
Abstract
Corrosion behaviour of Fe–Cr (15, 20, 25, or 30 wt.%) alloys in wet CO2 without and with HCl (0.05 or 0.1 vol%) at 750 °C was investigated. The results are compared with those at 650 °C to reveal the temperature effect. In general, weight gain kinetics and oxide scaling rates decreased with the increased chromium levels. The effect of temperature on weight gains and oxide scaling was strongly dependent on NCr. For the 15 and 20 Cr alloys, rates increased with temperature in all conditions. However, for the 25 Cr alloy, the rate increased with temperature in HCl free gas, but decreased with temperature when HCl was present. The 30 Cr alloy experienced very small weight gains which slightly increased with T. Cross-section analysis revealed the formation of thick multi-layered oxides for Fe–15Cr and Fe–20Cr, a semi-protective duplex scale of Fe2O3 and Cr2O3 on Fe–25Cr, and a thin protective chromia scale for Fe–30Cr at both temperatures. Both oxide growth rate and oxide constitution were affected by temperature, NCr, and \({p}_{HCl}\). The correlation of these factors is discussed by considering interactions between the oxides and chlorine inside the different scale types.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In several high temperature processes, including energy conversion plants, the presence of chlorine or hydrogen chloride can cause severe corrosion, reducing equipment life spans. It becomes a major problem in waste-to-energy plants, where hydrogen chloride is formed on the fire-side of superheater and/or boiler tubes [1,2,3,4].
Previous research [5,6,7,8] showed that the presence of HCl accelerates corrosion via an “active corrosion” process in which metal is volatilised as chlorides, which in turn are oxidised and redeposited to form a porous scale. Temperature is an important factor in corrosion reactions, and is expected to affect this active corrosion process. However, it is still not very clear what the temperature effect on this mechanism is in the oxyfuel process, where CO2 and water vapour are the major flue gases. Our previous results [9] showed that at 650 °C in CO2 and water vapour, Fe–Cr alloys experienced accelerated corrosion, and a higher critical Cr concentration was required for protective chromia formation when HCl was also present.
Based on that work, the present investigation focuses on the corrosion behaviour of Fe–Cr alloys in wet CO2 with (0, 0.05, 0.1%) HCl at 750 °C. The temperature 750 °C was chosen to represent an aim-point value in the design of advanced ultra-supercritical boilers. The results are compared with those at 650 °C to quantify the temperature effect with and without HCl, and determine its basis. This work was designed as a survey, to identify different corrosion mechanism regimes, rather than as a detailed kinetic study.
Materials and Experiments
Four binary ferritic alloys, Fe–15Cr, Fe–20Cr, Fe–25Cr, and Fe–30Cr (all alloy compositions in wt.%), were used. In alloy preparation, high-purity metals, Fe (99.97 wt.%) and Cr (99.95 wt.%), were melted together in a compact arc melter (MAM-1) under a varigon gas (Ar—5% H2), using non-consumable electrodes. The resulting 20 g button shaped ingots (approximately 23 mm in diameter and 8.5 mm in thickness) were subsequently homogenised by annealing in a horizontal tube furnace at 1150 °C for 50 h under flowing varigon gas. Alloy compositions were confirmed by an energy dispersive spectrometer (EDS) analysis and found to be close (± 1%) to the desired values.
As-annealed ingots were cut into rectangular samples of appropriate dimensions: (12.7 ± 1.3) × (5.5 ± 1.5) × (1.9 ± 0.4) mm, and suspension holes were drilled. The specimen surfaces were ground with 320-, 800-, and 1200-grit silicon carbide abrasive papers, and ultrasonically degreased in ethanol prior to exposure. Sample sizes were measured, and their weights were determined using an analytical balance (Mettler Toledo XP205) with an accuracy of ± 0.01 mg.
Alloys were isothermally oxidised in a horizontal tube furnace fitted with a quartz tube reactor at reaction temperatures of 650 and 750 °C for up to 160 h. All experiments were operated at approximately atmospheric pressure, and gas flow rates were adjusted to give a linear gas velocity of about 1 cm s−1 in the reactor tube at the reaction temperature. The reaction gas was N2–10CO2–10H2O–(0, 0.05, and 0.1) HCl (all in vol%). Table 1 lists the equilibrium partial pressures of oxidants calculated by FactSage 7.2.
Individual flow rates of CO2 and N2 were regulated using mass flow controllers (Brooks 5850E). Argon gas was employed to flush the reactor before and after the reaction. For HCl-free gas, the gas mixture of N2 (99.999%, < 2 ppm O2) + CO2 (99.997%) passed through demineralised water at a set temperature to generate an excess of water vapour, which was then partially condensed to obtain the required water vapour content. For the experiments using HCl, the gas mixture of CO2 and N2 passed through a thermostatted hydrochloric acid solution. The required partial pressures of H2O(g) and HCl(g) were achieved by adjusting the temperature and concentration of the solution [10].
Before reaction, alloy samples were placed in the cold zone of the reactor where samples were aligned with their flat surfaces parallel to the gas flow. Argon was then introduced to purge the quartz reactor for 1 h, and then replaced by the reaction gas. After the furnace temperature stabilised, the samples were carefully moved into the hot zone. After reaction, the samples were removed from the hot zone to the cold zone and the reactor flushed with argon.
A separate set of alloy samples was used for each reaction time. Reacted samples were weighed and then subjected to surface X-ray diffraction analysis (XRD: PANalytical). Sample cross-sections were metallographically prepared using alcohol-based DP-lubricant. Optical microscopy (Nikon-200 and Nikon-600), and a scanning electron microscope (SEM; Hitachi S3400 I) equipped with an energy dispersive spectrometer (EDS; Bruker) were used to determine reaction product morphologies and compositions. Raman spectroscopy (Renishaw (inVia 2) with a wavelength of 532 nm) was used to assist identification of oxides. Carbide formation was revealed by etching alloy cross-sections with Murakami’s solution (2 g K3Fe(CN)6 + 2 g KOH + 20 ml H2O) at room temperature.
Results
Weight Uptakes During Reaction
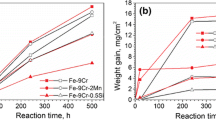
The weight gains of alloys in the gases with different HCl contents at 750 °C are compared with those at 650 °C for 30-h reaction in Fig. 1 and for 160-h reaction in Fig. 2. The same qualitative trends were evident after both reaction times. In general, increasing the Cr concentration decreases the rate of oxidation. On this basis, the alloys can be divided into two main groups: those with low Cr contents (15 and 20 wt.%) experienced large weight uptakes whereas alloys with Cr containing 25 and 30Cr reacted significantly more slowly.

