocellularity of 120 million/g liver (20) and HLW is the human liver weight of 21.4 g liver/kg bodyweight (21).
Determination of Apparent In Vivo Intrinsic Clearance
In vivo CLint was calculated from the in vivo CLp, fup and average hepatic blood flow (QH, 20.7 ml/min/kg (21)) using either the Well-Stirred (WS) or the Parallel Tube (PT) liver model (Eqs. 4 and 5). Blood clearance (CLb) and unbound fraction in blood (fub) were determined using the blood/plasma concentration ratio (Rb), where available (CLp/Rb or fup/Rb)), or assuming Rb = 1 for basic and neutral drugs and 0.55 for acidic drugs.
Accuracy and Precision of Predictions
The accuracy of a set of individual drug in vitro CL int/in vivo CL int predictions for each system was assessed using the average fold error (afe) metric (geometric mean error) determined using Eq. 6 (22).
The corresponding precision of the prediction sets was assessed using the root mean squared error (rmse) determined using Eq. 7 (23).
Assessment of in Vitro Predictions of Intrinsic Clearance
For each in vitro system, accuracy and precision of prediction of CL int was determined and compared between liver model. For each liver model, these metrics were determined with or without exclusion of predictions involving in vivo CL greater than 80% Q H to examine sensitivity of the model to values at the nonlinear extreme. Using the WS liver model for each system, the predictions were segregated according to arbitrary ranges for both fu b (<0.05, 0.05–0.2, 0.2–1) and in vivo CL int (<10, 10–100, 100–1,000, >1,000 ml/min/kg) to assess potential dependencies in prediction with extent of binding in blood and clearance, respectively. Microsomes and hepatocytes were directly compared for those drugs used in both systems (n = 46), using the WS liver model. An empirical relation between the systems was obtained by least squares regression of the following log linear function using Microsoft Excel:
The goodness of fit was assessed by examination of the residuals of the predicted values.
Empirical Relation of Predicted In Vitro Cl int and In Vivo
To provide an empirical correction of in vitro CL int to in vivo CL int,, where lack of proportionality was observed from the above analysis, the following log linear function was fitted by least squares regression using Microsoft Excel:
To provide a simple (log) nonlinear comparison, the following quadratic function was also fitted:
The goodness of fit was assessed and compared by examination of the residuals of the predicted values. These regression coefficients were then used to establish relationships between in vivo CL int and in vitro predictions.
RESULTS
Database of Human In Vitro and In Vivo Prediction of Intrinsic Clearance
Using the criteria described above, a net total of 89 and 67 drug predictions from hepatocytes and microsomes, respectively, was established (Table I). Individual predictions were based on an average of five source liver donors for hepatocytes and an average of 11 donors for microsomes. In vivo hepatic metabolic clearance following single intravenous dose (from several studies, in most cases) was obtained for each drug.
Liver Model Dependency in Prediction of Intrinsic Clearance and Effect of Exclusion of High Clearance Substrates
For each system, the total set of predictions was assessed for accuracy and precision using both the WS and PT liver models. In addition, to examine the impact of model sensitivity to CL b, an upper limit of 80% Q H was used to exclude the most rapidly cleared drugs. All predictions are shown graphically in Fig. 1. Bias (afe) was 5.2 and 3.9 for microsomes and hepatocytes, respectively, using the WS model and 3.5 and 2.9, respectively, using the PT model. Excluding drugs with CL b, >80% Q H, afe was 4.7, 3.4 and 3.6, 3.0, respectively, indicating minimal impact of proximity to Q H.

Prediction of CL int from the database for human hepatocytes (A and B) and liver microsomes (C and D) using the WS (A and C) or PT (B and D) liver model.
