
The aim of our study is to develop a simple, delicate, precise and economical protocol for the qualitative and quantitative determination of Chlorpheniramine using spectrophotometer. Our validation is distinctive and provides a precise picture of a very weak chromophore of chlorpheniramine (CPN) that had not been accurately detected before by traditional techniques. The blue color which formed by mixing pheniramine and Cobalt- thiocyanate solution was extracted with chloroform. CPN presented a linearity range of 25 – 125 μg at 628 nm. Limit of quantification (15 μg/mL) and limit of detection (08 μg/mL) of cobalt-chloride were analyzed. The resulting data was statistically significant. Because of the appropriate reproducibility and reliability of this proposed analysis, it can be used to screen CPN in pharmaceutical preparations. It is also validated through the International Council of Harmonization for accuracy, linearity, precision, LOD and LOQ. Various dosage forms of pheniramine were evaluated and have shown that they did not exceed their relative standard deviation by 1%. Their percentage purity was within 99.62 – 103.34%. The suspected sample was treated with 2 – 3 drops of cyanocobalamine after which violet coloration appeared. The percentage relative standard deviation of accuracy and precision was acquired as 1.57 and 1.15, respectively, exactly following the authentication criteria for newly established methods. Because of the profound potential of these developed methods, including precise, fast, flawless and inexpensive, it can easily be adopted in laboratories for the routine analysis of Chlorpheniramine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Pheniramine maleate is antihistaminic, belongs to the class of H1 receptor-antagonists and acts through the inhibition of capillary permeability, contraction and secretion of the smooth muscles of the GIT and bronchiole. Chemically it is recognized as N,N-dimethyl-3-methyl-3-phenyl-3-(2-pyridyl) propylamine hydrogen maleate. Pheniramine is freely soluble in water, alcohol and methylene and its molecular formula is C16H20N2-C4H4O4, as shown in Fig. 1.

Structure of Pheniramine.
Derivatives of pheniramine include dexchlorpheniramine, chlorphenamine, dexbrompheniramine, brompheniramine and triprolidine. The potency of halogenated products rises (20 times) in combination therapy effectively employed to manage different cancers and malarial disease. Pheniramine maleate (PNM) is usually indicated in various allergic situations such as asthma, angioedema, urticaria, hay fever, dermatitis, vertigo, eczema and rhinitis. It is available in various formulations including nasal solutions, injectables, oral dosage forms and ophthalmic preparations. The contraindications of PNM include prostatic hypertrophy, patient already on MAOI, hypersensitivity and combination with cortisol in long-term therapy [1, 2].
The most convenient and widely used analytical methods are the titrimetric modes. The recommended evaluation of Pheniramine (PN) is titrimetry (BP 2009), in which the sample is dissolved in anhydrous acetic acid and titrated against perchloric acid using a potentiometer for the detection of the equivalence point. In addition, non-aqueous titrimetric analysis of PN was also recommended by the United States of Pharmacopeia (USP). The strict requirement for use of these official methods is a robust anhydrous-medium to ensure accuracy.
The spectroscopic quantification of PN was accomplished via the use of the absorption spectra of a mixture (ternary) containing nephozoline, chloropheniramine and nephzoline derivative [3]. In addition, the estimation of the pheniramine derivative was reported in a mixture of chlorpheniramine maleate and naphazoline hydrochloride (binary mixture) through chlorpheniramine-derivative spectroscopic analysis [4]. The evaluation of the drug in a mixture at a wavelength of 528 nm (visible region) was determined to be composed of an ion pair complex of PN with quercetin and N-bromo succinimide [5]. The basic requirement for the spectroscopic analysis required pH-dependency or liquid/liquid extraction (organic solvents), which required lavish chemicals, and to ensure accuracy (official methods) an anhydrous medium is needed, which is also a challenging task.
The isolation and quantification of CPN were achieved via HPTLC using ethyl acetate:methanol:ammonia (8 : 2 : 0.5) as a solvent system in a naphazoline, CPN and naphazoline degradative mixture [3]. TLC with a densitometric protocol was used for the determination of pheniramine maleate (PNM) and chlorpheniramine maleate (CPNM) in liquid and solid dosage forms using cyclohexane : chloroform : methanol : diethylamine as the mobile phase on silica gel (F254 plates) and spots were observed under UV light [6]. High-performance liquid chromatographic technique was applied for the quantification of PN in various pharmaceutical preparations following the reversed-phase mechanism. The mobile phase was CH3OH and KH2PO4 buffer (45:55) with 0.7 ml/min flow rate and kept pH at 03 [7].