Weight gains at different temperature and in different HCl found in a Fe–15Cr, Fe–20Cr, and b Fe–25Cr, Fe–30Cr alloys reacted for 30 h, except for 50 h in HCl-free at 650 °C

Weight gains at different temperature and in different HCl found in a Fe–15Cr, Fe–20Cr, and b Fe–25Cr, Fe–30Cr alloys reacted for 160 h, except for 100 h in HCl-free at 650 °C
Weight gains for all alloys reacted at 750 °C were significantly higher than those at 650 °C, except for the 25Cr alloy which reacted in HCl-bearing gas to produce lower weight gains at 750 °C than at 650 °C. With increasing HCl content, weight uptakes increased at 650 °C for all alloys. At 750 °C, however, alloys containing 15 to 25Cr evidenced lower weight gains at higher PHCl values, whereas Fe–30Cr experienced the same rather low weight gain in HCl-free and HCl-bearing gases.
Reaction Product Characterisation
Corrosion Products in HCl-Free Gas at 750 °C
Surface XRD analyses of reacted samples after 30-h reaction in N2-10%CO2-10%H2O gas at 750 °C are shown in Fig. 3. The phases detected by XRD are listed in Table 2 where those formed at 650 °C after 50-h reaction are also listed for comparison. At 650 °C, XRD patterns obtained from Fe–15Cr and Fe–20Cr showed the formation of Fe2O3 and Fe3O4. However, no apparent products were detected for Fe–25, 30Cr alloys, only the α-Fe substrate being evident. At 750 °C, the corrosion products developed on the surface of 15-25Cr alloys were Fe2O3, Fe3O4, and FeO, whereas the oxide formed on the Fe–30Cr surface was Cr2O3.

XRD patterns of the Fe–Cr alloys exposed after 30 h in N2-10CO2-10H2O at 750ºC
Cross-sectional images and Raman spectra of the reaction products of Fe–15Cr alloy after 30-h reaction at 750 °C are presented in Fig. 4. The outer oxide layer contains a thick FeO layer, together with a thin non-continuous Fe3O4 at its surface. The inner oxide is composed of a thick spinel layer and a thin IOZ underneath. Phase identification of FeO in the outer oxide and spinel in the inner oxide was based on Raman spectroscopic analysis (Fig. 4b), while the scattered Fe3O4 at the top of the surface was identified by its metallographic appearance (Fig. 4a) and the XRD results.

Fe–15Cr alloy exposed after 30 h in HCl-free gas: a Optical microscopy cross-section image and b Raman spectra and c high magnification image of internal oxidation zone
External corrosion products formed on Fe–20Cr (Fig. 5a) after 30-h reaction indicated an outer layer of Fe3O4 with small regions of FeO, and an inner spinel layer, all identified by their Raman spectra (not shown).

Optical microscopy cross-section images of a Fe–20Cr, b Fe–25Cr, and c Fe–30Cr alloys reacted after 30 h in HCl-free gas
Non-continuous, dispersed particles of Fe2O3 above a thin Fe3O4 layer and an inner spinel layer were identified for Fe–25Cr (Fig. 5b). The Fe–30Cr alloy suffered the least attack among the specimens examined (Fig. 5c), forming a thin Cr-rich oxide scale (~ 1 μm thick), as indicated by the XRD result (Fig. 3, Table 2).
Figure 6 shows cross-sectional images of Fe–Cr alloys after 160-h reaction in HCl-free gas. As is seen, increasing the alloy chromium concentration decreased the oxide scale thickness. Comparison with Figs. 4 and 5 shows that both outer and inner scale layers on each of the 15Cr and 20Cr alloys thicken with times. After 160-h reaction, the outer oxide layer on Fe–15Cr was still mainly FeO, with a thin outermost Fe3O4 zone. The inner oxide layer consisted of spinel, together with a thin IOZ. Oxides developed on Fe–20Cr with a similar morphology but reduced thickness.

Optical microscopy images of oxide scales formed on a Fe–15Cr, b Fe–20Cr, c, d Fe–25Cr, and e Fe–30Cr alloys exposed after 160 h in HCl-free gas
Significant surface Fe2O3 layer spallation had occurred on some parts of the Fe–25Cr surface (Fig. 6c). Beneath the spallation site, traces of Cr-rich oxide remained, identified by EDS analysis (not shown). Chromium-rich oxide was also detected on the remainder of the surface (Fig. 6d). The Fe–30Cr alloy formed a thin (~ 1.5 μm) protective scale (Fig. 6e), identified by XRD as Cr2O3.
Corrosion Products in HCl Gas at 750 °C
05% HCl gas
Cross-sectional images in Fig. 7 show the oxide scale developed on Fe–Cr alloys after exposure to 0.05% HCl gas at 750 °C for 30 h. The 15% Cr alloy had grown an approximately uniform outer scale layer of Fe3O4, with an average thickness of 40 μm. Voids were found in the outer oxide at and near its interface with the inner oxide layer. Cracks within the outer Fe3O4 layer had facilitated oxidant entry and conversion of the adjacent oxide to Fe2O3. Cracks were also observed in the spinel inner layer (~ 40 μm thick) (Fig. 7a). The identification of different oxides was based on specific metallographic appearance of different oxides, using the results shown in Fig. 4.

Optical microscopy images of oxides formed on a Fe–15Cr, b Fe–20Cr, c Fe–25Cr, and d Fe–30Cr alloys after 30 h in 0.05%HCl gas
On the Fe–20Cr alloy, uniform outer and inner scale layers were of the same thickness (~ 25 μm). The outer layer consisted mainly of Fe3O4, with some Fe2O3 near the outermost surface and adjacent to the occasional cracks (Fig. 7b). The inner layer was spinel.
On Fe–25Cr, two types of scale were formed (Fig. 7c). Parts of the surface were converted by a thin protective oxide, likely Cr-rich oxide. Other parts developed mutilayered scales consisting of outer Fe2O3 above a thin layer of Cr-rich oxide, identified by SEM–EDS analysis shown in Fig. 8, below which oxide had grown into the alloy (Fig. 7c). The thin intermediate Cr-rich oxide layer appeared to be continuous with the adjacent protective oxide. The inward grown oxide beneath the thin Cr-rich layer consisted of spinel with a band of chromia, identified by EDX (not shown), at the alloy–oxide interface.

