
Abstract
The initial oxidation behavior of Fe–6Al alloys with different Cr contents (0–24 at.%) during heating to 1000 °C at 50 °C/min, followed by isothermal oxidation for 1 h, was investigated by means of synchrotron radiation in order to clarify the well-known beneficial effect of Cr on external Al2O3 scale formation. An external Al2O3 scale formed on the Cr-containing alloys but did not form on Fe–6Al. A corundum-type transient oxide scale, (Fe,Cr)2O3, formed on all alloys at about 500 °C, and an Al or (Al,Cr)-rich amorphous oxide layer was confirmed to form below the transient scale. This amorphous inner oxide layer was found to provide oxidation resistance up to at least 500 °C for the binary Fe–Al and about 900 °C for the ternary Fe–Cr–Al alloys. Internal precipitates of Al2O3 formed in the alloys when temperatures reached about 900 °C. The volume fraction of these internal precipitates increased with increasing alloy Cr content, which led to be inference that the outward diffusion flux of Al was enhanced by Cr addition.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chromium addition to a binary M–Al alloy (M = Fe, Ni, and Co) is known to decrease the critical Al content for external Al2O3 scale formation [1–6]. This beneficial Cr effect is often referred to as the third-element effect (TEE) [4–10]. In most previous studies, this beneficial Cr effect is explained by decreased inward diffusion flux of oxygen due to formation of an initial external Cr2O3 scale, which decreases the oxygen potential at the alloy/scale interface. The decrease in oxygen potential results in an increase in the relative outward diffusion flux of Al, which promotes establishment of an external Al2O3 scale [8]. Giggns and Pettit [1] observed formation of internal Al2O3 precipitates beneath a Cr2O3 scale on Ni–20wt%Cr–3wt%Al, which is the transient alloy to form an external Al2O3 scale, and suggested that internal Al2O3 precipitates can grow laterally to form a continuous Al2O3 layer. They also carried out the transient oxidation of Ni–6wt%Al with 5–30Cr alloys oxidized at 1000 °C in 0.1 atm O2 for 1 min in order to examine the establishment of steady-state condition. Because the multilayered scale consisting of NiO, NiCr2O4 and Cr2O3 layers was initially formed, they concluded that Al2O3 layer forms during the latter stage of the transient oxidation. Similar oxidation behavior, regarding a formation of the internal Al2O3 in the Fe–Cr–Al alloys, was also reported by Niu et al. [10]. They proposed that Cr addition cannot prevent the formation of internal Al2O3 but can suppress the growth of external Fe-containing oxide scale. Although small difference exists in the models proposed in those previous studies, all the mechanism suggested that decrease in the oxygen potential/inward oxygen flux by an initial Cr2O3 scale formation is very likely to explain TEE.
Although several transient oxidation studies were conducted, many of previous studies were mainly carried out under the isothermal oxidation condition. Moreover, those transient oxidation behavior was always interpreted based on the “ex situ” one by one short-term oxidation results for different time intervals. However, time to establish the steady-state oxidation period due to development of a protective Al2O3 scale and the transient oxidation phenomena relating to reach the steady-state oxidation could not be clearly understood by intermittent experimental method. If the development of stable Al2O3 scale via different transient oxides formation, which depends on alloy compositions, can be continuously observed from room temperature to higher temperatures, the effect of Cr on Al2O3 scale formation could be more clearly understood. However, it is difficult to observe development of a thin transient oxide scale and its fast transition to the external Al2O3 scale using conventional techniques, such as a laboratory XRD.
In our previous studies, we examined the metastable-to-stable Al2O3 phase transformation using an in situ high-temperature X-ray diffraction technique by means of synchrotron radiation with a two-dimensional detector, which has excellent time resolution [11, 12]. In those studies, not only fast transformation to α-Al2O3 from various transient oxides observed, but also changes in the lattice constant of α-Al2O3 during heating were able to be clearly resolved [11]. Accordingly, this technique can allow for experimental assessment of the TEE mechanism by observing structural changes of the transient oxide scale during heating and the eventual establishment of an Al2O3 scale. In this study, initial oxidation behavior of Fe–6Al alloys with different contents of Cr (0–24 at.%) was investigated and the effect of Cr on the development of Al2O3 scale was assessed.
Experimental Procedures
Fe–xCr–6Al (x = 0–24 at.%) alloys were prepared by argon arc melting using high-purity metals (99.99 at.%) as the starting constituents. The alloy ingots were then homogenized in vacuum at 1200 °C for 48 h. Approximately, 1-mm-thick samples were cut from the homogenized ingots and ground to 4000-grit finish using SiC abrasive paper. Samples were then polished using 3-μm diamond paste, followed by ultrasonic cleaning in acetone. Oxidation tests were conducted by heating a given sample to 1000 °C in air at a rate of 50 °C/min in a vertical furnace.
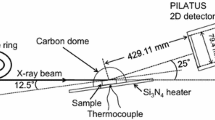
In situ high-temperature X-ray diffraction measurement was performed at the beam-line BL19B2 of the SPring-8 at Japan Synchrotron Radiation Research Institute. Figure 1 shows a schematic of the experimental setup used in this study. The sample was set in the heating stage (ANTON PARR, DHS110), which was installed in an 8-axis goniometer. The incident angle of the X-ray beam, α, was set to 12°. The two-dimensional X-ray detector was positioned to a 2θ angle, between sample surface and the center of the detector, of 24° with a camera length of 421.28 or 429.54 mm. After setting up the apparatus, the sample was heated to 1000 °C in air at a heating rate of 50 °C/min, followed by isothermal oxidation for 1 h in air. The sample temperature was monitored and controlled by a Pt–13Rh thermocouple embedded in the heating stage. During heating, isothermal oxidation and cooling, the diffracted signals from the sample surface were recorded every 10 s with a collection time of 6 s.

