
In defense of the internal and external environment of the lung airway, airway epithelial cells play important roles in resisting external microorganisms, toxic gases, sensitizing substances, and maintaining the stability of the internal environment of the airway. C57Bl/6 mice were exposed to ozone for 4 days (30 min/day) to induce lung injury. Airway responsiveness was measured using barometric whole-body plethysmography. Hematoxylin and eosin (HE) staining was used to detect pathological changes of the lung. Bronchoalveolar lavage (BAL) was performed to count the number of macrophages and neutrophils. 16HBE14o- cells were exposed to ozone for 30 min. 3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide was used to detect cell proliferation. Reactive oxygen species (ROS), lactate dehydrogenase (LDH), and catalase (CAT) in lung tissues and cells were also measured. The mRNA and protein expressions were detected by qPCR and western blot. We found that emodin attenuated ozone-induced airway hyperresponsiveness and lung inflammation in vivo. Emodin reduced the production of ROS, increased the expression of antioxidant enzymes, and attenuated the oxidative damage in vivo. In vitro studies showed that emodin also decreased ROS generation and LDH release and enhanced CAT activity. Emodin attenuates ozone-induced TLR4, MyD88, and NF-êB expression. Emodin has anti-inflammatory and antioxidant properties through the modulation of the TLR4-MyD88-NF-êB pathway in ozone-induced lung injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As the first line of defense of the internal and external environment of the lung airway, airway epithelial cells play important roles in resisting external microorganisms, toxic gases, sensitizing substances, and maintaining the stability of the internal environment of the airway [1, 2]. Repeated injury of airway epithelial cells and barrier dysfunction between epithelial cells increase the permeability of the epithelial layer, which promotes the entry of inhaled particulate matter and antigens into the subepithelial layer, triggers immune response, activates intracellular signal transduction, enhances secretory activity, causes inflammation, and leads to lung injury [3].
Oxidative stress and inflammatory response are the key events in pathogenesis [4]. Oxidative stress refers to the process of oxidative damage caused by the imbalance between the production and elimination of oxygen free radicals in organisms or cells, resulting in the accumulation of reactive oxygen species (ROS) in the body and cells. The stimulation of pollutants, allergens, bacteria, and ozone in the external environment can induce ROS production by airway epithelial cells [5,6,7]. Excessive ROS can increase the permeability of cell membranes, cause cell damage, and increase LDH release. Oxidative stress also stimulates redox-sensitive signaling pathways and drives the transcription of pro-inflammatory mediators [8, 9].
At present, glucocorticoids are widely used in the clinic for lung damage [10,11,12]. However, glucocorticoids are associated with many side effects, such as hypercortical syndrome, ulceration, osteoporosis, aggravated infection, and drug withdrawal reaction [13]. Exploring new medicines will bring great hope for patients with lung damage. Emodin is an anthraquinone derivative, which is the main effective monomer of rhubarb and many herbal plants. Studies show that it has many pharmacological effects, such as anti-inflammatory and bacteriostasis [14], immune regulation [15], antioxidation [16], and anticancer [17]. Emodin can attenuate bleomycin-induced pulmonary fibrosis via anti-inflammatory and anti-oxidative activities in rats [18]. Our previous studies also found that emodin has anti-inflammatory and antioxidative effects [19]. In this study, we establish an oxidative damage model with ozone in vitro and in vivo, observe the antioxidative and anti-inflammatory effects of emodin on air- way epithelial cells under ozone stress and explore the possible mechanisms.
Materials and Methods
Chemicals
The ozone exposure cabinet was self-made. The outlet of the ozone generator (Shandong Jinggong Haoyi Instrument Co., Ltd., Jinan, China) was shunted to the cabinet to create a uniform ozone environment. The concentration of O3 in the cabinet was determined by national standard 504-2009 ingot blue second reading acid vehicle spectrophotometry (once per hour). Emodin, dexamethasone injection, and red blood cell lysis buffer were ordered from Sigma (St. Louis, MO, USA). Pentobarbital was purchased from Abbott Laboratories (Abbott Park, IL, USA). Diff-Quick reagents were from Baxter Scientific (Miami, FL, USA). Lactate dehydrogenase (LDH) and catalase (CAT) assay kits were ordered from Nanjing Jiancheng Bioengineering Research Institute (Nanjing, China). DMEM/F-12 medium, newborn bovine serum, penicillin, and streptomycin were ordered from Hyclone. A RNeasy Fibrous Tissue Mini Kit was purchased from Qiagen (Hilden, Germany). A SuperScript First-Stand Synthesis System Kit was ordered from Invitrogen (Carlsbad, CA, USA). TLR4 monoclonal anti- body (Ab22048), MyD88 monoclonal antibody (ab135693), and NF-κB p65 (phospho S276) Polyclonal antibodies (ab194726) were ordered from Abcam. Enhanced chemiluminescence reagent was ordered from Pierce (Rockford, IL, USA).