Fe–25Cr alloy reacted for 30 h in 0.05%HCl gas: a BSE-SEM cross-section image, b EDX line profile along S-T and c EDX mapping of X
Oxide identifications are confirmed by the higher magnification BSE-SEM cross-section image of Fe–25Cr alloy reacted for 30 h in 0.05%HCl as shown in Fig. 8. It shows the multilayer oxide seen at low magnification at the left side of Fig. 7c. The outer Fe–rich oxide was adhered to a thin layer of Cr-rich oxide. Voids and Cr-rich spinel were identified by EDS line scan analysis (Fig. 8b) and confirmed by element mapping (Fig. 8c).
Only a protective Cr-rich oxide scale about 1.5 μm thick had formed on the surface of Fe–30Cr (Fig. 7d).
More severe corrosion attack was found for both 15Cr and 20Cr alloys reacted for longer, 160 h, in 0.05%HCl at 750 °C (Fig. 9a, b). A thick overall scale (~ 225 µm) had grown on the 15Cr alloy, consisting of two layers separately by a gap. On this alloy, the iron oxide outer layer consisted of Fe3O4 at the surface, with FeO beneath. These oxides and an inner spinel layer were identified by their Raman spectra (not shown). For Fe–20Cr, partial scale spallation occurred at the Fe3O4–FeO interface. Thick uniform outer and inner oxide layers (~ 80 μm each) were detected.

Optical microscopy images of oxides formed on a Fe–15Cr, b Fe–20Cr, c Fe–25Cr, and d Fe–30Cr alloys after 160 h in 0.05%HCl gas
High Cr alloys were much more successful. The Fe–25Cr alloy developed a thin Cr-rich oxide layer surmounted locally by Fe2O3 islands (Fig. 9c). A thin Cr-rich oxide scale subject to some local scale spallation formed on Fe–30Cr (Fig. 9d).
High magnification BSE-SEM images of Fe–15Cr and Fe–20Cr alloys after 160-h reaction in the 0.05% HCl gas are shown in Figs. 10 and 11, respectively. Local regions of IOZ had formed below the inner spinel layer in some areas of Fe–15Cr, illustrated in Fig. 10a. In addition, the higher magnification reveals the complex microstructure of the thick (~ 100 μm) inner spinel layer. As is evident from the EDS line scan analysis, the innermost region of this layer (Fig. 10a) is delineated by a series of Cr-rich oxide bands identified by point analysis shown in Fig. 10a, b, plus elemental maps (Fig. 10c–f).

Fe–15Cr alloy reacted for 160 h in 0.05%HCl gas: a BSE-SEM cross-section image b inner oxide, c–f element profile of enlarged image of L area in (a), and g line scan profile along M–N in (a)

a BSE-SEM cross-section image, b enlarged image of Z from inner oxide, and c line scan profile of Fe–20Cr alloy exposed after 160 h in 0.05%HCl gas
For Fe–20Cr, a uniform thick outer layer of Fe3O4 contained FeO islands near the prior scale–alloy interface. The latter is clearly imaged by the boundary between outer Cr-free oxide and inner Cr-bearing oxide layer. The inner oxide was a uniformly thick spinel layer about 40 μm thick (Fig. 11).
In addition to increasing the oxide layer thicknesses, increasing the reaction time led to changes in oxide composition and morphology. For both the 15 and 20 Cr alloys, the external oxides changed from Fe2O3 + Fe3O4 after 30-h reaction (Fig. 7) to Fe3O4 + FeO after 160-h reaction (Fig. 9). The fraction of Fe3O4 in the external oxide was found to increase with the increase of Cr content at both reaction times (Figs. 7, 10, 11).
1%HCl Gas
Surface XRD analysis results for Fe–Cr alloys after reaction in N2-10%CO2-10%H2O with 0.1%HCl for 30 h at 750 °C are shown in Fig. 12. The phases detected by XRD are listed in Table 3 where those at 650 °C after 50-h reaction are also listed for comparison. At 650 °C, the surface oxidation products developed by 15 to 25Cr alloys were mainly Fe2O3 and Fe3O4, while Fe–30Cr showed only the alloy, α-Fe [9], indicating that any oxide was too thin to be detected. At 750 °C, the near surface reaction products were identified as Fe2O3 and Fe3O4 for 15Cr and 20Cr alloys. For Fe–25Cr and Fe–30Cr, only Cr2O3 peaks were detected (Table 3).

XRD patterns of the tested alloys exposed after 30 h in N2–10CO2–10H2O–0.1%HCl at 750ºC
Reaction product morphologies and Raman spectra for the 15Cr alloy after 160-h reaction in 0.1% HCl gas at 750 °C are shown in Fig. 13. Local cracking and partial scale spallation are evident (Fig. 13a). A very thin layer formed on the outermost part of the oxide was identified as Fe2O3 by its metallographic appearance, and a relatively thick IOZ with Cr-rich band at its inner boundary is seen beneath the inner spinel oxide layer in some areas (Fig. 13c). From scale surface towards the substrate, oxide phases revealed by Raman spectroscopy (Fig. 13e) were magnetite, wüstite, and spinels.

Fe–15Cr alloy reacted for 30 h in 0.1%HCl gas a–c optical micrographs, d enlarged image of selected IOZ zone, and e Raman spectra of oxides from (c)
The oxide formed on the 20Cr alloy (Fig. 14) was similar to that formed in 0.05% HCl (Fig. 11). Its magnetite and spinel oxides were identified by Raman spectra. Thin (about 2 μm) protective scales of chromia were developed on the surfaces of both 25Cr and 30Cr alloys, as seen in Fig. 15 and Table 3.

Optical micrographs of Fe–20Cr alloy after reaction for 30 h in 0.1%HCl gas

Optical cross-section images of scales formed on a Fe–25Cr and b Fe–30Cr alloys after reaction for 30 h in 0.1%HCl gas
Similar oxide structures developed on Fe–15Cr alloy after 160-h reaction in both 0.05% HCl (Fig. 10) and 0.1% HCl atmosphere (Fig. 16): outer Fe3O4, intermediate FeO, and an inner spinel layer. Increasing alloy Cr to 20% led to a decreased FeO layer thickness after 160-h reaction (Fig. 17a), but left the phase constitution of the scale unchanged. Both Fe–25Cr and Fe–30Cr alloys formed the same protective scale thickness (~ 2 μm) as shown in Fig. 17b, c. In each case, the scale thickness was approximately the same after 160-h reaction as it was after 30-h reaction. Analysis by EDS (Fig. 18) showed the protective scale on Fe–25Cr alloy exposed to 0.1%HCl for a period of 160 h to be Cr-rich, containing very little Fe. This indicates that the Cr2O3 scale formed on this alloy in the first 30 h of reaction was retained after 160-h exposure.