Schematic of the experimental setup
After oxidation testing, cross sections of the oxide scale were observed by transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM). Element distributions were analyzed by energy dispersive X-ray spectrometer (EDS) attached to the STEM.
Results
Oxidation Kinetics
Figure 2 shows the oxidation mass gains of Fe–xCr–6Al alloys at different temperatures during heating to 1000 °C. The oxidation mass gains of all alloys were almost the same during heating to 500 °C, but the mass gain of Fe–6Al increased rapidly after further heating. By contrasts, above 500 °C the mass gains of the Fe–Cr–Al alloys remained similar and much smaller than that of the binary alloy up to at least 800 °C. When the temperature reached 1000 °C, the mass gain of Fe–24Cr–6Al became the highest amongst the alloys with Cr addition.

Oxidation mass gains of Fe–6Al, Fe–4Cr–6Al, Fe–12Cr–6Al, and Fe–24Cr–6Al during heating to 1000 °C in air at a heating rate of 50 °C/min
In Situ X-ray Diffraction Measurements
Figure 3 shows the in situ high-temperature X-ray diffraction patterns of Fe–6Al, Fe–4Cr–6Al, and Fe–24Cr–6Al alloys during heating, followed by isothermal oxidation for 10 min at 1000 °C in air. Diffraction peaks from a transient oxide, which was identified to be the corundum-type oxide (Fe,Cr)2O3, were initially detected on all alloys at about 500 °C. The peak position of transient oxide formed on binary Fe–6Al alloy shifted toward a lower 2θ angle with increasing temperature due to thermal expansion. Diffraction signals from Al2O3 were not observed on this binary alloy under these experimental conditions.