Mice and Grouping
Forty-eight female C57Bl/6 mice (6–8 weeks old) were purchased from the animal experimental center of Central South University (Changsha, People’s Republic of China) and maintained in the animal facilities on a 12-h light/dark cycle with food and water available. All experiments were performed according to the institutional guidelines of the animal ethics committee of Central South University (Changsha, People’s Republic of China).
The mice were divided into six groups randomly: control group, ozone group, ozone +10 mg/kg emodin group, ozone + 40 mg/kg emodin group, ozone + 80 mg/kg emodin group, and ozone + dexamethasone group. Eight mice were used for each group (n = 8). The concentration of ozone was controlled at 1.5 ± 0.2 ppm; the modeling condition was established. Mice were stressed with ozone (1.5 ppm) for 4 days, 30 min a day. Control mice were exposed to normal air. Emodin dissolved in normal saline (10, 40, or 80 mg/kg) or dexamethasone injection (5 mg/kg) was given by daily intraperitoneal bolus injection 1 h after ozone exposure [20]. The lung tissues and BALF were collected 6 h after the last ozone exposure.
Measurement of Airway Responsiveness In Vivo
Airway responsiveness was measured using barometric whole-body plethysmography by recording airflow and respiratory pressure curves (Buxco; EMKATechnologies, Paris, France) in response to inhaled histamine. Airway responsiveness was expressed in pulmonary resistance (RL), which was determined by multiple linear regressions from transpulmonary pressure and airflow [21].
Hematoxylin and Eosin Staining
Mice were sacrificed with an overdose of pentobarbital (100 mg/kg intraperitoneally. The lungs were inflated to a pressure of 25 cm H2O with 10% formalin, harvested en bloc, fixed overnight, embedded in paraffin, and sectioned at 5 ìM. Hematoxylin and eosin staining was performed. Histological analysis was performed under an optical microscope (OLYMPUS BX41, OLYMPUS, Tokyo, Japan) and photographed at ×400 magnification.
Bronchoalveolar Lavage
Mice were injected intraperitoneally with 100 mg/kg of pentobarbiturate (Abbott Laboratories), and then sacrificed by exsanguination. The trachea was cannulated by using a 20-gauge catheter. Bronchoalveolar lavage (BAL) was performed twice with 0.8 mL of ice-cold phosphate-buffered saline (PBS; pH 7.4) each. The BAL fluid was spun at 1500 rpm for 5 min at 4°C, and supernatant was collected. The pelleted cells were harvested, and red cells were lysed by incubating the cell pellet for 5 min at room temperature in red blood cell lysis buffer. Cells were washed and resuspended in cold PBS. The total cells were determined by counting on a hemocytometer. For differential cell counting, cells (50,000 per slide) were spun onto glass slides, air-dried, fixed in ethanol, and stained with Diff-Quick reagents. The number of macrophages and neutrophils in 500 cells was counted based on morphology.
Assay of ROS
Fresh lung tissue was immediately cut into 10-micron-thick frozen sections and dyed with nuclear fluorescent dye (Hoechst) and reactive oxygen fluorescent probe dihydroethidium at 37°C for 20 min, and then rinsed with 0.01 M PBS three times, for 30 sec each time. Photographs were observed under fluorescence microscope.
Assay of LDH and CAT Activity in the Lung
Mouse lung tissues were homogenated. LDH and CAT assay kits were used to determine the concentration of LDH and CAT. As the substrate, TMB was transformed into blue under the catalysis of peroxidase and finally yellow under the action of acid. The absorbance (OD value) was determined at a wavelength of 450 nm by Tecan Sunrise, and the sample concentration was calculated [22].