The concurrent separation and valuation of chiral-PNM, CPNM and brompheniramine maleate was accomplished using Cyclodextrin as a reference drug through Capillary-electrophoresis. Different parameters such as pH and concentration of buffer as well as chiral selections were considered during the separation process. The assay of dex-chlorpheniramine maleate in pharmaceuticals was also resolute with other similar electrophoresis procedures [8, 9]. Gas chromatography – mass spectrometry (GC-MS) study was used to analyze the PN in human serum under a basic medium [10, 11].
The precise and fast estimation of CPN was described using Isotope Dilution Gas-Chromatography/Mass-Spectrometry (ID-GC/MS). The fluorimetry and colorimetry were also used to estimate the CPN in the sample [12, 13]. Gas chromatography is another suitable technique to quantify chlorpheniramine via other additional compounds [14, 15].
Various drawbacks were confronted with the reported analysis of PN, including multiple steps, requirement of expensive chemicals, massive clean-up procedure in HPLC, lack of reproducibility, time intensive and the requirement to use toxic solvents to perform the investigation. The objective of the study was to adopt a method that would be simple, need only freely available chemicals and materials as well as needing less and inexpensive equipment. As modern automated machinery was not accessible, the reproducibility, accuracy and cost-effectiveness offered by QC Lab were appreciated.
EXPERIMENTAL
Reagents and Formulations
Chloroform (ACS-grade Fisher Scientific), Cobalt thiocyanate (99.9% Sigma Aldrich Germany), Pheniramine maleate Reference (Sigma-Aldrich), Cyanocobalamin (Sigma-Aldrich), Sodium hydroxide (ACS grade Acros Organics USA). Deionized water was used for the extraction and preparation of reagents. Commercially available formulations used for drug estimation (Table 1).
Instruments
Thermo scientific UV/Vis Spectrophotometer (xenonlamp), analytical-balance (AL-204 Mettler Toledo China made), Whatman-filter paper # 41, TurboVap (Model-10430 Harris oaks-USA), Eppendorf centrifuge (Mod-5810).
Methodology
Evaluation of Pheniramine Tablets
Tablets (10) were crushed to a fine powder via pestle and mortar and 50 mg of powder was poured into a centrifuge tube followed by 1 ml of 0.5 N sodium hydroxide solution and mixed to form a slurry. The drug was extracted with 2 ml chloroform after centrifugation (1500 rpm) for 4 min and then the chloroform was transferred to a vessel. The same procedure of extraction was repeated three times and all three organic layers were collected in the same vessel. The solvent was evaporated in a dry-bath until a purple-blue coloration formed in the chloroform layer after the addition of a mixture of Cobalt-thiocyanate and chloroform (1 ml each). The mixture was again centrifuged until the absorbance of the colored organic layer was 629 nm. The quantification of the active drug was achieved via the calibration curve by comparison with the standard.
Determination of Pheniramine Syrup
Accurately measured 3 ml syrup of Pheniramine was put in a centrifuge tube and mixed for 3 min with 0.5 N NaOH (1 ml). Chloroform (2 ml) was added and shaken gently (15 min) and then centrifuged (4 min) for the purpose of extraction. The extract was removed from the aqueous layer and transferred to a separate test tube. A similar procedure of extraction was repeated 3 times and extracts were collected in 1 container. The solvent was allowed to vaporize until the absorbance rate was at 629 nm (after making a solution with cobalt-thiocyanate in a similar way as already done with the tablet-protocol) and finally graphed.
Assessment of Pheniramine Injection
Ten brands of Pheniramine injections were randomly selected from the local market and 01 ml of the solution of injection was precisely transferred into a centrifuge tube. The added 1 ml NaOH (0.5 N) was allowed to vortex for 3 min. The processes of extraction and absorption procedure were executed in previous methods.