X-ray diffraction patterns of a Fe24Cr–6Al, b Fe–4Cr-6Al, and c Fe–6Al
The peak position of the transient oxide formed on the Fe–24Cr–6Al alloy also showed peak shifting toward lower a 2θ angle during heating, and diffracted signals from α-Al2O3 were observed just before the sample temperature reached 900 °C.
The peak position of the transient oxide formed on Fe–4Cr–6Al was initially located at slightly higher a 2θ position than those formed on the lower Cr alloys. This peak further shifted toward higher 2θ position during further heating, and finally approached that of Cr2O3. After formation of Cr2O3, signals from α-Al2O3 were also detected. The temperature at which α-Al2O3 was detected on this high Cr alloy was about 900 °C. Moreover, the diffracted spectra from α-Al2O3 on this alloy clearly consisted of two peaks, but those from alloys with lower Cr content consisted of just one peak. The lower 2θ angle of α-Al2O3 observed during the isothermal oxidation stage was due to the formation of a Cr2O3–α-Al2O3 solid solution, α-(Al,Cr)2O3. Just after the formation of α-Al2O3, diffraction peaks from α-Fe2O3 were observed on this alloy. This Fe2O3 reacted with Cr2O3 during isothermal oxidation to form a Fe2O3–Cr2O3 solid solution, (Fe,Cr)2O3.
Figure 4 shows change in lattice spacing of the various oxide phases formed on Fe–6Al, Fe–4Cr–6Al, and Fe–24Cr–6Al with oxidation time and temperature. The lattice spacing of the transient oxides formed on Fe–6Al and Fe–4Cr–6Al alloys was initially similar and close to Fe2O3, but that formed on Fe–24Cr–6Al tended to be slightly smaller. This is because on the higher Cr-containing alloy the transient oxide, (Fe,Cr)2O3, also had higher Cr content. The lattice spacing of transient oxide formed on Fe–24Cr–6Al decreased continuously during the heating stage, reaching a constant value at about 800 °C, which corresponded to Cr2O3. This change in lattice spacing of the transient oxide with time or temperature suggests selective oxidation of Cr during heating, followed by dissolution into the transient oxide. Such a decrease in lattice spacing of the transient oxide was not clearly observed on Fe–6Al or Fe–4Cr–6Al. On these alloys, the increase in lattice spacing of the transient oxide during heating was due to thermal expansion. In the specific case of Fe–4Cr–6Al, the lattice spacing became almost constant after heating to about 650 °C (see arrow in Fig. 4b). This change in lattice spacing may also indicate dissolution of Cr into the transient oxide. In all cases of oxidation of Cr-containing alloys, the signals from α-Al2O3 began to be observed at about 900 °C. The lattice spacing of initially formed α-Al2O3 agreed with that of pure α-Al2O3 when thermal expansion was taken into account [13, 14]. However, about 5 min after initial α-Al2O3 formation on Fe–24Cr–6Al, an additional signal from α-Al2O3 was detected which had a higher lattice spacing. The α-Al2O3 with larger lattice spacing would have been formed by a reaction between the initially formed (Cr,Fe)2O3 and Al2O3.

Change in the lattice spacing of different oxide phases formed on a Fe–24Cr–6Al, b Fe–4Cr–6Al, and c Fe–6Al with oxidation time and temperature
Scale Microstructures
TEM cross sections of oxide scales formed on Fe–6Al, Fe–4Cr–6Al, and Fe–24Cr–6Al after heating to 650 °C are shown in Fig. 5. Although only corundum-type oxide, (Fe,Cr)2O3, was detected by X-ray diffraction at this oxidation temperature, the duplex oxide scales consisted of outer (dark contrast) and inner (bright contrast) layers. The outer layer was confirmed to be (Fe,Cr)2O3 and tended to decrease in thickness with increasing alloy Cr content. The inner layer also became thinner with increasing alloy Cr content.

TEM cross sections of a Fe24Cr–6Al, b Fe–4Cr–6Al, and c Fe–6Al after heating to 650 °C
Figure 6 shows higher magnification images and FFT patterns near the interface between the inner and outer layers formed on Fe–4Cr–6Al. The structure of the inner oxide layer was identified as being amorphous, and EDS analysis shown in Fig. 7 revealed that this inner amorphous layer was rich in Al. Since such an amorphous layer was not formed on a binary Fe–20Cr alloy, as shown in Fig. 8, the formation of this amorphous oxide layer was due to Al oxidation. EDS analysis found that Cr was also contained in this layer on ternary Fe–Cr–Al alloys, but the Cr/Al ratio seemed to be independent of alloy composition, as indicated by the data in Fig. 7. The outer layer contained both Cr and Fe, and Cr content in the outer oxide was higher for alloys with higher Cr content.

Higher magnification image of oxide scale formed on Fe–4Cr–6Al after heating to 650 °C

STEM-EDS analysis of oxide scales formed on a Fe–24Cr–6Al, b Fe–4Cr–6Al, and c Fe–6Al after heating to 650 °C

TEM cross section of oxide scale formed on Fe–20Cr after heating to 650 °C
Figure 9 shows cross sections of the oxide scales formed on Fe–6Al, Fe–4Cr–6Al, and Fe–24Cr–6Al alloys after heating to 900 °C. The outer oxide layer had grown thicker by this temperature and was much thicker on Fe–6Al than that formed on the alloys with Cr addition. Al2O3 was found to develop as internal oxide precipitates in the subsurface region of all alloys. The thickness of the internal oxidation zone formed on alloys with Cr addition was similar, but the volume fraction of internal Al2O3 precipitates was higher in Fe–24Cr–6Al than Fe–4Cr–6Al, as shown in Fig. 10.