Cells Culture and Ozone Stress In Vitro
16HBE14o- cells, a generous gift from Dr. Dieter C Gruenert, University of California San Francisco, is an immortalized human brain microvascular endothelial cell line. The cells were cultured in DMEM/F-12 medium (Hyclone) supplemented with 10% newborn bovine serum (Hyclone), 100 U/mL penicillin, and 100 U/mL streptomycin (Hyclone). Cells were cultured at 37°C with 5% CO2. When cells reached 80% confluence, they were stressed with ozone (1.5 ppm) for 30 min with or without incubation by emodin or dexamethasone (20 µmol/l) for 12 h [23].
Cell Proliferation Analysis
3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide (MTT) is cleaved by mitochondrial dehydrogenases to formazan crystals in metabolically active cells and this method is used to detect viable cells. MTT diluted ten times from a stock solution of 5 mg/mL was added to the 96-well plate. The cells were then incubated for 4 h and solubilized in dimethylsulfoxide. Absorption was measured at a wave- length 490 nm. The proportion of viable cells was expressed as a percentage of the control [23].
Measurement of Intracellular ROS, LDH Release and CAT Activity In Vitro
We used DCFH-DA Molecular Probes to measure the intracellular ROS according to the manufacturer’s protocol with a few modifications. Briefly, after the proper treatments, cells were washed with PBS and incubated in pre-warmed PBS containing the probes in a final working concentration of 5 μM for 30 min at 37°C. Intracellular ROS fluorescence intensity (488 nm/518 nm) was detected by a fluorometer [23].Levels of LDH in the culture media were determined using an LDH assay kit, LDH concentrations were measured by enzyme-linked immunosorbent assay (ELISA; Tecan Sunrise) at 440 nm. CAT activity was measured by the rate of decrease in hydrogen peroxide absorbance at 240 nm by ELISA.
Reverse Transcription Polymerase Chain Reaction
Total RNA was extracted from the isolated mouse lung tissues by the RNeasy Fibrous Tissue Mini kit. The concentration of RNA was determined by optical density at 260 nm (GeneQuant RNA/DNA calculator; Pharmacia LKB Biochrom, Cambridge, UK). RNA was reverse transcribed into complementary DNA with a SuperScript First-Stand Synthesis System for reverse transcription polymerase chain reaction (RT-PCR) kit. The sequences of oligonucleotide primers are shown in Table 1. The PCR cycles were performed in a thermal cycler (iCycler; Bio-Rad, Hercules, CA, USA). After the amplification reaction, PCR products were subjected to electrophoresis in a 1% agarose gel and stained with ethidium bromide. Densitometry was performed using the Quantity One TM software package (Bio-Rad) and normalized against β-actin.
Western Blotting
After treatment with different reagents, the cells were washed with ice-cold PBS, and lysed in lysis buffer (50 mM Tris–HCl [pH 8.0], 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1% Triton X-100, and 100 mg/mL phenylmethylsulfonyl fluoride) on ice for 20 min. After centrifugation at 16,0000 g for 5 min at 4°C, the supernatants (50 µg proteins) were analyzed by 10% SDS-PAGE and then electrophoretically transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA). After blocking for 2 h with 5% fat-free milk at room temperature, the membranes were incubated with either anti-human TLR4 monoclonal antibody (1:500 dilution) or anti-human MyD88 monoclonal antibody (1:500 dilution), or anti-NF-κB p65 (phospho S276) polyclonal antibody (1:1000 dilution) and then reacted with HRP-conjugated secondary antibody. Protein bands were visualized with an enhanced chemiluminescence reagent. Relative protein expression abundance was carried out by normalizing to the amount of β-actin.
Statistical Analysis
All data were expressed as mean and SE. The numerical data were analyzed using unpaired Student’s t test. The differences between groups were analyzed using the one-way ANOVA with Student–Newman–Keuls multiple comparison tests. *P < 0.05 or **P < 0.01 was considered statistically significant.
