
Abstract
The decomposition kinetics of sulfonol (surfactant) in water solutions as well as the formation kinetics of decomposition products under the action of an oxygen dielectric barrier discharge (DBD) at atmospheric pressure in the presence or absence of TiO2, NiO and Ag2O catalysts in the plasma zone was studied. The DBD discharge was shown to have high decomposition efficiency (phenol-up to 98%, sulfonol-up to 80%). In a plasma-catalytic hybrid process, the efficiency of organic substances decomposition was higher than efficiency for the DBD treatment without catalyst. The catalysts application resulted in a change in the ratio and yield of decomposition products.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The water purification problem from organic pollutants is of great concern. Previously, applications of gas discharges for the solution of this problem were widely studied. Up to date the decomposition processes of water solutions of the following substances were investigated: various organic dyes [1, 9], phenol [2, 4, 6, 8] and its derivatives (resorcin, pyrocatechol, hydroquinone) [6], chlorine benzene derivatives [5, 7, 10], organics of real surface waters [3] and some others. At the same time, it would be interesting to check the capabilities of such methods for purification of water solutions from surfactants. As far as we know, that type of pollutants was not tested with respect to discharge action. As an example of a surfactant, sulfonol was chosen. Sulfonol, which refers to sodium salts of linear alkyl benzene sulfonates of common formula CnH2n+1-Ar-SO2ONa (n = 12–18), is widely used for the increase in oil measures efficiency and for production of various synthetic detergents. For comparison purpose the phenol solution was used because decomposition of this compound is the most studied.
Experimental
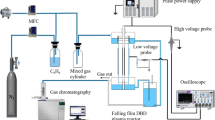
The DBD discharge was used for the solution treatment. The scheme of the experimental set-up was the same as in study [11]. A discharge was excited in a coaxial type reactor. The reactor body with a 12 mm internal diameter was fabricated from a Pyrex tube. This tube served as the dielectric discharge barrier. The external electrode was located on the tube glass surface in a cylindrical form. The length of this electrode was 12 cm. An 8 mm diameter internal electrode made from aluminum alloy was covered with an about 1 mm thick hydrophilic glass fiber fabric with the catalyst. NiO, TiO2 (anatase phase), Ag2O and mixed NiO–TiO2 were used as catalysts. For the comparison, the treatment without catalyst was performed as well. The methods used for preparing the catalysts were described elsewhere [6, 11]. The solution was flowed through the fabric along the internal electrode with film flow. The solution flow rate could be adjusted and the residence time of the solution, τL, varied from approximately 2–12 s. Technical grade oxygen (99.8%) was passed in the opposite direction. All experiments were carried out under applied voltage up to 16 kV with a frequency of 50 Hz. The discharge power was measured with the V–Q Lissajous method.
The initial and treated samples were chemically analyzed after having reached stationary conditions at reactor outlet. The total concentrations of phenol, surfactant and aldehydes were measured using fluorescent method (spectro-fluorimeter Fluorat—02, Russian production). The sulfonol was extracted into the mixture of the hydrochloric acid and the chloroform with addition of fluoresceine. The phenols were extracted into the butyl acetate. Then re-extraction into a water solution and acidation were accomplished. In the case of aldehydes the fluorescent substance was formed as the interaction product of formaldehyde and 1,3-cycloxehanedione in the presence of the ammonium ions. The total concentration of carboxylic acids was obtained by measuring the absorbance of a color reaction of acids with m-ammonium vanadate at the wavelength, λ, of 400 nm [12]. A Hitachi U-2001 spectrophotometer was used for this purpose. A determination of CO2 in a gas phase was carried out by means of passing a gas through the absorbing vessel containing ammonia solution of barium chloride followed by titration of formed barium carbonate with hydrochloric acid. The ozone concentration of the gas just passing through discharge zone in the quartz cell was measured using Hg-lamp irradiation absorption at a wave length of 254 nm. We attempted to measure the H2O2 concentration using the method of I− ion oxidation with hydrogen peroxide followed by iodine reduction with sodium thiosulfate. No H2O2 formation was discovered in the limits of this method sensitivity. Probably, the forming hydrogen peroxide is intensively decomposed under discharge active agent action (for example, ion bombardment and UV-radiation) due to the small thickness of solution layer (~0.1 mm). The relative measurement errors of concentrations were determined in series of five measurements with a confidence limit of 0.95. Curves on graphs were plotted using the treatment with the B-spline.
Results and Discussion
Experiments showed that at a fixed solution residence time the sulfonol decomposition efficiency (α) depended on the discharge power (Fig. 1), oxygen flow rate (Fig. 2) and sulfonol initial concentrations (Fig. 3). The sulfonol decomposition efficiency (α) was calculated as follows.
where Cin and Cout are the initial and final concentrations of sulfonol, respectively.

