
Abstract
In this comprehensive study, we investigate the properties of Cs2NaBiX6 (X = I and Br) lead-free double perovskites using density functional theory (DFT). This is the first in-depth DFT-based analysis that delves into the substantial characteristics of these materials, addressing a significant gap in the existing literature. Our analysis focuses on the optical and electronic properties of these materials. Additionally, we examine the impact of strain (tensile and compressive) on their characteristics. The negative values of formation energy and lack of negative frequencies in phonon dispersion band structures provide evidence for the thermodynamic and dynamic stability of considered compounds, respectively. The results reveal that Cs2NaBiBr6 is the most thermodynamically and moisture-stable variant, as corroborated by the narrower energy fluctuation range, indicating superior thermal stability. The adsorption energy of water molecule on Cs2NaBiBr6 and Cs2NaBiI6 are − 0.109 and − 0.114, respectively.Using PBE approximent, bandgap values for Cs2NaBiBr6 and Cs2NaBiI6, are 2.09 and 3.14 eV, respectively. Furthermore, the DFT analysis suggests that the mechanical moduli increase as the halide changes from iodide to bromide. According to the computed Pugh's ratio, these materials are ductile. These findings highlight the promising potential of lead-free double perovskites Cs2NaBiX6 (X = I and Br) for applications in optoelectronic devices.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The perovskite crystal structure is characterized by the general formula ABX3, in which A denotes a larger cation, B signifies a smaller cation, and X can be a halogen or an oxygen atom (Qiu et al. 2020; Ramdane et al. 2024). Various oxide perovskites have demonstrated remarkable properties, exemplified by the significant ferroelectric response and the substantial magnetoresistance, which have resulted in numerous practical applications. While perovskite materials are also known as metal oxides or chalcogenides (Bairwa et al. 2023; Bairwa et al. 2024; Pandit et al. 2024a, b), a metal halide perovskite demonstrates exceptional photovoltaic characteristics. Halide perovskites, unlike oxide perovskites, have high ionic conductivity due to their highly ionic crystal structure (Li et al. 2024).
In the past few years, there has been extensive research on perovskite solar cells (Babaei et al. 2022a; Chu et al. 2019). In 2012, Grätzel and colleagues achieved a power conversion efficiency (PCE) of 9.7% by using methylammonium lead iodide (MAPbI3) as the perovskite absorber material in solar cells (Kim et al. 2012). Currently, the record PCE for perovskite solar cells has reached an impressive 25.5% (Zheng, et al. 2019), which has sparked significant interest and optimism in the field. During this period, various perovskite materials have been investigated and studied for their potential use as absorbers in perovskite solar cell technology (Babaei et al. 2022b; Alidaei et al. 2022; Wu et al. 2023; Zhu et al. 2024a; Zhu et al. 2024b; Wang et al. 2018; Saadatmand et al. 2023a). Researchers have explored alternative perovskite materials to address the stability issues of MAPbI3, which has a high-power conversion efficiency but is not stable at room temperature and in ambient moisture conditions (Guo et al. 2024). One approach has been to investigate the use of Formamidinium (FA), Cesium (Cs), and Rubidium (Rb) instead of Methylammonium (MA) (Han et al. 2016). Additionally, efforts have been made to find lead-free perovskite materials to mitigate the potential environmental concerns associated with the toxicity of lead. As a result, compounds such as MASnX3, CsSnX3, and FASnX3 have also been extensively studied as alternative perovskite absorbers (Liu et al. 2018; Zhang et al. 2019).
In recent years, researchers have studied a new class of perovskites with the chemical formula A2(B′B″)X6, known as double perovskites. The formula presented here utilizes A to denote a cation of moderate size, typically Cs+ or MA, while B′ and B″ are used to represent either monovalent or trivalent cations, and X can be either oxygen or halogen (Babaei et al. 2022b; Rani et al. 2022; Rani et al. 2024). Recently, there has been a significant surge in interest within the field of photovoltaic research towards halide double perovskites, as they offer the potential to address the instability and toxicity challenges associated with Pb-based perovskites (Xiao et al. 2017). By exploring different combinations of B′ and B″ in A2B′B″X6, it is possible to find a viable substitute for Pb-based halide perovskites (Lee et al. 2020; Tang and Tang 2023a; Tang et al. 2022; Yaseen et al. 2022; Qi et al. 2024; Hu et al. 2022). Mc. Clure et al. (McClure et al. 2016) Introduced double perovskites Cs2AgBiX6 (X = Cl and Br) with band gaps of 2.77 and 2.19 eV, respectively. However, these compounds had an indirect band gap, which could limit their optical capabilities. Volonakis et al. succeeded in synthesizing Cs2InAgCl6 double perovskite with a direct band gap of 3.3 eV (Volonakis et al. 2017).