Overall, bias was marginally less from the PT liver model than from the WS model. Precision (rmse) was similar (within 20% difference) between in vitro systems; rmse was 3740/3850 and 3140/3230 including/excluding drugs with CL b, >80% Q H, for microsomes and hepatocytes, respectively, using the WS model. However, precision was considerably greater (>2-fold difference in rmse) using the PT model (rmse was 1540/1620 and 1300/1350 including/excluding drugs with CL b, >80% Q H, for microsomes and hepatocytes, respectively).
Blood-Binding Dependency in Prediction of Intrinsic Clearance
The database of predictions of CL int from hepatocytes and microsomes was segregated according to the level of fu b to assess if prediction was dependent on the extent of binding in blood. There was little variation in afe between low and high binding for microsomes using either the WS or PT liver model (although afe was less in the mid-range); however, for hepatocytes, underprediction increased with decreasing fu b with both models (Table II), indicating a difference in prediction between the systems associated with binding in blood. The distribution of binding level with CL int is shown if Fig. 2.

Prediction of CL int from the database for human hepatocytes (A) and liver microsomes (B), according to range of fu b, using the WS liver model.
Comparison of Prediction of Intrinsic Clearance Between Hepatocytes and Microsomes
Predictions of CL int for substrates common to both the hepatocyte and microsome databases (n = 46) were directly compared to assess the relationship between systems according to clearance level. A trend of decreasing relative CL int from hepatocytes with increasing CL int was clearly evident, further indicating a fundamental system difference (Fig. 3). The relationship between the systems was adequately described (via least squares regression analysis) by the log linear function: \( { \log }C{L_{{\text{int}},{\text{ hepatocytes}}}} = 0.{53}\,{ \log }C{L_{{\text{int}},{\text{ microsomes}}}} + 0.{59}\left( {{r^2} = 0.{68}} \right) \). This is supported by low standard errors (SE) of the slope (0.008) and intercept (0.014).

Comparison of prediction of CL int from the database between human hepatocytes and liver microsomes, using the WS liver model. Dashed lines represent unity and power function fit.
Clearance Dependency in Prediction of Intrinsic Clearance
The log ratio of predicted to observed CL int indicated a clear distinction in trend between the systems: prediction from microsomes appeared to be constant across the range of in vivo CL int, whereas prediction from hepatocytes appeared to be dependent on in vivo CL int (Fig. 4). To quantify the dependency of predictions according to estimated in vivo CL int, the database predictions were segregated according to four arbitrary levels of in vivo CL int. For hepatocytes, there was a clear shift from an in vitro–in vivo correspondence for drugs at in vivo CL int < 10 ml/min/kg to an underprediction of about 3-fold at 10–100 ml/min/kg increasing to about 30-fold at >1,000 ml/min/kg (Fig. 5a); afe increased disproportionally over this range, approximately equally for either liver model (Table III). In contrast, for microsomes, underprediction was relatively constant, in the range 3–6-fold, from <10 to 1,000 ml/min/kg (Fig. 5b; Table III).

Ratio (log) of predicted to observed CL int, as a function of observed CL int, for human hepatocytes (A) and liver microsomes (B).

Bias (afe) of predicted CL int with level of in vivo CL int from the database for human hepatocytes (A) and liver microsomes (B) using the WS and PT liver models.
The relationship between in vivo and in vitro CL int was adequately described by a log linear function (Eq. 9) for both systems (r 2 = 0.54 and 0.64, respectively; p < 0.01, by t-test), as indicated by an unbiased distribution of residuals. The data were also described by a quadratic function (Eq. 10), but as the distribution of residuals was very similar to that of the power function, the latter, as the simpler model, was considered the most justifiable representation. There was a considerable difference in parameter values between the systems: the slope of fitted predictions from hepatocytes diverged significantly from unity (Eq. 11, Fig. 6a), whereas the slope for predictions from microsomes was only marginally divergent from unity (Eq. 12, Fig. 6b). This is supported by low SE of the slope (hepatocytes, 0.005; microsomes, 0.009) and intercept (hepatocytes, 0.011; microsomes, 0.020).