Optical microscopy images of Fe–15Cr exposed after 160 h in 0.1%HCl gas

Optical microscopy images of a Fe–20Cr, b Fe–25Cr, and c Fe–30Cr alloys exposed after 160 h in 0.1%HCl gas

Fe–25Cr alloy reacted after 160-h reaction in 0.1%HCl gas: a BSE-SEM image and b line scan profile along A-B in (a)
Carburisation
Etching with Murakami’s reagent revealed carbide formation for Fe–15Cr and Fe–20Cr reacted in CO2-H2O gas (without HCl) at 650 °C and 750 °C, as seen in Fig. 19. At 650 and 750 °C, both intergranular and intragranular carbides were observed, with higher precipitate number densities and deeper penetration at higher temperature. For Fe–15Cr, the depth of carbon penetration reached ~ 60 μm as intragranular carbides, as measured from the original alloy surface to intragranular carbide precipitation front, after 100 h at 650 °C (Fig. 19a). The corresponding depth was ~ 600 μm after 160 h at 750 °C (Fig. 19b), again measured from the prior scale–alloy interface. Similarly, in the 20Cr alloy, intragranular carbides were detected in a shallow subsurface zone beneath Fe–rich oxide nodules at 650 °C (Fig. 19c), but to a depth of approximately 330 μm at 750 °C (Fig. 19d) under a continuous, uniform scale. In HCl-free gas, carburisation was not detected in the higher Cr containing alloys, 25Cr, and 30Cr alloys at 650 °C [9]. At 750 °C, a small number of scattered carbides precipitated in the alloy matrix of Fe–25Cr (Fig. 20a), but no carbides were found in the 30Cr alloy (Fig. 20b).

Metallographic cross section of carbide formation found in Fe–15Cr alloys: a after 100 h at 650ºC, b after 160 h at 750ºC, and in Fe–20Cr alloys: c after 100 h at 650ºC, and d after 160 h at 750ºC exposed in HCl-free gas

Metallographic cross section of carbide formation observed in a Fe–25Cr and b Fe–30Cr alloys exposed after 160 h in HCl-free gas at 750ºC
Internal carbide formation in Fe–15Cr and Fe–20Cr reacted for 160 h in N2–10CO2-10H2O-0.1%HCl at 650 and 750 °C is shown in Fig. 21. As with the HCl-free gas at 650 °C, carburisation was revealed by etching in both alloys (Fig. 21a, c) where denser and deeper carbide precipitation was found after 160-h reaction than in alloys reacted for 100 h in HCl-free gas (Fig. 19a, c). At 750 °C, addition of HCl to the reaction gas significantly reduced the amount of carbide formed. There was no significant intragranular carburisation in any alloy, even for 15 and 20Cr alloys (Fig. 21b, d) at this temperature. At 650 °C, no carbide was found in Fe–30Cr alloys but intergranular carbides developed in the Fe–25Cr alloy [9]. At 750 °C, a few scattered carbides formed along alloy grain boundaries in both high Cr alloys (Fig. 22).

Metallographic cross section of carbide formation found in Fe–15Cr alloys: a at 650ºC, b at 750ºC, and in Fe–20Cr alloys: c at 650ºC, d at 750ºC exposed after 160 h in 0.1%HCl gas