Cross sections of a Fe–24Cr–6Al, b Fe–4Cr–6Al (TEM images), and c Fe–6Al (SEM image) after heating to 900 °C

Volume fraction of internal precipitates of Al2O3 and thickness of internal oxidation zone
Figure 11 shows TEM cross sections of oxide scales formed on Fe–6Al, Fe–4Cr–6Al, and Fe–24Cr–6Al alloys after isothermal oxidation for 1 h at 1000 °C. An external Al2O3 scale was formed on the alloys with Cr addition and the transition from internal oxidation to external scale formation occurred in the isothermal oxidation stage. An external Al2O3 scale was not formed on Fe–6Al.

Cross sections of oxide scale formed on a Fe–24Cr–6Al, b Fe–4Cr–6Al (TEM images), and c Fe–6Al (SEM image) after isothermal oxidation for 1 h at 1000 °C
Discussion
Initial Transient Oxide Scale and Formation of the Amorphous Oxide Layer
The initially detected transient oxide scale in this study was a corundum type oxide, M2O3 (M = mainly Fe and/or Cr, Al), and the lattice spacing of this oxide changed depending on the alloy Cr content. Since the ionic radius decreases in the order Fe3+ > Cr3+ > Al3+ [15], the variation in lattice spacings of the initially formed transient oxides resulted from different amounts of Cr and/or Al dissolution in Fe2O3. Based on the STEM-EDS analysis, Al was not present in the most outer transient oxide scale after heating to 650 °C, so that this outer layer can be identified as a (Fe,Cr)2O3 solid solution. The decrease in lattice spacing of the transient scales formed on higher Cr alloys with time or temperature indicates selective oxidation of Cr and its continuous dissolution in the transient oxide. Cr dissolution also occurred to some extent in the low Cr alloy, since the lattice spacing of the transient oxide became constant during the heating stage, counteracting the increase from thermal expansion. The lattice spacing of transient oxide formed on the binary Fe–6Al continuously increased due to thermal expansion.
Because of the higher Cr content in the transient oxide scale formed on alloys with more Cr, it may be expected that the growth rate of that scale will tend to decrease with increasing alloy Cr content. Indeed, TEM observation of scales formed at 650 °C showed that the thickness decreased with increasing alloy Cr content. However, oxidation mass gain was similar for all alloys for up to about 800 °C, except for the binary alloy which had much higher mass gain at temperatures above 500 °C (Fig. 2). This discrepancy could be due to the oxidation mass gain being too small to accurately measure by an analytical balance. The dependence of alloy Cr content on thickness of the transient oxide scales, which contains different Cr contents, tended to decrease after heating to 900 °C except a binary alloy (Fig. 9). Thus, composition of the transient oxide scale is considered to affect the growth rate only very initial oxidation period before the amorphous oxide layer was developed as we will discuss below.
Since growth of the outer transient oxide scale was similar and slow among the alloys up to 500 °C for binary and up to 900 °C for ternary, even though α-Al2O3 scale had not yet developed at this temperature, and the Al- or (Al,Cr)-rich amorphous oxide layer was found to develop on all alloys below the outer transient oxide scale at lower temperature ~650 °C, it is reasonable to consider that this amorphous oxide acts as a protective layer and control the overall oxidation process of alloys at lower temperatures during heating up to at least about 500 °C for binary and about 900 °C for Fe–Cr–Al alloys.
Cr in the alloy apparently enhances the oxidation resistance of the amorphous layer. However, comparing the amorphous layers formed on ternary alloys, the Cr content in these layers was found to be independent of alloy Cr content. Thus, the effect of alloy Cr content on the resistance of the amorphous layer against oxidation was not strong in this experimental condition. In our separate study, we found that alloy Cr content affects the life time of the amorphous layer strongly from our separate study of oxidation of Fe–Cr–Al alloy at 650 °C [16].
Effect of Cr on Transition from Internal to External Al2O3 Scale Formation
When the temperature reached about 900 °C, diffraction signals from α-Al2O3 were observed on all alloys with Cr addition, as shown in Fig. 4. TEM observation shown in Fig. 9 revealed that the initially formed Al2O3 was in the form of internal precipitates, even though the lattice spacing of transient oxide scale formed on the alloy with higher Cr content was similar to that of Cr2O3. Same result was also reported by Niu et al. [10] on their oxidation study of Fe–Cr–3at.%Al at 1000 °C in O2. They mentioned that TEE observed in their study on Fe–xCr–3Al differs from Wanger’s model [8]. The result obtained in this study also strongly indicates that Al content, 6 at.% in Fe–Cr–Al alloys is essentially insufficient to form an external Al2O3 scale at the very initial stage of oxidation.
The thickness of the internal oxidation zone was independent of alloy Cr content (Fig. 10), but the volume fraction of internal Al2O3 precipitates increased with increasing alloy Cr content. This result indicates that Cr addition enhanced Al enrichment as an oxide in the internal oxidation zone, which may promote the transition from internal to external Al2O3 scale formation.
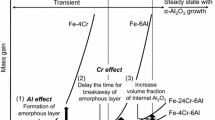
Figure 12 presents the TEE mechanism proposed in this study. At the very initial stage of oxidation, an (Al,Cr)-rich amorphous oxide layer develops below a transient (Cr,Fe)2O3 or (Fe,Cr)2O3 scale on the alloys with both high and low Cr contents. As we discussed earlier, the amorphous layer is considered to be a protective layer; therefore, the oxygen potential at the amorphous/alloy interface is inferred to be sufficiently low to suppress Fe oxidation. Additionally, because of similar Cr content in the amorphous oxide layer, it is expected that both the oxygen potential and Cr content at the interface are similar among the alloys if we assume that “metastable” equilibrium between amorphous oxide and the alloy substrate is established. Thus, the inward diffusion flux of oxygen in alloys should be similar with different alloy Cr contents and smaller (Fig. 12a, b). However, the volume fraction of internal Al2O3 precipitates increased with increasing alloy Cr content (see Fig. 10), which strongly indicates that the outward diffusion flux of Al is enhanced by higher alloy Cr composition.