Validation Parameters
The methods for the validation procedures were derived from Current Good Manufacturing Practice, Code of Federal-Regulations, National Drug Administration, United States Pharmacopeial Convention, Food and Drug Administration, the American Public Health Association and the International Conference on Harmonization” [16]. In addition, the European Network of Forensic Science Institutes, ScientificWorking Group for the Analysis of Seized Drugs and the International Union of Pure and Applied Chemistry (IUPAC) have recommended these in detailed publications [16, 17].
The spectrophotometric calibration parameters included repeatability and the wavelength accuracy of UV/Vis spectrophotometry was ±1.0 nm deviation (maximum).
The validation constraints regarding the quantitative analysis of drugs included: Accuracy, Precision, Linearity, Limit of detection (LOD), Limit of quantification (LOQ), Stability, Selectivity/Specificity and Recovery.
Accuracy
The accuracy of the proposed procedure was evaluated by comparing the determined amount with the known quantity which pointed to 3 levels of sample concentrations (percentages).
Precision
The precision is a degree of confidence of the analytical results attained from a series of replicates using the same method under similar conditions. It reveals the random errors which occurred during the analytical procedure. Repeatability and reproducibility are usually recognized in precision, which is imperative, because a linear relationship is not assured without precision. It is generally measured as the coefficient of variation or relative standard deviation of analytical results. Moreover, it is concentration-dependent and should be measured at different concentrations for the assortment being used. Standard deviation was determined by the following:
Intra-day precision
Intra-day precision was accomplished by sampling the same concentration of analyte and evaluating it at 3 different intervals per day.
Inter-day precision
In this mode, the inter-day precision was performed in 3 different days.
Linearity
The linearity of the experimental model in a given range should always be directly proportional to the concentration (concn) of analytes in the sample. The employed concentrations (25, 50, 75, 100 and 125 μg/ml) were centrifuged in tubes and allowed to dry. Then Cobalt-thiocyanate (1 ml) was added to each aliquot until the appearance of a blue color. Chloroform (1 ml) was poured into each tube, which extracted the drug successfully. A standard-calibration curve was acquired from the resulting solutions by plotting concentrations vs. absorbance. As a principle of linearity, generally, a high correlation coefficient (r2) of 0.999 is required. The validation of the linearity of the proposed method was usually determined by taking a blank sample and two known concentrations of the sample, marked as QC1 and QC2 of 15 and 35 μg/ml, respectively, and checking their concentrations with the calibrated curve.
Limit of detection (LOD)
The recognition of the lowermost concentration in the sample is termed as LOD, which is associated with the determination of the impurity and the placebo (least drug) during experimental situations. LOD is assessed from the response through the standard deviation and the slope of the calibration curve statistically by employing the Hubaux and Vos method [18].
Limit of quantification (LOQ)
This represented the lowest analyte concentration was determined with adequate precision and accuracy. It was quantified from the slope of the calibration curve and standard deviation (SD) using the Hubaux and Vos method.
Repeatability
When the same analyst investigates the sample in the same laboratory with the same instruments on the same day, this is known as the occurrence of repeatability. It is calculated using 10 replicates with the same concentration of the sample. The measurement of absorbance is done three times in 1 day. The presented result (table) is the average of the three values.
Reproducibility
Any dissimilarity pointed out, other than repeatability, such as different analysts, different instruments, different days and in different laboratories is characterized as reproducibility. The reproducibility of the proposed method was tested by the absorbance of 10 imitations of 50 μg/mL of PN.
Specificity
The extent of interference of other substances (endogenous or exogenous) in an analyte sample run during identification and quantification lined to the specificity. Specificity was recognized by the investigation of the absorbance (UV/Visible) of the placebo and diluents in Pharmaceutical formulations (tab/syp/inj). It is concentration-dependent and should be estimated at the low end of the calibration series. Therefore, drug absorbance was determined at lower levels within the working range of the PN concentration.
Stability
The assessment of the stability of a solution stored at room temperature for a particular time was made. Stability determination is vital for the evaluation of drug products when accompanied by degradation products.
Wavelength and photometric accuracy as well as repeatability
Different known concentrations of PN, such as L1, L2, L3, L4 and L5, were prepared (1ml each) in test tubes. Cobalt-Thiocyanate solution (1 ml) was added to all aliquots, turning the concentration purple-blue. After the addition of 1 ml chloroform, the blue color that had formed shifted to the organic layer. The chloroform portion was carefully isolated in separate test tubes and their maximum absorbance was checked (ëmax) in 400 to 700 nm ranges. The whole practice was done 3 times at different intervals of times.