Metallographic cross section of carbide formation found in a Fe–25Cr and b Fe–30Cr alloys exposed after 160 h in 0.1%HCl gas at 750ºC
Discussion
Discussion is facilitated by use of Tables 4 and 5, which summarise scale types—breakaway, semi-protective or protective—and scale thicknesses. Here “semi-protective” refers to thin, duplex scales such as that shown in Fig. 9c. The measured average thicknesses do not correspond exactly with the weight changes shown in Figs. 1 and 2, because of occasional, randomly distributed instances of minor spallation. In addition, as discussed in "Effects of HCl" section, volatilisation of oxide as metal chloride is possible.
Alloy weight uptakes in Figs. 1 and 2 reveal similar patterns of dependence on alloy chromium concentration (NCr), temperature, and \({p}_{\text{HCl}}\) at the two reaction times. Closely similar patterns seen for oxide scale thicknesses in Tables 4 and 5 indicate that weight uptakes reflect principally external scale growth, weight changes associated with internal carburisation and oxidation being relatively small.
In general, scaling rates decrease with increased chromium levels. However, the effect of temperature on scaling rate is strongly dependent on NCr. For the 15 and 20 Cr alloys, rates increase with temperature, the effect being somewhat diminished when HCl is added to the gas. In the 25 Cr alloy case, the scaling rate increases with temperature in HCl free gas, but decreases with T when HCl is present. The 30 Cr alloy experiences small scaling rate increases with T, the extent of the increase being less as HCl levels are increased.
The effects of HCl on scaling vary with both NCr and T. At 650 °C, the rate increases for all alloys as HCl is added to the gas. This effect is large for alloys containing 15 – 25 Cr, but relatively small for 30 Cr. In contrast, at 750 °C the scaling rate decreases for alloys containing 15–25 Cr as HCl is added to the gas. For the 30 Cr alloy, rates are low, and small increases accompany HCl additions.
Evidently, there are interactions among temperature, NCr, and \({p}_{\text{HCl}}\) as factors affecting oxide scale growth. The discussion commences with a correlation of scale morphologies with growth rates. It then goes to an examination of the conditions under which a protective Cr-rich oxide scale can form. Finally, the ways in which HCl can interact with growth of the different possible oxides are assessed.
Effect of Temperature on Oxidation Behaviour in CO2–H2O Gas
The thick oxide scales grown by many of the Fe–Cr alloys are typical of scales grown in CO2-rich gases [11]. The outer iron oxide layers grow by outward diffusion of iron, whereas the porous inner spinel layer grows inward via oxygen transport [12]. Alloy Cr, immobilised as spinel within the inner layer, serves to mark the space occupied by the alloy before reaction.
In general, increasing temperature enhances oxidation rates of Fe–Cr alloys in CO2-H2O gas, as evidenced by the increased oxide thickness shown in Table 4 (short time reaction) and Table 5 (long time reaction). The reported total scale thickness includes both outer Fe oxide and inner spinel layers. This accelerating effect reflects more rapid diffusion of iron and oxygen, leading to an enhanced outer and inner oxide thickening for Fe–15Cr and Fe–20Cr alloys.
Increasing temperature also changes the oxide constitution in the external oxide scale. For Fe–15 and 20Cr alloys, at 650ºC, the outermost oxide was Fe2O3 and the inner part of the Fe–rich oxide was Fe3O4 [9]. However, at 750 °C, the outermost layer became Fe3O4 and the inner part FeO (Figs. 4, 5 and 6). This result confirms that increasing temperature significantly accelerates iron cation diffusion in the underlaying oxides. Thus, FeO grows more rapidly and the increased supply of iron at higher temperatures converts Fe2O3 to Fe3O4. Similar observations of these oxide compositional changes with temperature have been reported before [13, 14]
Alloy Cr concentration affects both the growth rate and composition of the oxide scale. Increasing Cr concentration from 15 to 20 wt.% led to a decrease in relative amounts of FeO in the outer scale layer at 750 °C (Fig. 5a). Further increasing alloy Cr to 25 wt.% led to the formation of Fe2O3 at the scale surface. No FeO developed, and Fe3O4 made up the balance of the outer layer (Fig. 5b). These effects reflect a slower rate of outward iron diffusion in the scale resulting from the higher volume fraction of slow-diffusing spinel in the inner scale layer.
The effects of temperature and alloy Cr content on growth rates of nonprotective (breakaway) scales are clear. Alloys containing sufficient chromium form protective chromia scales, which grows at a much slower rate, almost independent of the alloy Cr/Fe ratio. This rate is temperature-dependent, but present results are insufficient to permit analysis. What the results do show is that the boundary between breakaway and protective scaling is temperature-dependent.
Effect of Temperature on Critical Cr Concentration
At 650 °C, an Fe–20Cr alloy is partially protective and Fe–25Cr forms a fully protective scale [9] in HCl-free gas. However, a Cr level > 25 wt.% is required for protection at 750 °C (Table 5). To understand this difference, the use of Wagner’s diffusion analysis is explored.
The critical concentration of Cr, \({N}_{Cr}^{(1)}\) for converting chromia formation from internal to external oxidation can be calculated using Wagner’s equation [15].
where g is a critical volume fraction of oxide precipitate, generally approximated as about 0.3 [16], ʋ the stoichiometric coefficient of CrOʋ (1.5), Vm and VCrOʋ are the molar volumes of the alloy (7.3 cm3/mol) and oxide (14.6 cm3/mol), respectively, DO represents the diffusion coefficient of oxygen in the alloy [17, 18], and \({\tilde{D }}_{Cr}\) is the alloy interdiffusion coefficient [19].
The quantity \({N}_{O}^{(S)}\) is the oxygen solubility (mole fraction) in the alloy in equilibrium with the oxygen activity at the scale–alloy interface, which is calculated from Sievert’s equation:
Here K is the equilibrium constant for the dissolution of oxygen taken from available data [20], and pO2 is the partial pressure of oxygen determined by the Fe/FeO equilibrium at the scale–alloy interface [9].
In addition, the Cr concentration in the alloy must exceed another critical value, \({N}_{Cr}^{(2)}\) required for chromium diffusion to the alloy–scale interface to maintain the chromia growth [21]:
Here \(k_{p}\) represents the parabolic rate constant for chromia scale thickening.
where \(X\) is the chromia layer thickness. At 750 °C, the value of kp = 2.0 × 10−14 cm2/s was calculated approximately, using the chromia scale thickness of about 1.5 μm formed on Fe–30Cr alloy after 160-h reaction in HCl-free gas (Fig. 6e). Similarly, the value of kp = 3.5 × 10−14 cm2/s was estimated from the 2.0 μm thickness of chromia grown in the same reaction time in 0.1% HCl environment (Fig. 17c).