TEE mechanism proposed in this study. a, c High Cr alloy b, d low Cr alloy at a, b lower temperatures c, d higher temperatures
The diffusion flux of Al in ternary Fe–Cr–Al system can be expressed by the following Eq. (1):
Here, \(\tilde{D}_{\rm AlAl}^{\rm Fe}\) and \(\tilde{D}_{\rm AlCr}^{\rm Fe}\) are the main-term and cross-term interdiffusion coefficients of Al, respectively, and \(\frac{{\partial C_{\rm i} }}{\partial x}\) (i = Al, Cr) is the concentration gradient of element i. Nesbitt [17, 18] predicted minimum Al concentration for protective Al2O3 scale formation on Ni–Cr–Al system using the main-term and positive cross-term interdiffusion coefficients (\(\tilde{D}_{\rm AlAl}^{\rm Ni}\) ≫ \(\tilde{D}_{\rm AlCr}^{\rm Ni}\)) at 1200 °C [19]. He took only Al to be oxidized element into account for his model. According to his prediction, the cross-term coefficient has little effect on the critical Al content, \(N_{\rm Al}^{\rm min}\). In the present study, however, Cr oxidation was initially observed, and after heating to at least 650 °C, formation of both the Al and Cr depletion zones accompanied with (Al,Cr)-rich amorphous oxide layer was observed. Moreover, in the Fe–Cr–Al system at 900 °C, the cross-term coefficient of Al, \(\tilde{D}_{\rm AlCr}^{\rm Fe}\), was reported to be positive (about 5 × 10−11 cm2/s) and is same order of magnitude to main-term coefficient, \(\tilde{D}_{\rm AlAl}^{\rm Fe}\), (about 7 × 10−11 cm2/s) [20]. Different from Nesbitt’s analysis of Ni–Cr–Al system, the effect of the cross-term coefficient on Al diffusion flux is expected to be high in Fe–Cr–Al system. Additionally, relatively steeper concentration gradient of Cr near the internal oxidation front in Fe–24Cr–6Al after heating to 800 °C was observed as shown in Fig. 13. Although Cr concentration profile in Fe–4Cr–6Al was not able to be obtained in this study, it is reasonable to expect that the concentration gradient of Cr in Fe–24Cr–6Al is much steeper than that of Fe–4Cr–6Al, as it is schematically described in Fig. 12c, d. Although the effect of cross-term on outward diffusion flux of Al is not evaluated quantitatively, both cross-term diffusion coefficient (comparable order to main-term coefficient) and steeper concentration gradient of Cr are inferred to enhance the outward diffusion flux of Al, which promotes the transition from internal to external Al2O3 scale formation.