Results
Emodin Attenuates Ozone-Induced Airway Hyperresponsiveness In Vivo
To verify the success of animal models, airway responsiveness to aerosolized histamine was tested. We measured the changes in airway resistance after treatment with different concentrations of histamine challenge in control and OVA-sensitized mice. As shown in Fig. 1, the value of RL for the ozone-induced mice was higher than for the control mice. Thus, ozone-induced mice showed airway hyperresponsiveness in vivo. Emodin decreased the ozone-induced airway hyperresponsiveness dose-dependently. However, there was no significant difference between emodin group and dexamethasone group (p > 0.05).

The assessment of airway reactivity. Airway resistance was measured after different concentrations of methacholine stimulation (dose 1: 0.39 mg/mL; dose 2: 0.78 mg/mL; dose 3: 1.56 mg/mL; dose 4: 3.12 mg/mL) stimulation to evaluate airway reactivity. *p < 0.05 vs control; #p < 0.05 vs ozone.
Emodin Attenuates Ozone-Induced Lung Inflammation In Vivo
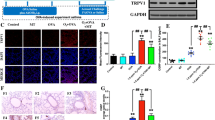
The lung tissues in the control group showed normal architecture with clear alveolar spaces. Inflammatory cells infiltrated into the lung interstitium and alveolar spaces after exposure to ozone. Treatment with emodin decreased the inflammatory infiltration in ozone-treated mouse lungs, especially at the dose 40 mg/kg (Fig. 2A). Ozone exposure also induced significant increases in total BAL cells, compared with those in control mouse lungs (Fig. 2B). The BAL cells were attenuated after treatment with emodin, especially at the dose 40 mg/kg. However, there was no significant difference between the emodin group and the dexamethasone group (p > 0.05).

The assessment of lung inflammation. A shows the pathological changes and the lung; B shows the bronchoalveolar lavage cells. **p < 0.01 vs control; ##p < 0.01 vs ozone. DXM dexamethasone.
Emodin Protects Against Ozone-Induced Oxidative Damage on Lung In Vivo
Reactive oxygen species, LDH, and CAT were used to assess the effects of emodin on ozone-induced oxidative injury. Ozone stress resulted in increased release of LDH and ROS (Fig. 3A and B). Emodin relieved the release of LDH and ROS induced by ozone. Interestingly, the mean levels of LDH were lower in 10 mg/kg and 40 mg/kg of the emodin group. As shown in Fig. 3C, ozone stress decreased the activities of CAT in mouse lung homogenates. Emodin reversed the reduction in the activities of CAT, especially at the dose of 40 mg/kg. The efficacy in the emodin 40 mg/kg group was stronger than that of the dexamethasone group (p < 0.05).

The effects of emodin on oxidative damage in vivo. A shows the reactive oxygen species (ROS) level in each group; B shows the lactate dehydrogenase (LDH) release in each group. C shows the catalase (CAT) activity in each group. *p < 0.05 vs control; #p < 0.05 vs ozone; **p < 0.01 vs control; ##p < 0.01 vs ozone.
The Effect of Emodin on 16HBE14o- Cell Viability
To explore the effect and mechanism of emodin on oxidative damage, we cultured 16HBE14o- cells. We first observed the effect of different concentrations of emodin on cell viability through MTT. We found that 0.1 μM, 1 μM, 10 μM, and 50 μM emodin had no influence on cell viability. 100 mM emodin decreased the cell viability; p < 0.01 (Fig. 4).

The effects of emodin on 16HBE14o- cell viability. 3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide assay was used to assess the influence of different concentrations of emodin (0.1 mM, 1 mM, 10 mM and 50 mM) on cell viability. **p < 0.01 vs control.
Emodin Decreases ROS Generation, LDH Release, and Enhances CAT Activity in 16HBE14o-cells
We examined the ROS, LDH, and CAT levels after treatment with different concentrations of emodin. We found that different concentrations of emodin had no effects on ROS production and LDH release in normal 16HBE14o-cells, but enhanced the CAT activity at 50 μM (Fig. 5A–C). 1 μM and 10 μM emodin significantly inhibited the ozone-induced ROS generation and LDH release. 10 μM and 50 mM emodin reversed the inhibition of catalase activity induced by ozone (Fig. 5D–F). We also found that 10 μM emodin had a similar effect to dexamethasone on ROS generation, LDH release, and CAT activity (Fig. 5G–I). However, there was no significant difference between the emodin group and the dexamethasone group (p > 0.05).