The decomposition efficiency (α) of sulfonol (1) and phenol (2) as a function of total inputted power. Initial concentration is 5 mg/L. Gas flow rate is 3.2 sm3/s. Solution residence time is 8 s. No catalysts

The decomposition efficiency (α) of sulfonol as a function of gas flow rate. Initial concentration is 5 mg/L. Inputted power is 10 W. Solution residence time is 8 s. No catalyst

The decomposition efficiency (α) of sulfonol as a function of initial concentration. 1—no catalysts, 2—TiO2, 3—TiO2+NiO, 4—NiO, 5—Ag2O. Inputted power is 10 W. Solution residence time is 8 s. Gas flow rate is 3.2 sm3/s
The discharge power growth was carried out with the increase in applied voltage and, as a result, in discharge current. This raises the increase in electron density and in rates of ionization and excitation. The increase in discharge radiation intensity was visually observed at discharge current growth. Therefore, the ion bombardment of solution surface as well as the fluxes of other possible active plasma agents, for example, UV-radiation is to be more intensive as well. As a result, the rate of non-equilibrium water dissociation in solution with reactive species formation has to increase. The dependence of α on the gas flow rate is a partly unexpected. Usually, concentrations of active particles being formed in flow discharge (for example, ozone concentration [13], see, also, Fig. 4) are decreased with the flow rate increase. Blank experiments (no discharge) showed that oxygen itself doesn’t practically react with sulfonol. Probably, the explanation can be connected with the influence of gas flow on the composition of plasma-forming gas. In study [14] it was shown that the water vapor content in oxygen–nitrogen mixture influences strongly on the electron energy function distribution (EEDF). The water vapor content growth leads to the decrease in EEDF “tail” part and, as a result, to the decrease in rate constants and rates of high threshold processes. The gas flow rate increase has to decrease in water vapor content and to growth in rates of activation processes in gas phase.

The ozone concentration (1) and the average ozone consumption rate (2) as a function of gas flow rate. Points are experiment. Line 1 is fitting. Line 2 is calculation using fitted curve 1
The dependence of α on the initial concentration is shown in the Fig. 3 for different catalysts. It can be seen that in all cases the decomposition efficiency is decreased under the growth of the initial concentration. The existence of the dependence of the degradation degree on the initial concentration indicates that, for example, formation back reactions could take place along with the degradation reactions. If an organic substance decomposed irreversibly, the degree of its decomposition would be a non-decreasing function of the initial concentration. Another reason can be connected with the of active species formation rates. These rates can be not enough to provide the complete decomposition. Particularly, the decomposition efficiency dependence on the inputted power (Fig. 1) confirms that supposition.
We do not know the exact mechanism of catalyst action. It is a subject of separate study. At least, blank experiments (no discharge) showed that the sulfonol concentration at outlet and inlet was the same after having reached stationary conditions. We suppose that there is a limitation step connected with the formation rates of active species at a plasma action only. Catalyst plasma activation provides an additional way of these particles formation. There is a correlation between initial decomposition rates (τL → 0) and energy gap width (0.8, 3.2 and 4 eV) for Ag2O, TiO2 and NiO, respectively. The more is the gap, the less is the rate.
The mechanism of catalyst action can be as follows. Under energy action a generation of electrons (e) and holes (h+) occurs:
For TiO2 and NiO the source of excitation can be the ultra-violet irradiation. The Ag2O should be additionally excited with the radiation of discharge spectrum visible part. The holes are the effective oxidizer and can form the hydroxyl radicals which are strong oxidizers of organics
Also, for the catalyst on the Ag2O base it is possibly an electro catalytic enhancing the destruction. During annealing at catalyst preparing a superstoichiometric silver can be formed. The oxidation potential of AgO+/Ag+ and AgO++/Ag+ systems (1.77 and 1.98 V/SHE, respectively) is rather high and organics decomposition is quite possible. Further experiments were carried out at 10 W of inputted power, 3.2 sm3/s of gas flow rate and 5 mg/L of sulfonol initial concentration. These conditions correspond to the maximal efficiency of decomposition (Figs. 1, 2).
The alterations in the discharge power and solution flow rate can result in changes in solution temperature and, as a result, in the changes of reaction rates. Figure 5 presents the solution temperature difference between discharge outlet and inlet at maximal inputted power. This difference does not exceed seven degrees. For this reason, the temperature changes can not influence strongly on the process kinetics.