Recently, there has been research conducted on A2NaB″X6 double perovskite materials, as a novel type of double perovskite materials. The double perovskite materials are anticipated to exhibit a narrow and distinct band structure due to the significant difference in electronegativity between Na+ and B″(B3+) cations. Computational analyses have indicated that the band-edge molecular orbitals of NaB3+ double perovskite materials, such as Cs2NaBiCl6, and Cs2NaInBr6, are primarily composed of [B3+X6]3− orbitals. As a result, NaB3+ double perovskite materials possess distinct optical transition characteristics (Lee et al. 2020; Shi and Du 2015). More recently, Ma and colleagues were able to synthesize a new double perovskite, Cs2NaBiI6, which has a bandgap of 1.6 eV (Zhang et al. 2018). Theoretical calculations on the structure of Cs2NaBiI6 and Cs2NaBiBr6 suggest band gaps of 2.23 and 3.07 eV, respectively (Zhao et al. 2018). In addition to having a suitable band gap for optoelectronic applications, the lead-free Cs2NaBiI6 is also very stable against moisture and oxygen in the air (Zhang et al. 2018).
The Cs2NaBiI6 bandgap is close to the MAPbI3 bandgap, but on the contrary, it is lead-free and does not have the problems of MAPbI3 instability. In addition, the mechanical stability and adequate flexibility of perovskites are very important in the design and fabrication of heterostructures of optoelectronics. Elastic coefficients are one of the most important parameters in studying the elastic properties of materials. The elastic coefficient C11 of MAPbX3 structures (X = Cl, Br, and I) is of the order of 30 GPa (Saadatmand et al. 2023b; Laamari et al. 2019). Thus, MAPbI3 is softer than MAPbCl3 and can be used in heterostructures under the influence of inter-layer mismatch.
In the existing research on Cs2NaBiI6 double perovskites, the focus has primarily been on their optical properties, while their stability and mechanical characteristics have received relatively less attention. This paper aims to address this gap by examining the thermodynamical, dynamical, moisture, and thermal stability and mechanical properties of Cs2NaBiI6 and Cs2NaBiBr6 materials, which are promising candidates for optoelectronic applications. We employed Density Functional Theory (DFT) to evaluate the potential of alternative materials as substitutes for lead-based perovskites and to assess their stability in optoelectronic devices. The phonon dispersion and formation energy related to degradation are determined through DFT to analyze dynamic and thermodynamic stability, respectively. We calculate the elastic constants and mechanical parameters to investigate the mechanical stability of the materials. Additionally, we examined the effect of water molecules on the proposed perovskites by determining the adsorption energy of water on the perovskite surface. This research aims to provide a more comprehensive understanding of the properties of stable, lead-free perovskites, which are essential for advancing optoelectronic devices.
2 Computational methods
In this work, the DFT calculations were performed using the Quantum ESPRESSO software (Giannozzi et al. 2009). After converging the input parameters, the cut-off energy and Monkhorst–Pack scheme (Monkhorst and Pack 1976) of k-point sampling is set to 500 eV and 10 × 10 × 10, respectively. The Perdew-Burke-Ernzerhof (PBE) approximation (Perdew et al. 1996) is used for the exchange–correlation term in the DFT calculations. Additionally, the projector-augmented wave (PAW) pseudo-potentials (Blöchl 1994) are employed to accurately calculate the electronic properties (Babaei et al. 2010). The convergence criteria are set to an energy change less than 10–6 eV and forces less than 10–5 eV/Å. Crystal elastic constants are estimated with a maximum strain amplitude of 0.003 and steps of 5.The calculations are carried out on the primitive unit cell of Cs2NaBiX6 (X = I and Br) structures, each containing 10 atoms (Fig. 1).

Primitive and conventional unit cells of Cs2NaBiX6 (X = I and Br) compounds
3 Results and discussion
After performing geometry optimization using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm, the lattice vectors of the Cs2NaBiX6 (X = I and Br) compounds are reported in Table 1. The obtained lattice vectors are in good agreement with the previously reported results (Yin et al. 2019). The Cs2NaBiX6 (X = I and Br) compounds have a cubic structure and belong to the Fm3̅m space group. The lattice parameters decrease when moving from I to Br due to the decrease in the size of the halide ions (Saadatmand et al. 2023c).