The effects of emodin on oxidative damage in vitro. A–C showed the effect of different concentrations of emodin on reactive oxygen species (ROS), lactate dehydrogenase (LDH), and catalase (CAT) in 16HBE14o- cells; D–F showed the effect of different concentrations of emodin on ROS, LDH, and CAT in 16HBE14o- cells induced by ozone; G–I showed the effect of 10 mM emodin on ROS generation, LDH release and CAT activity. *p < 0.05 vs control; #p < 0.05 vs ozone; **p < 0.01 vs control; ##p < 0.01 vs ozone. DXM dexamethasone
Emodin Attenuates Ozone-Induced TLR4, MyD88, and NF-κB expression
As shown in Fig. 6, ozone stress significantly enhanced the expression of TLR4, MyD88, and NF-κB at both mRNA (Fig. 6A–C) and protein levels (Fig. 6D–F). Emodin reduced ozone-induced upregulation of TLR4, MyD88, and NF-κB mRNA and protein in 16HBE14o- cells.

The effects of emodin on TLR4, MyD88, and NF-κB expression. Real-time polymerase chain reaction and western blotting were used to assess the relative expression of TLR4, MyD88, and NF-κB. A, B, and C show the relative mRNA level in each group. D, E, and F show the relative protein level in each group. **p < 0.01 vs control; ##p < 0.01 vs ozone.
Discussion
In this study, we explore the effect of emodin on lung injury induced by ozone in vivo and in vitro. Our results suggest that emodin might reduce the production of ROS, increase the expression of antioxidant enzymes, and reduce the oxidative damage caused by ozone. The TLR4-MYD88- NF-κB signaling pathway may be involved in this process.
Emodin is an anthraquinone derivative. A great number of studies showed that emodin can reduce cellular oxidative stress induced by various insults and drugs. Emodin attenuates acetaminophen-induced hepatorenal oxidative stress and glutathione status [24]. Emodin acts as a potent free radical scavenger and provides nephroprotection against cisplatin- induced oxidative stress by reversing cisplatin-mediated suppression of antioxidant enzymes in HEK 293 cells [25]. Emodin protects mouse lung from CS-induced lung inflammation and oxidative damage [26]. In this study, we assessed the effect of emodin on oxidative stress lung damage induced by ozone and found that emodin decreased the oxidative stress by increasing the activities of antioxidant enzymes (CAT) and decreasing the production of ROS and the release of LDH.
Bronchial epithelial cells play a critical role in the maintenance of homeostasis in the airway local microenvironment through antioxidation, exocrine/endocrine secretions, mucus production, and antigen presentation [27]. When the lung and airway are exposed to environmental pollutants, ozone, and other stressors for a long time, they produce oxidative damage [28] and inflammation [29, 30]. Ozone is highly oxidative. When the ozone is inhaled directly into the airway, the airway epithelial cells are oxidized and damaged, accompanied by increased ROS release [31]. Excessive ROS can cause lipid peroxidation, damage the structure and function of the cell membrane, increase the permeability of the cell membrane, and increase the release of LDH [32, 33]. So, we employed ozone to induce oxidative stress in this study to observe the effect of emodin on lung damage induced by ozone. The experiment of pulmonary resistance (RL) showed high RL after treatment with ozone, indicating that the model was successful. Emodin dose-dependently decreased the ozone-induced airway hyperresponsiveness. Our results suggested that ozone stress resulted in the increased release of LDH and ROS. Emodin relieved the release of LDH and ROS induced by ozone. The BAL cells were attenuated after treatment with emodin. Therefore, emodin can attenuate the ozone-induced oxidative injury and inflammation.