Critical chromium concentrations calculated in this way are shown in Table 6. The Cr concentration (\({N}_{\text{Cr}}\sim \)0.21) in Fe–20Cr alloy is higher than \({N}_{\text{Cr}}^{(1)}\) and \({N}_{\text{Cr}}^{(2)}\) values at both oxidation temperatures in the absence of HCl, and the alloy is predicted to form and maintain a chromia scale. This is in fact partly correct at 650 °C, as the surface is covered by a protective scale interrupted by Fe–rich oxide nodules [9]. At 750 °C, however, present results clearly show breakaway oxidation (Tables 4 and 5).
All such predictions are to greater or less extent inaccurate because of uncertainties in the data on which they are based. Nonetheless, qualitative effects of temperature on the competition between chromia passivation and the different breakaway processes should be predictable. Passivation is predicted in Table 6 to be achieved at lower NCr levels as the temperature is increased, because the temperature sensitivity of \({\tilde{D }}_{\text{Cr}}\) is so high [18]. However, as seen in Table 5, this prediction also fails. At this point, it needs to be recognised that the high chromium alloys used here are subject to \(\sigma \)-phase formation at the relatively low temperatures employed.
Precipitation of the \(\sigma \)-phase is slow and was not metallographically detectable in the present case. However, should it have occurred at a submicroscopic level, the effect would be to lower the value of NCr in the α-matrix, thus leading to slower chromium diffusion to the alloy surface. Increasing T from 650 to 750 °C leads to an increase in the solubility of Cr in the α-phase from 17.5 to 25 at. % [22]. Thus, not only is \({\tilde{D }}_{Cr}\) increased, but so too is the concentration of mobile chromium, making the T-effect on \({N}_{\text{Cr}}^{(1)}\) and \({N}_{\text{Cr}}^{(2)}\) even stronger. A more detailed analysis of the conditions leading to selective oxidation of 2-phase alloys is available [23], but it leads to the same conclusion regarding the T-effects. Discussion returns to this point in "Carburization" section.
Addition of HCl to the gas is not expected to affect \({N}_{\text{Cr}}^{(1)}\), dependent as it is only on alloy properties. However, HCl does change the predicted values of \({N}_{\text{Cr}}^{(2)}\) by accelerating chromia scaling rates. On this basis, it is predicted that higher NCr values are required to achieve protection in HCl-bearing gas, a prediction which is borne out at 650 °C (Table 5). At 750 °C, however, the prediction fails for the marginal Fe–25Cr alloy. In this case, the alloy is semi-protective in the absence of HCl, failing, as predicted, by growing external Fe2O3 as well as chromia. Additions of 0.5% HCl show the growth of this duplex scale, and 0.1% HCl prevents iron oxide formation entirely. Clearly, other factors are in effect.
Effects of HCl
As seen in Figs. 1 and 2, additions of HCl to wet CO2 gas had different effects for different alloys, and those effects changed with temperature. In considering possible interactions between HCl and the oxides, it is useful to distinguish between the different scale types: breakaway, semi-breakaway and protective.
Breakaway scales form on Fe–15 Cr in all gases at both temperatures. They consist of thick outer layers of iron oxides and inner layers of spinel. The presence of HCl accelerated the rate of oxidation of all Fe–Cr alloys at 650 °C, a phenomenon attributed to the vaporisation of iron chloride by “active oxidation” [9].
The active oxidation process [7] is one of volatilisation of metal oxides as chlorides, for example
The chlorine required for this reaction derives from the reaction
Chlorine has access to the scale surface and, if it can diffuse into the oxide, to the scale interior. At the low oxygen potentials prevailing within the scale, reaction (5) is favoured, leading to partial loss of oxide and outward gas phase transport of metal chloride. In the outer parts of the scale, oxygen activities are much higher and the reverse of reaction (5) becomes thermodynamically possible, leading to redeposition of oxide.
Breakaway Oxidation
Active oxidation accelerates outward transport of iron within the scale. As this is the rate controlling process in breakaway scale growth on Fe–Cr alloys [12], more rapid oxidation results (Fig. 2, Table 5) from the presence of HCl. This accounts for the behaviour of Fe–15Cr and Fe–20Cr at 650 °C shown in Fig. 2, but not for the opposite effect found at 750 °C. In considering this, attention is directed first to the relative volatility of iron and chromium chlorides.
Chromium is present in the inner layer of breakaway scales in the form of spinel, so the relevant volatilisation reaction can be written
To evaluate \({p}_{{FeCl}_{2}}\) and \({p}_{{CrCl}_{2}}\), knowledge of both \({p}_{{O}_{2}}\) and \({p}_{{Cl}_{2}}\) is required. Both can be calculated for gas phase equilibrium at the scale surface, and are shown in Table 1. The oxygen potential at the scale–alloy interface can be calculated from the local metal–metal oxide equilibrium. In the case of breakaway scales, this is assumed to be
leading to the values for \({p}_{{O}_{2}}\) shown in Table 7.
The value of \({p}_{{Cl}_{2}}\) at the scale–alloy interface is unknown, depending as it does on the transport processes within the scale. Its maximum value would be that of the gas phase, of order 10–10 atm at both temperatures. However, use of this value in the equilibrium expression for reaction (5) leads to estimates of \({p}_{{FeCl}_{2}}\) > 1 atm. Proceeding instead on the basis that \({p}_{{Cl}_{2}}\) cannot be estimated, one can nonetheless calculate the ratio \({p}_{{FeCl}_{2}}\)/\({p}_{{CrCl}_{2}}\) by eliminating \({p}_{{Cl}_{2}}\) between the equilibrium expressions for (5) and (7). In this way, it is found that
where Ki is the equilibrium constant for the indicated reaction. Values for this ratio are seen in Table 7 to be extremely high at both temperatures. Whilst it is clear that iron is much more readily volatilised from the scale than is chromium, the thermodynamic analysis provides no explanation for the slowing in oxidation rate at the higher temperature when HCl is present.
The effects of HCl on breakaway oxidation morphologies are illustrated in the case of Fe–15Cr in Fig. 23. At 650 °C, the addition of HCl to the gas leads to more rapid thickening of both the spinel and Fe–oxide layers, but has little effect on the IOZ. At 750 °C, in the presence of HCl, the growth of both scale layers is slowed and IOZ formation is suppressed. Significantly, an additional outermost layer of bright contrast iron oxide is formed in the presence of HCl at 650 °C, but no such oxide is present at 750 °C. Closer examination of this oxide [9] has confirmed that it is highly porous Fe2O3.