Relation of predicted CL int in vitro and CL int in vivo for hepatocytes (A) and microsomes (B). Dashed lines represent unity, fitted power functions and (A) upper and lower limits of bias correction for hepatocytes.
DISCUSSION
An extensive database of published predictions of clearance from human hepatocytes and liver microsomes was compiled for analysis of some potential causes of the prevailing uncertainties of negative bias and imprecision in current methodology. Focus was maintained on systemic hepatic extraction by exclusive use of predictions involving intravenous CL b. The influence of two alternative liver models (WS and PT), plasma protein binding and clearance level was examined.
Variation in drug metabolising capacity between hepatocytes or microsomes from different donors, possibly reflecting the source phenotypes, will manifest as part of the imprecision in a database such as this. The relatively small number of source individuals (average 6) for hepatocytes compared with microsomes (average 11) contributing to individual drug predictions might also affect the measured system average prediction (bias). Methodological differences between laboratories may also contribute to overall imprecision as well as bias. However, these potential effects are minimized by the relatively large number of predictions in each system dataset enabling trends with in vivo and between the systems to be resolved.
Reduced bias in prediction for high clearance drugs has been reported for the PT liver model, which, accordingly, has been advocated in preference to the widely used WS liver model (7,11). The sensitivity of either liver model to the value of CL b increases with convergence on Q H, but is relatively greater for the WS liver model. The effect is proportional with respect to fu b and hence independent of this parameter, which can have a major influence on apparent CL int in vivo. Consequently, for the WS liver model, an uncertainty of 10% in CL b at 80% Q H leads to an uncertainty in estimated CL int of about 2-fold. This should not be a cause of prediction bias, as any error in CL b is expected to be random. The relatively greater precision with the PT model observed in the database analysis reported here appears to reflect this assumption. This study also shows that the PT model offers a marginal reduction of bias, compared with the WS model, and that this appears to be dependent on clearance, hence the previous assertions that the PT model is more appropriate for high clearance drugs. However, the overall reduction in bias using the PT model compared with the WS model is marginal (30%), including clearance up to 1,000 ml/min/kg (which includes the vast majority of drugs). Considering the level of underprediction by either in vitro system, the difference between liver models cannot be considered to have a major impact on prediction.
A high degree of binding to plasma protein (fu p < 0.1) is common among drugs, and therefore the fu b term frequently has a large influence on the estimation of CL int from CL b. The established WS and PT liver models accommodate only a steady state estimate of binding (fu), although there may exist unequilibrated levels of free drug in blood during extraction of rapidly cleared drugs in the liver. Consequently, dynamic modelling of hepatic extraction and blood binding has been advocated to provide a rationale to explain the underprediction of clearance observed using current methodology (12). Any tendency of the WS or PT liver model to overestimate unbound CL int from CL b (due to false assumption of equilibration of binding in blood) might be expected to correlate with fu b because this would tend to (inversely) reflect high association/low dissociation binding rate constants. There was evidence of a dependency of bias on fu b with hepatocytes in this study, but this was not reflected with microsomes and therefore does not support the concept of hepatic modelling inadequacies as a source of underprediction of CL int.
Differences in dependency of prediction bias on fu b (and presumably, by correlation, with clearance) between hepatocytes and microsomes indicate fundamental system differences. When the systems were directly compared—using only predictions involving drugs common to both systems—a nonlinear relationship was observed. This difference is supported by the greater dependency of prediction bias for hepatocytes on in vivo CL int, compared with microsomes (for the entire databases). Accuracy of predictions from microsomes may be considered effectively independent of in vivo CL int, so, by comparison, hepatocytes appear to respond nonlinearly to a ‘subcellular’ CL int; the cause of this can only be speculative but may involve endogenous cofactor depletion, enzymatic activity loss (24) or permeability limitation (25).