RESULTS AND DISCUSSION
Determination of λmax of Pheniramine
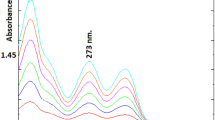
Isolated organic layers from the aqueous phase were analyzed through their absorbance. The detector exhibited a linear rise with an increase in concentration as shown in Fig. 2A, 2B, 2C, 2D and 2E, respectively. The L1 level of drug showed a wavelength (628 nm) at which the maximum absorbance was observed (Fig. 2A). The λmax (628 nm) was noticed with the L2 concentration of drug (Fig. 2B). The perceived absorbance maximum (λmax) of other concentrations (L3, L4, L5) also showed the same wavelength (628 nm) presented in Fig. 2C, 2D and 2E.

λmax determination of: L1 (A), L2 (B), L3 (C), L4 (D), L5 (E).
Repeatability and wavelength accuracy played a fundamental role in the validation method for specific wavelengths and the spectrophotometer readings should not differ more than ±1 nm for various concentrations of the drug (specific) within a wavelength range. Results showed that λmax of all drug concentrations exhibited 628 nm, indicating a significant response, whereas no wavelength deviation was detected with PN. The established response of the detector was found in a linear fashion with various levels of drug and absorption response from the detector.
Standard Calibration-curve
By plotting the absorption vs. concentrations (25, 50, 75, 100 and 125 μg/ml) of PN (Table 2), validation of the proposed method was carried out using a blank and two known concentrations of samples (QC1: 36 and QC2: 65 μg/mL) (Table 3). The response of the detector in the form of absorbance vs. dilutions was recognized to be linear with a correlation coefficient (0.999) at 628 nm (Fig. 3). Evaluation of the raw material of PN with the proposed method of analysis was also found to be adequate and within the official limits (Table 4).

Standard Calibration Curve between concentration (μg/mL) and absorbance (A).
Limit of detection
The limit of detection for Pheniramine-cobalt thiocyanate with our proposed method was 08 μg/mL (Table 4). The accuracy limit was found to be significant compared to the formerly reported spectrophotometric analysis, where “LOD” values were higher than the suggested technique.
Limit of quantification
Limit of quantification (LOQ) was recognized (15 μg/mL) for Pheniramine-cobalt-thiocyanate in a more accurate form in comparison to reported spectroscopic methods (Table 4). The LOQ values were recognized to be higher with reported techniques than with our proposed method.
Evaluation parameters
Specificity
The specificity was calculated by measuring the absorbance (UV/Vis) of the placebo and diluents used in various formulations of Pheniramine such as syrups, tablets and injectables. The statistical data proved that deviation did not deviate more than ±1 %, which significantly followed the recommendations of the International Conference on Harmonization (ICH). A number of diluents/placebos were used—Maize starch, Magnessium-sterate, Lactose, Silicon-dioxide, Propyl hydroxyl-benzoate, Sucrose, Ammonium-chloride, Dextromethorphan, Menthol, Diazepam, Glucose, Lorazepam, Sorbitol and Amaranth.
Precision
Repeatability and reproducibility parameters were employed to evaluate the precision of the suggested method of analysis. In this regard, the absorbance of 10 replicates of Pheniramine (70 μg/mL) samples was used simultaneously. Statistical analysis presented the percentage relative standard deviation (0.68), which proved to be “good precision” for the selected method (Table 5).
Accuracy
The accuracy of the proposed method was assessed (triplicate) by the comparison of the spiked amount (2, 4 and 6 mg) of the drug vs. the determined amount. Substantial results such as %RSD (1.93) and mean % recovery (98.49), were also achieved with this proposed technique (Table 6).