For the mechanism of emodin, studies showed that emodin can inhibit the TLR4 signaling pathway to attenuate induced by LPS in several tissues, such as liver and the mesentery [34, 35]. Both oxidative damage and inflammation are closely related to the activation of Toll-like receptor 4 (TLR4) pathway. TLR4 is the key receptor of airway epithelium after ozone stress. Steven R. Kleeberger’s research showed that TLR4 was the most sensitive gene to ozone stress after the whole genome quantitative trait locus analysis [36]. TLR4 ligands can be divided into exogenous and endogenous ligands according to their source. Exogenous ligands mainly come from pathogenic microorganisms and are conservative components in the evolution of microorganisms, such as bacterial lipopolysaccharides, mural acids, peptidoglycan, etc. Endogenous ligands come from host cells, such as degradation components of extracellular matrix, some necrotic cell debris, defensins, etc. Endogenous ligands are released during stress or tissue damage. TLR receptor dimerization occurs after recognition of ligands, which triggers the activation of downstream signaling pathways and biological effects [37]. We found that emodin protected against ozone-induced lung injury by decreasing ozone-induced inflammatory infiltration into mouse lungs, and attenuating TLR4.
NF-κB is a central nuclear transcription factor that regulates the expression of several genes, which play a fundamental role in the regulation of inflammation. Numerous stimuli, including ozone-induced lung injury can activate NF-κB signaling. The NF-κB activated by reactive species can translocate to the nucleus, regulating the transcriptional activation of the target genes, such as the expression of proinflammatory cytokines. Studies showed that ozone-induced airway hyperresponsiveness is dependent on the TLR4-MyD88-TIRAP signaling pathway. By activating TLR4, MyD88, and TIRAP, the levels of tumor necrosis factor-α (TNF-α), interleukin-1 β (IL-1β), monocyte chemotactic protein-1 (MCP-1) were increased, which increased inflammation and oxidative damage [38]. Thus, we observed the effect of emodin on signaling. We found that ozone increased the expression of the p65 subunit of TLR4, MyD88, and activated NF-κB complex in airway epithelial cells. Emodin decreased the expression of TLR4, MyD88, and NF-κB.
In conclusion, ozone stress can induce lung injury, which leads to activation of TLR4-MyD88-NF-κB signaling pathway. Emodin reduced the production of ROS, increased the expression of antioxidant enzymes, and attenuated the oxidative damage of airway epithelial cells, which were induced by ozone. The TLR4-MyD88-NF-κB signaling pathway contributes to the process.
Declarations
Ethics approval and consent to participate: All experiments were performed according to the institutional guidelines of the animal ethics committee of Xiangya School of Medicine, Central South University (Changsha, People’s Republic of China).
Funding: This study was funded by the Hunan Natural Science Foundation (grants #2013JJ4030,#2015JJ3170, #2015JJ2147,#2017JJ2402, and #2018JJ2463); the Open Foundation of Hunan College Innovation Program (grants #16K097, #14K109); the Young Elite Scientists Sponsorship Program by CAST (grant #2015QNRC001); the Fundamental Research Funds for the Central Universities of Central South University (grants #2017zzts354, #2018zzts813, #2018zzts812); and the Innovation Guidance Project of Hunan Science and Technology Department (grants #2017SK50507).
Conflicts of Interest Statement
The authors declare that there are no conflicts of interest.
Authors’ Contributions: DZ is responsible for the study concepts, data analysis, statistical analysis, manuscript preparation, editing review; WX is responsible for the literature research, experimental studies; YX is responsible for the study design, definition of intellectual content; MLT is responsible for the data acquisition; QQQ is responsible for the guarantee of integrity of the entire study; all the authors read and approved the final manuscript.
Acknowledgements: Not applicable.