Currently, as there is no mechanistic basis for incorporating the extent of underprediction into prediction modelling, bias in CL int may be minimized empirically. As indicated above, this study provides a large dataset on which to base an empirical correction which incorporates the widest range of drugs and liver donors. For hepatocytes, using Eq. 11 and assuming the WS model for extrapolation to in vivo CL, average predicted in vivo CL int can be expressed as
Applying this equation to the hepatocyte database, afe of prediction of in vivo CL int was 1.0 (rmse = 4,270); hence, bias was eliminated with only a marginal effect on precision. It may be recognized that this relationship for hepatocytes differs from what would be determined from datasets from individual laboratories; a different slope and intercept might appear to apply to a particular laboratory. However, it is not feasible to distinguish any such apparent methodological effects from differences in drugs (clearance range) and source liver activity. Nevertheless, correction of clearance-dependent bias for hepatocytes is an important step in reduction of uncertainty, and use of a ‘global’ correction may be realistic. A suggested working range for (unbound) hepatocyte CL int is approximately 5–100 ml/min/kg, which would be suitable for the vast majority of established drugs; it has been shown that the practicable lower limit for this system with the substrate depletion method is between 1 and 10 ml/min/kg (7). For microsomes, correction of bias in a similar range of CL int may be achieved using Eq. 12; however, given the proximity of the prediction slope to unity for this system, an adequate approach would be multiplication of the (unbound) microsomal CL int by 5 (afe of whole dataset). Correction of the microsome database this way eliminated bias (afe CL int = 1.0; rmse = 3,610).
There are issues with both hepatocytes and microsomes concerning variability between donors and variable quality of tissue available. Hence, there is a need for novel cellular systems that are devoid of this limitation and display the advantages of both systems. Cell lines which are genetically engineered to provide activities comparable to what is believed to be occurring in vivo should be the aspiration (14,26). The large database of in vivo numbers enclosed here indicates the range that is required and provides the necessary in vivo correlates to allow a top-down assessment of any novel in vitro system for clearance prediction. Such considerations are particularly timely, as the quality of human tissue available will inevitably continue to decline with the increase in transplantation success and associated procedures. There have been several reports of genetically engineered cell lines being successful in predicting P450 induction (27,28). Although human hepatocytes are regarded as the gold standard for induction, variability both in basal level and response presents a major challenge, and the advantages of a more stable system providing adequate response are obvious. Such systems would address the ethical concerns associated with the use of human tissue and in addition address the scientific needs of having a robust in vitro system where the capacity is comparable to that observed under the in vivo situation.
Despite the high degree of uncertainty inherent in predictions of clearance from human liver-derived in vitro systems, this study has further distinguished the relative quantitative prediction capabilities of microsomes and hepatocytes, most notably in terms of bias. Bias is an important component of prediction uncertainty, and this report has shown that both clearance-dependent (hepatocytes) and clearance-independent (microsomes) bias can be corrected empirically. For a large set of drugs undergoing early evaluation, average prediction should be improved, although for individual cases, caution is required because a high level of imprecision in prediction remains.
Abbreviations
- CL int :
-
intrinsic clearance
- CL b :
-
blood clearance
- WS:
-
‘Well-stirred’ (liver model)
- PT:
-
‘Parallel tube’ (liver model)
- fu b :
-
fraction unbound in blood
- CYP:
-
Cytochrome P450
REFERENCES
Hallifax D, Houston JB. Methodological uncertainty in quantitative prediction of hepatic clearance from human experimental systems. Curr Drug Metab. 2009;10:307–21.
Hewitt NJ, Gomez Lechon MJ, Houston JB, Hallifax D, Brown HS, Maurel P, et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39:159–234.
Li AP. Human hepatocytes: isolation, cryopreservation and applications in drug development. Chem Biol Interact. 2007;168:16–29.
Soars MG, McGinnity DF, Grime K, Riley RJ. The pivotal role of hepatocytes in drug discovery. Chem Biol Interact. 2007;168:2–15.
Riley RJ, McGinnity DF, Austin RP. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab Dispos. 2005;33(9):1304–11.