Solution Stability
The calorimetric analysis (proposed method) was employed to determine the drug concentration after shifting the blue-color to the organic layer from the aqueous phase. As per ICH recommendation, the evaluation of the solution’s stability was compulsory. The analysis of Pheniramine (35 μg/mL) was assessed by plotting a standard-calibration curve, which resulted from the absorbance after every six hour. Results showed that a significant result was noticed after 3-days of storage, which might be due to the degradation of the blue color. Moreover, no further color transition was observed after 1 month but a quantity deficit was noticeable. It was clear from observations that the drug deficit was negligible up to the first 12 hours. The absorbance parameters dwindled after 24 hrs and exhibited a percentage error of around 0.34%. On days 2 and 3, the calculated percentage error and quantity-deficit values were 2.55 and 3.12% and 0.87 and 1.06, respectively (Table 7). The stability parameters rendered a significant picture after a specified period of time.
Evaluation of Commercially Available Products
Various commercially available formulations of Pheniramine, such as tablets, injections and syrups were evaluated with the proposed method of analysis. The mean recovery from 50 mg Avil® tablet was 101.08% and SD was 1.20. Coldene and Enril tablets exhibited a percentage recovery of the drug at 100.39 ± 0.79 and 101.97 ± 1.13, respectively. Three randomly selected brands of tablets from the local market were quantitatively assessed indicating 102.09 ± 1.19, 100.39 ± 0.79 and 99.97 ± 0.45, respectively. The commercially available injectables such as Pheniramine (102.78 ± 0.81), Afrovil (99.85 ± 0.229), Ann-Vil (103.34 ± 1.07), Alergifen (99.62 ± 0.519), Pheniral (100.55 ± 0.79), zafvilare (102.78 ± 0.81) and Antivil (102.36 ± 0.82) were magnificently determined with our proposed method (Table 8).
CONCLUSION
This work can easily be adapted by quality control laboratories for the routine analysis of CPN without dependence on pH adjustment, solvent selection and other associated problems.
References
C. B. In, S. J. Lee, T. Y. Sung, et al., Int. J. Med. Sci., 18(4), 1075 – 1080 (2021).
D. Beasley and W. Glass, Occup. Med., 48(7), 427 – 431 (1998).
S. N. H. Azmi, S. S. K. Al-Hadhrami, B. M. R. Al-Marhoubi, et al., J. Mol. Liq., 243, 750 – 760 (2017).
K. M. Kelani, J. AOAC Int., 81(6), 1128 – 1134 (1998).
F. A. Mohamed, A. M. I. Mohamed, H. A. Mohamed, et al., Talanta, 43(11), 1931 – 1939 (1996).
S. P. Subramaniyan and S. K. Das, J. AOAC Int., 87(6), 1319 – 1322 (2004).
A. Marýn, E. Garcýa, A. Garcýa, et al., J. Pharm. Biomed. Anal., 29, 701 – 714 (2002).
H. L. Wu, C. H. Huang, S. H. Chen, et al., J. Chromatogr. Sci., 37(1), 24 – 30 (1999).
J. Martens, J. Chromatogr. B. Biomed., 673(2), 183 – 188 (1995).
J. O. Egekeze and F. W. Oehme, J. Anal. Toxicol., 3(3), 119 – 124 (1979).
P. Lawson-Smith, E. C. Jansen, and O. Hyldegaard, Scand. J. Trauma. Resusc. Emerg., 19, 1 – 5 (2011).
P. Yadav and M. Goutam, Asian. J. Pharm. Pharmacol., 6(3), 150 – 163 (2020).
P. Lundquist and B. Sörbo, Clin. Chem., 35(4), 617 – 619 (1989).
A. Sano, N. Takimoto, and S. Takitani, J. Chromatogr. B. Biomed., 582(1 – 2), 131 – 135 (1992).
J. Zamecnik and J. Tam, J. Anal. Toxicol., 11(1), 47 – 48 (1987).
J. Vessman, R. I. Stefan, and J. F. Van Staden, Pure Appl. Chem., 73, 1381 – 1386 (2001).
V. Abbate, M. Schwenk, C. Presley, et al., Pure Appl. Chem., 90, 1255 – 1282 (2018).
S.Wendelken, L. Vanatta, D. Coleman, et al., J. Chromatogr. A, 1118, 94 – 99 (2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abbas, M., Ansari, M.T. & Abbas, M. Improved and Validated Quantitative Spectroscopic Analysis of Chlorpheniramine in Pharmaceutical Formulations. Pharm Chem J 58, 538–545 (2024). https://doi.org/10.1007/s11094-024-03176-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-024-03176-0