STEM-EDS analysis across the oxide scale formed on Fe–24Cr–6Al after heating to 800 °C. a STEM image, b concentration profiles of Fe, Cr, and Al
In order to evaluate the effect of cross-term coefficient on the critical Al content for an external Al2O3 scale formation, a diffusion study in Fe–Cr–Al system at 650 °C and 1000 °C is currently in progress. The effect of cross-term coefficient and concentration gradient of Cr on the Al diffusion flux will be shown quantitatively in this journal.
Conclusions
The transient stage of oxidation of Fe–6Al alloys with different Cr contents (0–24 at.%) was investigated to better understand the effect of Cr on the development of an external Al2O3 scale. The results obtained may be summarized as follows:
-
(1)
The initially formed transient oxide scale was corundum type (Fe,Cr)2O3, and the Cr content in the transient oxide increased with increasing alloy Cr content. An Al- or (Al,Cr)-rich amorphous oxide layer formed below the transient oxide scale. This amorphous oxide layer provided oxidation resistance at least up to about 500 °C for binary Fe–Al and about 900 °C for ternary Fe–Cr–Al alloys.
-
(2)
Internal Al2O3 formed in all alloys even if the alloy Cr content was high, ~24 %, at about 900 °C. The volume fraction of internal Al2O3 precipitates increased with increasing alloy Cr content.
-
(3)
Enhanced outward diffusion flux of Al due to both the positive cross-term coefficient and steeper Cr depletion in the subsurface region is proposed to promote an external Al2O3 scale formation.
References
C. S. Giggins and F. S. Pettit, J. Electrochem. Soc. 118, 1971 (1782).
P. Tomaszewicz and G. R. Wallwork, Rev. High Temp. Mater. 4, 75 (1978).
P. Tomaszewicz and G. R. Wallwork, Oxid. Met. 20, 75 (1983).
E. Airiskallio, E. Nurmi, M. H. Heinonen, I. J. Väyrynen, K. Kokko, M. Ropo, M. P. J. Punkkinen, H. Pitkänen, M. Alatalo, J. Kollár, B. Johansson and L. Vitos, Corr. Sci. 52, 3394 (2010).
P. F. Tortorelli and K. Natesan, Mater. Sci. Eng. A258, 115 (1998).
R. Prescott and M. J. Graham, Oxid. Met. 38, 73 (1992).
F. H. Stott, G. C. Wood and J. Stringer, Oxid. Met. 44, 113 (1995).
C. Wagner, Corros. Sci. 5, 751 (1965).
Z. G. Zhang, F. Gesmundo, P. Y. Hou and Y. Niu, Corros. Sci. 48, 741 (2006).
Y. Niu, S. Wang, Z. G. Xhang and F. Gesmundo, Corros. Sci. 50, 345 (2008).
S. Hayashi, I. Saeki, Y. Nishiyama, T. Doi, S. Kyo and M. Segawa, Mater. Sci. Forum 696, 63 (2011).
S. Hayashi, Y. Takada, I. Saeki, A. Yamauchi, Y. Nishiyama, T. Doi, S. Kyo and M. Sato, Mater. Corros. 63, 682 (2012).
F. Bondioli, A. M. Ferrari, C. Leonelli and T. Manfredini, J. Am. Ceram. Soc. 83, 2036 (2000).
T. Fujimura and S.-I. Tanaka, J. Mater. Sci. 34, 425 (1999).
R. D. Shannon and C. T. Prewitt, Acta Cryst. B25, 925 (1969).
S. Yoneda and S. Hayashi, to be published in Oxid. Met.
J. Nesbitt, J. Electrochem. Soc. 136, 1511 (1989).
J. Nesbitt, J. Electrochem. Soc. 136, 1518 (1989).
J. A. Nesbitt and R. W. Heckel, Metall. Trans. A 18A, 2075 (1989).
H. C. Akuezue and J. Stringer, Metall. Trans. A 20A, 2767 (1989).
Acknowledgments
The authors greatly appreciate Dr. S. Ford for his useful suggestion and comments on the structure and contents of this manuscript. This work was primarily supported by “Research and development of oxide dispersion strengthened (ODS) ferritic steel fuel cladding for maintaining fuel integrity at high temperature accident conditions” entrusted to Hokkaido University by the Ministry of Education, Culture, Sports, Science and Technology, MEXT, Japan. The synchrotron radiation experiments were performed at the BL19B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal Nos. 2014A1550 and 2014B1646). TEM characterization was conducted under the program of NIMS microstructural characterization platform as a program of “Nanotechnology Platform” of the MEXT.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yoneda, S., Hayashi, S., Saeki, I. et al. Investigation of Initial Transient Oxidation of Fe–xCr–6at.%Al Alloys Using Synchrotron Radiation During Heating to 1000 °C in Air. Oxid Met 86, 357–370 (2016). https://doi.org/10.1007/s11085-016-9641-0
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-016-9641-0