Brown HS, Griffin M, Houston JB. Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos. 2007;35:293–301.
Stringer R, Nicklin PL, Houston JB. Reliability of human cryopreserved hepatocytes and liver microsomes as in vitro systems to predict metabolic clearance. Xenobiotica. 2008;38:1313–29.
Barter ZE, Bayliss MK, Beaune PH, Boobis AR, Carlile DJ, Edwards RJ, et al. Scaling factors for the extrapolation of in vivo metabolic clearance from in vitro data: reaching a concensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr Drug Metab. 2007;8:33–45.
Ito K, Houston JB. Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm Res. 2004;21(5):785–92.
Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23.
Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP-glucuronosyltransferase cofactors in alamethicin-activated human liver microsomes. Drug Metab Dispos. 2009;37:82–9.
Baker M, Parton T. Kinetic determinants of hepatic clearance: plasma protein binding and hepatic uptake. Xenobiotica. 2007;37(10–11):1110–34.
Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F, Guguen-Guillouzo C. The human Hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem Biol Interact. 2007;168:66–73.
Aoyama K, Yoshinari K, Kim HJ, Nagata K, Yamazoe Y. Simultaneous expression of plural forms of human cytochrome P450 at desired ratios in HepG2 cells adenovirus-mediated tool for cytochrome P450 reconstitution. Drug Metab Pharmacokinet. 2009;24:209–17.
McGinnity DF, Soars MG, Urbanowicz RA, Riley RJ. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab Dispos. 2004;32(11):1247–53.
Stringer R. PhD Thesis. The University of Manchester (2006).
Ito K, Houston JB. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 2005;22:103–12.
Hallifax D, Houston JB. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab Dispos. 2006;34:724–6.
Kilford PJ, Gertz M, Houston JB, Galetin A. Hepatocellular binding of drugs: correction for unbound fraction incubations using microsomal binding or drug lipophilicity data. Drug Metab Dispos. 2008;36:1194–7.
Hakooz N, Ito K, Rawden H, Gill H, Lemmers L, Boobis AR, et al. Determination of a human hepatic microsomal scaling factor for predicting in vivo drug clearance. Pharm Res. 2006;23:533–9.
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–5.
Obach RS. Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab Dispos. 1997;25:1359–69.
Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9(4):503–12.
Jones HM, Houston JB. Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos. 2004;32(9):973–82.
Lu C, Li P, Gallegos R, Uttamsingh V, Xia CQ, Miwa GT, et al. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos. 2006;34(9):1600–5.
Donato MT, Hallifax D, Picazo L, Castell JV, Houston JB, Gomez-Lechon MJ, et al. Metabolite formation kinetics and intrinsic clearance of phenacetin, tolbutamide, alprazolam and midazolam in adenoviral P450 transfected HepG2 cells, and comparison with hepatocytes and in vivo. (submitted for publication).
McGinnity DF, Zhang G, Kenny JR, Hamilton GA, Otmani S, Stams KR, et al. Evaluation of multiple in vitro systems for assessment of CYP3A4 induction in drug discovery: human hepatocytes, pregnane X receptor reporter gene, and Fa2N-4 and HepaRG cells. Drug Metab Dispos. 2009;37:1259–68.
Turpeinen M, Tolonen A, Chesne C, Guillouzo A, Uusitalo J, Pelkonen O. Functional expression, inhibition and induction of CYP enzymes in HepaRG cells. Toxicol In Vitro. 2009;23(4):748–53.
ACKNOWLEDGEMENTS
J. A. Foster was supported by funding from the EU 6th Framework Programme for Optimisation of Liver and Intestine In Vitro Models for Pharmacokinetics (Liintop/STREP 037499).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hallifax, D., Foster, J.A. & Houston, J.B. Prediction of Human Metabolic Clearance from In Vitro Systems: Retrospective Analysis and Prospective View. Pharm Res 27, 2150–2161 (2010). https://doi.org/10.1007/s11095-010-0218-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0218-3