The solution temperature difference between the discharge outlet and inlet as a function of reverse solution flow rate. The solution temperature at inlet is 292 K. Inputted power is 10 W
Main products of sulfonol decomposition were carboxylic acids (Fig. 6), aldehydes (Fig. 7), carbon dioxide (Fig. 8) and phenol (Fig. 9). The same products except phenol were formed under the phenol processing in the oxygen DBD discharge [6]. It is clear that phenol forms from aromatic ring of sulfonol molecule over the reaction with ·OH radicals. And one of the pathways of destruction is sulfonol molecule decay with the phenol molecule formation. From the shape of kinetic curves for carboxylic acids formation (at least, for TiO2 and TiO2+NiO) it follows that these products are intermediate ones. Also, in study [15] it was shown that under the action of oxygen DBD discharge on the solution of formic acid the formaldehyde and CO2 were formed only. At the same time, a formaldehyde degradation in the same conditions resulted in the formation of carbon oxides only [16]. These results are partly unexpected ones because usually carboxylic acids are products of aldehydes oxidation. Since the decomposition rate of aldehyde itself being determined in study [16] was by a factor 3 smaller than that of phenol at the conditions of our experiments it can be supposed that the aldehyde concentration accumulation occurs due to that formation rate is more than aldehyde transformation rate to carbon dioxide.

The formation kinetics of carboxylic acids in terms of acetic acid. 1—DBD only, 2—TiO2, 3—NiO, 4—TiO2+NiO

The formation kinetics of aldehydes in terms of formaldehyde. 1—DBD only, 2—TiO2, 3—NiO, 4—Ag2O, 5—TiO2+NiO

The formation kinetics of carbon dioxide. 1—DBD only, 2—TiO2, 3—NiO, 4—Ag2O, 5—TiO2+NiO