References
C. Xiao, S. M. Puddicombe, S. Field, et al. J Allergy Clin Immunol., 128(3): 549-56.e1-12 (2011).
S. N. Georas and F. Rezaee. J Allergy Clin Immunol., 134(3):509 – 520 (2014).
A. Kato and R. P. Schleimer. Curr Opin Immunol., 19(6): 711 – 720 (2007).
C. A. Lerner, I. K. Sundar, H. Yao, et al. PLoS One., 10(2):e0116732 (2015).
O. Escaffre, H. Halliday, V. Borisevich, et al. J Gen Virol., 96(10): 2961 – 2970 (2015).
J. S. Brown, T. F. Bateson, W. F. McDonnell. Environ Health Perspect.,. 116(8):1023 – 1026 (2008).
E. Koike and T. Kobayashi. Toxicology., 196(3):217 – 227 (2004).
P. E. Pfeffer, H. Lu, E. H. Mann, et al. PLoS One., 13(8):e0200040 (2018).
A. J. Ghio, M. S. Carraway, M. C. Madden. J Toxicol Environ Health B Crit Rev., 15(1):1 – 21 (2012).
C. B. Magalhaes, N. V. Casquilho, M. N. Machado, et al. Respir Physiol Neurobiol., 259: 30 – 36 (2019).
Y. Weifeng, L. Li, H. Yujie, et al. PloS one., 11(3):e0151672 (2016).
S. Faller, R. Seiler, R. Donus, et al. PloS one., 2017; 12(4): e0176649(2017).
T. Nissly and S. Prasad. J Fam Pract., 62(9):500 – 502 (2013).
J. K. Hwang, E. M. Noh, S. J. Moon, et al. Rheumatology (Oxford)., 52(9):1583 – 1591 (2013).
S. Iwanowycz, J. Wang, D. Altomare, et al. J Biol Chem., 291(22):11491 – 11503 (2016).
G. Yang, L. Zhou, X. H. Xie, et al. Chinese Journal of Public Health., 23(22):601 – 602 (2007).
E. Haque, M. Kamil, S. Irfan, et al. Int J Biochem Cell Biol., 96: 90 – 95 (2018).
S. L. Tian, Y. Yang, X. L. Liu, et al. Med Sci Monit., 24: 1 – 10 (2018).
D. Zhang, X. Q. Qin, C. Liu, et al. Cell Biology International., 32(3):S42 (2013).
C. Liu, Y. Xiang, H. J. Liu, et al. Biochem Biophys Res Commun., 397(2):290 – 295 (2010).
Y. Xiang, Y. R. Tan, J. S. Zhang, et al. J Cell Biochem., 103(3):920 – 930 (2008).
H. Aebi. Methods in enzymology., 105(105):121 (1984).
X. L. Zhu, X. Q. Qin, Y. Xiang, et al. Peptides., 40: 34 – 41 (2013).
M. Bhadauria. Exp Toxicol Pathol., 62(6):627 – 635 (2010).
M. I. Waly, B. H. Ali, I. Al-Lawati, et al. J Appl Toxicol., 33(7):626 – 630, (2013).
W. H. Xue, X. Q. Shi, S. H. Liang, et al. J Biochem Mol Toxicol., 29(11):526 – 532 (2015).
X. Q. Qin, Y. Xiang, C. Liu, et al. Sheng li xue bao., 59(04): 454 – 464 (2007).
A. F. Behndig, A. Blomberg, R. Helleday, et al. Inhal Toxicol., 21(11):933 – 942 (2009).
I. S. Mudway and F. J. Kelly. Am J Respir Crit Care Med., 169(10): 1089 – 1095 (2004).
G. Pizzino, N. Irrera, M. Cucinotta, et al. Oxid Med Cell Longev., 2017: 8416763 (2017).
K. Z. Voter, J. C. Whitin, A. Torres, et al. Inhal Toxicol., 13(6): 465 – 483 (2001).
Y. M. Kim, S. J. Kim, R. Tatsunami, et al. Am J Physiol Cell Physiol., 312(6):C749-C764 (2017).
H. Yao, S. R. Yang, A. Kode, et al. Biochem Soc Trans., 35(Pt 5): 1151 – 1155 (2007).
Y. Ding, P. Liu, Z. L. Chen, et al. Front Pharmacol., 9: 962 (2018).
D. Shrimali, M. K. Shanmugam, A. P. Kumar, et al. Cancer Lett., 341(2):139 – 149 (2013).
S. R. Kleeberger, R. C. Levitt, L. Y. Zhang, et al. Nat Genet., 17(4):475 – 478 (1997).
Z. Meng, C. Yan, Q. Deng, et al. Acta Pharmacol Sin., 34(7):901 – 911 (2013).
Z. Li, E. N. Potts-Kant, S. Garantziotis, et al. PLoS One., 6(11):e27137 (2011).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zeng, D., Xie, W., Xiang, Y. et al. Emodin Attenuates Ozone-Induced Lung Injury Via TLR4/MYD88/NF-κB Signaling Pathways. Pharm Chem J 57, 957–964 (2023). https://doi.org/10.1007/s11094-023-02971-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-023-02971-5