Cross-section of oxide reaction products formed by Fe–15Cr after, a–c 100 h in HCl-free gas [9] and d–f 160 h in HCl-bearing gases
The formation of porous Fe2O3 at 650 °C results from “active oxidation” [9] and reflects an acceleration in scaling rate. The disappearance of Fe2O3 at the higher temperature can be understood in terms of the reaction
which can occur at the Fe3O4–Fe2O3 interface. Here, \({p}_{{O}_{2}}\) is controlled by the local equilibrium between the two oxides and its value is shown in Table 7. For the case where the Fe2O3 layer is very thin and highly porous, \({p}_{{Cl}_{2}}\) is approximated as the gas phase value (Table 1), leading to the \({p}_{{FeCl}_{2}}\) values shown in Table 7 for this interface.
As is seen, \({p}_{{FeCl}_{2}}\) is relatively high, and destruction of the thin outermost layer of Fe2O3 might be anticipated. The difficulty, however, is that \({p}_{{FeCl}_{2}}\) values are very similar at the two temperatures, and no acceleration with temperature in the rate of hematite loss can be accounted for in this way.
The net rate of iron oxide layer growth reflects the balance between the rates of outward iron diffusion and oxide volatilisation. Comparison of the inner spinel layer grown on Fe–15Cr at 750 °C in the different gases at the two temperatures (Fig. 23) reveals that semi-continuous Cr-rich bands had developed within this layer parallel to the alloy surface, during exposure to HCl bearing gas, but not in HCl-free gas. Similar bands are seen in Fig. 11 to have formed in the inner oxide layer grown on Fe–20Cr under these conditions, but not in the absence of HCl (Fig. 6b).
These Cr-rich oxide bands, aligned as they are normal to the diffusion direction, will slow outward iron diffusion. This in turn accounts for the slower growth of the inner layer. In combination with the iron oxide volatilisation process, it explains why the outer layer thickness is smaller at the higher temperature, in the presence of HCl. Reasons for the formation of the Cr-rich oxide bands are examined below in the discussion of carburisation.
Semi-protective Oxidation
This regime of behaviour is characterised by formation of a chromia layer which is nonetheless penetrated by outwards diffusion of iron. The resulting duplex scales consisting of outer Fe2O3 and inner Cr2O3 layers grow on the marginal Fe–25Cr alloy at 750 °C in HCl-free gas, and also in 0.05% HCl gas but to a reduced thickness (Table 5). In 0.1% HCl gas; however, no outer Fe2O3 layer is seen and the alloy appears to have passivated.
Passivation of this alloy is predicted to occur in HCl-free gas (Table 6), but its failure by iron oxide growth in HCl gas is seen in Table 5 to be marginal. Adding HCl evidently diminishes the extent of Fe2O3 formation, and at the 0.1% level prevents its growth completely.
The reaction of interest is now (10), and the fate of an Fe2O3 nucleus on the surface of an otherwise protective chromia scale is considered. At the interface between the two oxides, local equilibrium between them [24] yields an estimate of \({p}_{{O}_{2}}\approx \) 10–13 atm at 627 °C. The value of \({p}_{{Cl}_{2}}\) at this early stage of nodule nucleation would be close to that of the surrounding gas, and the \({p}_{{FeCl}_{2}}\) values in Table 8 are calculated from (10). The value of \({p}_{{FeCl}_{2}}\) at 750 °C is ~ 40 times that at 650 °C. It is reasonable then to suppose that increasing levels of HCl will lead to the diminution and ultimately suppression of Fe2O3 formation at 750 °C, but the effect is unimportant at the lower temperature.
Verification of this proposal is desirable. Whilst the mechanism succeeds in explaining the observations in Table 6, it implies that the apparent protection afforded by the chromia scale in HCl gas is somewhat illusory: iron continues to be removed from the alloy as chloride, and loss of component section is predicted to result.
Protective Oxidation
Only the Fe–30Cr alloy passivates at both temperatures, regardless of whether or not HCl is present. This is a consequence simply of the high NCr level, which is sufficient to form and sustain an external chromi scale (Table 6). The questions of interest concern the possible effects HCl might have on the stability of the protective scale.
Approximating the scale as pure Cr2O3, its interactions with Cl2 can be written as
At a chromia scale–gas interface, calculation for the equilibrium (12) yields \({p}_{Cr{O}_{2}{Cl}_{2}}\) = 5 × 10–14 and 1 × 10–13 atm at 650 and 750 °C, respectively. At the scale–alloy interface, \({p}_{{Cl}_{2}}\) is unknown, but has a maximum value equal to that of the gas phase. The corresponding maximum values of \({p}_{Cr{O}_{2}{Cl}_{2}}\) are 10–20 and 10–19 atm at 650 and 750 °C, respectively. Volatilisation of Cr via reaction (12) is concluded on this basis to be unimportant.
Chromium chloride formation at the scale–alloy interface is much more favourable. Again taking the maximum value of \({p}_{{Cl}_{2}}\) equal to that of the gas phase, the maximum values of \({p}_{{CrCl}_{2}}\) shown in Table 8 are calculated. Such high values are inconsistent with the observations of apparently adherent, non-porous chromia scales, which implies that chromia scales are better barriers to chlorine ingress than iron oxides. However, the possibility of some Cr volatilisation by this mechanism cannot be fully excluded if defects are present inside the chromia scale.
Carburisation
Alloys forming protective or semi-protective oxide scales suffered very little carburisation at either 650 [9] or 750 °C, in any of the gas mixtures. The small amounts of intergranular carbide form as a result of carbon penetration through chromia via their grain boundaries, as shown by atom probe tomography [25].
A more complex pattern of behaviour was observed for alloys developing breakaway oxide scales. Both intragranular and intergranular carbides were observed in lower Cr content alloys reacted in HCl-free gas at 650 °C (Fig. 19a, c) and 750 °C (Fig. 19b, d), with a greater depth of carbon penetration at 750 °C. The faster carburisation kinetics observed at higher temperature in alloys forming breakaway oxide scales are consistent with rate control by inward carbon diffusion.
For Fe–15Cr and Fe–20Cr alloys at 650 °C, the addition of HCl to the gas accelerated the oxidation, causing the extensive formation of iron-rich, multilayer, porous oxide scales plus IOZ (Fig. 21a, c). These scales more readily permit carbon diffusion to the underlying alloy, leading to the formation of more intragranular carbides and intergranular carbides than in HCl-free gas [9].
In contrast, at 750 °C, no apparent carbide can be identified even for low Cr alloys (Fig. 21b, d) in the presence of HCl. Clearly, carburisation was suppressed at the higher temperature in HCl-containing gas, an unexpected outcome.
In the absence of internal carburisation, these alloys are subject to very little depletion in Cr because its selective oxidation does not occur. As the rate at which the scale–alloy interface advances slows with time, the probability of Cr-rich oxide aggregates forming as a result of alloy chromium diffusion at the interface increases. The banded microstructure, involving alternate Cr-rich and Cr-lean oxide seen in Figs. 10, 11 and 16, is in fact a common observation in marginal alloys [26, 27]. On the other hand, when internal carbides precipitate, chromium is removed from the alloy matrix, concentrated and immobilised in the Cr-rich precipitates. These are subsequently oxidised in situ to form an IOZ. As reaction proceeds, these are incorporated into the advancing spinel layer as dispersed particles, and no banded structure can form.
Whether or not a banded structure develops is important, as the semicontinuous Cr-rich oxide provides a partial barrier not only to outward iron diffusion, but also to inward carbon transfer. Thus once the bands form, they prevent the onset of carburisation and allow growth of additional bands.