The formation kinetics of phenol. 1—DBD only, 2—TiO2, 3—NiO, 4—Ag2O, 5—TiO2+NiO
Experimental data on the O3 and ·OH interaction with sulfonol are unknown. But the possibility of ozone participation in decomposition processes can be estimated according to the following way. Supposing that the ozone is formed with the constant rate W and consumption follows the first-order rate law Kc[O3] the following expression can be obtained for ozone concentration at discharge outlet and for ozone consumption rate \( {\bar{\text{W}}}_{\text{c}} \) averaged over the discharge length
where L is discharge length, S is annular gap square for gas flow, q is gas flow rate. This expression describes the experimental curve quite well under W = 2.2 1019 molecules × L−1 × s−1 and Kc = 0.13 s−1 (Fig. 4). As it can bee seen from the Table 1 the ozone is able to ensure the sulfonol destruction. But the ozone consumption rate is decreased with gas flow rate whereas the decomposition efficiency is increased (Fig. 2). At least, it means that the ozone is not the main reagent responsible for sulfonol decomposition.
According to studies [4, 17, 18] the main active species in liquid phase are ·OH, ·H, O, HO2· O2, H2O2 and O3. The pathways of these particles formation are discussed in details in study [18]. The first way is formation in gas phase followed by particles penetration into solution. And the second is formation in a near-surface thin layer. Specifically, it is supposed that the ·OH radicals recombine fast in the surface layer forming the relative stable hydrogen peroxide. The H2O2 diffusion into a solution bulk accompanied by H2O2 decomposition provides the formation of various radicals. For example:
Taking into account the active species mentioned above the reactions of carboxylic acids formation can be as follows.
Linear part of sulfonol molecule reacts with ·OH, O· HO2· and H· radicals forming alkyl radicals.
The alkyl radical reactions result in the formation of alkoxy radicals followed by their destruction accompanied by chain scission and carboxyl radicals formation.
The reactions of the carboxyl radicals can result in the formation of carboxylic acids.
Further, carboxylic acids can be transformed to aldehydes and gaseous products with the following reaction pathways.
According to the studies [4, 19, 20] the decomposition process of aromatic hydrocarbons and phenol as a whole involves three steps: the first step is the attack of the hydroxyl radical on the aromatic rings, where the predominant products are o-dihydroxybenzene, p-dihydroxybenzene and p-benzoquinone:
The second step is the breaking of benzene rings in which the major products are maleic acid, oxalic acid and formic acid. And the last step is further oxidation where the above acids form CO2 and H2O. From view point of results mentioned above the formation of CO2 and H2O has to include another step of acids transformation to appropriate aldehydes.
According to these data the total scheme of sulfonol transformation can be as follows (Scheme 1).

Total pathway of sulfonol transformation
It is necessary to point out that catalysts application results not only in changes in decomposition rate of initial substance but in changes in formation rates of intermediate and final destruction products and in a different products ratio (Table 2). Thus, carboxylic acids were not observed at the Ag2O catalyst usage in a method detection limit. We suppose that this is due to the differences in active species formation rates which are provided by different catalysts. For this reason the ratios between rates of same reaction channels differ for various catalysts. In particular, the fact that the Ag2O provides the higher phenol concentration at residence time of ~2 s (Fig. 9) confirms our supposition mentioned above that this catalyst accelerates the ·OH radical formation rate. Probably, in this case the fast acids transformation to aldehydes provides the low concentrations of acids. It can be seen that the carbon balance is satisfied excepting of DBD discharge without catalyst and TiO2+NiO catalyst application. Probably, in this case the additional products which we can not measure are formed. For example, it can be the carbon monoxide presented in the reaction scheme mentioned above.
Table 1 shows the initial decomposition rates (τL → 0), which were obtained by analyzing the kinetic curves in Fig. 10. It can be seen that the catalyst on the Ag2O base provides both the highest decomposition rate and energetic yield. At energetic yield calculation we supposed that the discharge energy dissipates in the liquid layer. Actually, the inputted energy is divided between the gas gap and liquid phase. It is necessary to note that the calculated energetic yield is not a single-valued parameter of process since there are no linear relations between decomposition rate, discharge power and initial concentration (Figs. 1, 3). For these reasons the results on energetic yields determination obtained in various studies differ strongly. Thus, the values of 0.035, 35 and 0.18 molecules per 100 eV were obtained for the phenol water solution decomposition in studies [8, 21, 22], respectively.

The kinetics of sulfonol decomposition. Initial concentration is 5 mg/L. 1—DBD only, 2—TiO2, 3—NiO, 4—Ag2O, 5—TiO2+NiO
From viewpoint of water purification process implementation there are two important tasks. The first is a decomposition rate of target substance. The higher the rate, the better. The second is a composition of destruction products. The Ag2O and TiO2 have showed the highest decomposition rates (Table 1), the stability to the initial concentration change (Fig. 3) and maximal yield of gas (CO2). At the same time two toxic products are formed: phenol and aldehyde. For the Ag2O the aldehyde concentration is higher whereas the phenol concentration is lower than for TiO2. Therefore, the catalyst choice will depend on pollutant real initial concentration and appropriate standards on maximum permissible concentrations.
Conclusions
It was shown that the application of an oxygen dielectric barrier discharge under atmospheric pressure provides rather high decomposition efficiency for surfactants in water solution. The plasma-catalyst hybrid process demonstrated a higher rate of decomposition in comparison with usual DBD discharge action. Among all tested catalysts the catalysts on the base of Ag2O and TiO2 showed the best results. Independently on whether the catalyst was applied or not, the formation of undesirable decomposition products (phenol, aldehydes) occurs.
References
Janca J, Kuzmin S, Maximov A, Titova J, Czernichowski A (1999) Plasma Chem Plasma Proc 19(1):53–67
Tomizawa S, Tezuka M (2007) Plasma Chem Plasma Proc 27(4):486–495
Njoyin E, Ghogomu P, Laminsi S, Nzali S, Doubla A, Brisset JL (2009) Ind Eng Chem Res 48(22):9773–9780
Grymonpre DR, Sharma AK, Finney WC, Locke BR (2001) Chem Eng J 82(1–3):189–207
Bian W, Zhoi M, Lei L (2007) Plasma Chem Plasma Proc 27(3):337–348
Bubnov AG, EYu Burova, Grinevich VI, Rybkin VV, Kim JK, Choi HS (2007) Plasma Chem Plasma Proc 27(2):177–187
Guang-Zhou Qu, Lu Na, Jie Li, Yan Wu, Guo-Feng Li, Li Duan J (2009) Hazard Mater 172(1):472–478
Ogmer S, Iya-sou D, Fourmond C, Cavadias S (2009) Plasma Chem Plasma Proc 29(4):261–273
Malic MA (2003) Plasma Sources Sci Tech 12(1):26–33
Wang L (2009) Plasma Chem Plasma Proc 29(3):241–250
Bubnov AG, EYu Burova, Grinevich VI, Rybkin VV, Kim JK, Choi HS (2006) Plasma Chem Plasma Proc 26(1):19–30
Simonov VA, Nekhoroshaeva EV, Zavorovskaya NA (1988) Analysis of air environment at treatment of polymeric materials. Leningrad. Khimiya. (in Russian)
Grinevich VI (2004) Theoret Found Chem Eng 38(1):56–60
Titov VA, Rybkin VV, Smirnov SA, Kulentsan AL, Choi HS (2006) Plasma Chem Plasma Proc 26(6):543–555
Grinevich VI, Plastinina NA, Rybkin VV, Bubnov AG (2009) High Energy Chemistry 43(2):138–142
Bobkova ES, Grinevich VI, Isakina AA, Rybkin VV (2011) Izv Vyssh Uchebn Zaved Khim Khim Tekhol 54(8):55–58 (in Russian)
Bobkova ES, Grinevich VI, Isakina AA, Rybkin VV (2011) Izv Vyssh Uchebn Zaved Khim Khim Tekhol 54(6):3–17 (in Russian)
Locke BR, Shih Kai-Yuan (2011) Plasma Sources Sci Technol 20(3):034006
Gao J, Lin Y, Yang W, Pu L, Yu J, Lu Q (2003) Plasma Sources Sci Tech 12(4):533–538
Yan JH, ChM Du, Li XD, Cheron BG, Ni MJ, Cen KF (2006) Plasma Chem Plasma Proc 26(1):31–41
Lukes P, Locke BR (2005) J Phys D Appl Phys 38(22):4074–4081
Grabowcki LR, Van Veldhuizen EM, Pemen AJM, Rutgers WR (2006) Plasma Chem Plasma Proc 26(1):3–17
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bobkova, E.S., Grinevich, V.I., Ivantsova, N.A. et al. A Study of Sulfonol Decomposition in Water Solutions Under the Action of Dielectric Barrier Discharge in the Presence of Different Heterogeneous Catalysts. Plasma Chem Plasma Process 32, 97–107 (2012). https://doi.org/10.1007/s11090-011-9326-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-011-9326-z