The remaining question concerns the mechanism whereby HCl or Cl2 prevents the onset of internal carburisation. The answer presumably involves interactions between Cl species and both the detailed mechanisms of carbon transport through the complex scale, and the oxide–metal interfacial reactions
Present results provide no answers to these questions.
Conclusions
The effect of temperature on the corrosion process is strongly dependent on both alloy Cr concentration and gas phase pHCl in the gas. The 15 and 20 Cr alloys underwent breakaway corrosion, at rates which increased with temperature in all gases.
A 25 Cr alloy was borderline developing either protective chromia scales or semi-protective chromia plus hematite. The rate increased with temperature in HCl free gas, but decreased in HCl-containing gases. The 30 Cr alloy was always protective, and its corrosion rate increased slightly with T. The weight uptake kinetics reflect principally external oxide growth, but internal oxidation and carburisation were also observed in some cases.
Increasing temperature also changes the oxide constitution in the external scale. On the Fe–15 and 20Cr alloys breakaway scales consisted of an outer iron oxide layer and inner layer containing spinel at both temperatures. At 650 °C, the outermost oxide was Fe2O3 and the inner part of the Fe-rich oxide was Fe3O4. However, at 750 °C, the outermost layer became Fe3O4 and the inner part FeO. This results from accelerated iron cation diffusion in the underlaying oxides, converting Fe2O3 to Fe3O4 and Fe3O4 to FeO at higher temperature.
Alloy Cr concentration affects both the growth rate and composition of the oxide scale. Increasing Cr concentration from 15 to 20 wt.% led to decreased relative amounts of FeO in the outer scale layer at 750 °C. Further increasing alloy Cr to 25 wt.% led to the formation of Fe2O3 at the scale surface and no FeO developed. These effects reflect a slower rate of outward iron diffusion in the scale resulting from the higher volume fraction of slow-diffusing spinel in the inner scale layer.
The presence of HCl affects both the morphologies and growth rates of oxides by chemical interactions which differ with the types of scale. In the case of breakaway oxidation (15Cr and 20Cr alloys), chlorine can penetrate through thick multi-layered iron-rich oxide scales which are partly volatilised as iron chloride, which is reoxidised near the scale surface by “active oxidation.” In the situation of semi-protective scales (25Cr alloy), chlorine attacks the thin outer layer of Fe2O3 at its interface with the inner Cr2O3 layer. In the protective case (30Cr alloy), only Cr2O3 forms, and its surface vaporisation was demonstrated to be negligible.
Carburisation was observed for alloys forming non-protective oxide scales. The addition of HCl led to increased carburisation of these alloys at 650 °C due to enhanced oxide porosity which allowed greater permeability by carbon species. In contrast, HCl suppressed carburisation of the breakaway scales at 750 °C, by the development of chromia bands within the inner spinel layer.
Almost no carburisation, just sparsely distributed intergranular carbides developed in alloys forming semi-protective or protective oxide scales. This was the consequence of the superior barrier properties of chromia layers.
References
D. Mudgal, S. Singh, S. Prakash, Corrosion problems in incinerators and biomass-fuel-fired boilers, International Journal of Corrosion, 2014 (2014).
T.T. Sharobem, Mitigation of high temperature corrosion in waste-to-energy power plants, Columbia University, 2017.
D.J. Young, High temperature oxidation and corrosion of metals, Elsevier (2016).
G. Lai, A. International, Waste-to-Energy Boilers and Waste Incinerators, High-Temperature Corrosion And Materials Applications, (2007) 335–358.
L.W. McNallan MJ, Kim SH, Kang CT High Temperature Corrosion, NACE-6, Houston TX (1983) 316–321.
H. P. Nielsen, F. Frandsen, K. Dam-Johansen, and L. Baxter, The implications of chlorine-associated corrosion on the operation of biomass-fired boilers. Progress in energy and combustion science 26, 2000 (283–298).
H. Grabke, E. Reese, and M. Spiegel, The effects of chlorides, hydrogen chloride, and sulfur dioxide in the oxidation of steels below deposits. Corrosion science 37, 1995 (1023–1043).
J. Liu and M. McNallan, Effects of temperature variations on oxidation of iron–20% chromium alloys at 1200 K in Ar–20% O2-Cl2 gas mixtures. Materials and Corrosion 50, 1999 (253–260).
K. K. Aye, J. Zhang, and D. J. Young, Effect of Hydrogen Chloride on Corrosion Behaviour of Fe–Cr Alloys in Wet CO2 Gas at 650°C. Oxidation of Metals 94, 2020 (51–80).
G.H. Peter EL, Daniel GF, Thomas ED, Evan B, Physical and Chemical data, in: Chemical Engineering, The McGraw-Hill Companies, Inc., 2008.
D. Young, P. Huczkowski, T. Olszewski, T. Hüttel, L. Singheiser, and W. Quadakkers, Non-steady state carburisation of martensitic 9–12% Cr steels in CO2 rich gases at 550°C. Corrosion science 88, 2014 (161–169).
M. Taylor, J. Calvert, D. Lees, and D. Meadowcroft, The mechanism of corrosion of Fe-9% Cr alloys in carbon dioxide. Oxidation of Metals 14, 1980 (499–516).
T. Gheno, Oxidation and carburisation of model chromia-forming alloys in carbon dioxide, in, (Institute National Polytechnique of Toulouse and University of New South Wales, Ph.D diss., 2012).
T. D. Nguyen, J. Zhang, and D. J. Young, Effects of silicon on high temperature corrosion of Fe–Cr and Fe–Cr–Ni alloys in carbon dioxide. Oxidation of metals 81, 2014 (549–574).
C. Wagner, Reaktionstypen bei der Oxydation von Legierungen, Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für physikalische. Chemie 63, 1959 (772–782).
R. Rapp, The transition from internal to external oxidation and the formation of interruption bands in silver-indium alloys. Acta Metallurgica 9, 1961 (730–741).
J. Takada and M. Adachi, Determination of diffusion coefficient of oxygen in α-iron from internal oxidation measurements in Fe-Si alloys. Journal of materials science 21, 1986 (2133–2137).
J. Takada, S. Yamamoto, S. Kikuchi, and M. Adachi, Determination of diffusion coefficient of oxygen in γ-iron from measurements of internal oxidation in Fe-Al alloys. Metallurgical Transactions A 17, 1986 (221–229).
R. Braun, M. Feller‐Kniepmeier, Diffusion of chromium in α‐iron, physica status solidi (a), 90 (1985) 553–561.
J. Swisher and E. Turkdogan, Solubility, permeability, and diffusivity of oxygen in solid iron. AIME Met Soc Trans 239, 1967 (426–431).
C. Wagner, Theoretical analysis of the diffusion processes determining the oxidation rate of alloys. Journal of the Electrochemical Society 99, 1952 (369–380).
A. Jacob, E. Povoden-Karadeniz, and E. Kozeschnik, Revised thermodynamic description of the Fe-Cr system based on an improved sublattice model of the σ phase. Calphad 60, 2018 (16–28).
F. V. F. Gesmundo, Y. Niu, and D. L. Douglass, Oxidation of Metals 40, 1993 (374–393).
H. Davies and A. Dinsdale, Theoretical study of steam grown oxides as a function of temperature, pressure and p (O2). Materials at High Temperatures 22, 2005 (15–25).
D. J. Young, T. D. Nguyen, P. Felfer, J. Zhang, and J. M. Cairney, Penetration of protective chromia scales by carbon. Scripta Materialia 77, 2014 (29–32).
I.G. Wright, R. Barry Dooley, Morphologies of oxide growth and exfoliation in superheater and reheater tubing of steam boiler, Materials at High Temperatures, 28 (2011) 40–57.
I. G. Wright, M. Schutze, P. F. Tortorelli, and R. Dooley, Towards improved prediction of scale exfoliation from steam tubes. Materials at High Temperatures 24, 2007 (265–274).
Acknowledgements
Financial support by the Australian Research Council under the Discovery Scheme is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aye, K.K., Cai, Y., Zhang, J. et al. Effect of Temperature on Corrosion Behaviour of Fe–Cr Alloys in Wet CO2 With and Without HCl Gases. Oxid Met 97, 371–400 (2022). https://doi.org/10.1007/s11085-021-10092-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-021-10092-1