3.1 Structural, thermodynamical, moisture, and thermal stability
The stability of the proposed perovskite crystal phase can be assessed using two empirical parameters: the Goldschmidt tolerance factor (t) and the octahedral factor (μ). The tolerance factor is calculated as (Qiu et al. 2020):
where rCs, rNa, rBi, and rX are the ionic radii of Cs, Na, Bi, and the halide ion (I or Br), respectively. The octahedral factor is calculated as:
The formability criterion for perovskites is 0.8 ≤ t ≤ 1.0 and 0.41 ≤ μ ≤ 0.9. For the Cs2NaBiI6, the values of t and μ are 0.894 and 0.465, respectively. For the Cs2NaBiBr6, the values are 0.909 and 0.522, respectively. So, these materials have structural stability (Babaei et al. 2022b).
The new tolerance factor (τ) is also used to confirm the stability of the perovskites. This factor is calculated as (Babaei et al. 2022b):
where nCs is the oxidation state of Cs. The perovskite stability is indicated by τ < 4.18. The τ values for the Cs2NaBiX6 (X = I and Br) are 4.17 and 3.93, respectively, which suggests that these compounds are within the perovskite stability range.
Furthermore, to ensure the thermodynamic stability of the double perovskite Cs2NaBiX6 (X = I and Br), the formation energy is calculated using DFT. The Cs2NaBiX6 (X = I and Br) structures can potentially decompose when exposed to light, as shown by the reaction:
The formation energies (Ef) of the considered structures are calculated using the following equation (Babaei et al. 2022a, b; Zhang et al. 2016):
where \({E}_{{Cs}_{2}AgSb{X}_{6}}\), \({E}_{\text{CsX}}\), \({E}_{\text{NaX}}\), and \({E}_{\text{Bi}{X}_{3}}\) represent the optimized crystal energies of the respective compounds.
A negative formation energy indicates that the structure is thermodynamically stable, and a higher negative value suggests greater stability. The computed formation energies of the double perovskites Cs2NaBiI6 and Cs2NaBiBr6 are − 0.209 and − 0.33 eV, respectively. Since both perovskites have negative formation energy values, it can be concluded that they are thermodynamically stable. It should be noted that the proposed materials are more stable than MAPbI3 (Saadatmand et al. 2023b).
The moisture stability of the Cs2NaBiX6 (X = I and Br) perovskites is investigated by determining the adsorption energy (Eads) of these materials concerning water molecules (H2O) (Saadatmand et al. 2023c).
where Eadsorbate/sub is the total energy of the H2O molecule with the double perovskite. A positive value for Eads indicates that the double perovskite is hydrophobic, while a negative value shows that the substrate is hydrophilic.
Figure 2 illustrates the method used to calculate the adsorption energy by placing a water molecule on the Cs2NaBiX6 (X = I and Br) surfaces. We focused on the (001) crystal surface (Crystal Facet Index) of the Cs2NaBiX6 perovskites. The results are presented in Table 2. Based on the calculated adsorption energies, it can be concluded that the Cs2NaBiI6 perovskite exhibits lower stability against water compared to the Cs2NaBiBr6 perovskite. The results show that the stability of these materials against moisture is better than MAPbI3.

Representation of H2O placed on a Cs2NaBiI6 and b Cs2NaBiBr6
Using ab initio molecular dynamic (AIMD) simulations, the thermal stability of the Cs2NaBiX6 (X = I and Br) is investigated. The simulations are performed for 10 ps at a temperature of 298.15 K, with 10,000 simulation steps taken at intervals of 1 fs (Shokouhi et al. 2024). In the AIMD simulations, we employed the NVT ensemble, which maintains a constant number of particles (N), volume (V), and temperature (T) throughout the simulation. To control the temperature within the NVT ensemble, we utilized a Nosé–Hoover thermostat. This thermostat effectively regulates the temperature of the system by coupling the system to a heat bath, ensuring that the temperature remains stable at 298.15 K during the simulation. The simulation results, as depicted in Fig. 3, illustrate the variations in total energy and temperature over time at 298.15 K. Notably, the ground state energy exhibits very low fluctuations despite significant temperature changes. The range of energy fluctuation for Cs2NaBiBr6 is lower than Cs2NaBiI6, suggesting greater thermal stability. Importantly, these energy fluctuation values are an order of magnitude smaller than the formation energies of these materials. This indicates that the chemical bonds remain intact, and the lattice parameters remain constant throughout the simulation, suggesting stability at room temperature.

AIMD simulations of the total energy and temperature for a Cs2NaBiI6 and b Cs2NaBiBr6
In the following step, it is crucial to assess the dynamic stability of these materials. We utilize a standard approach to analyze the phonon dispersion relations along the high-symmetry directions within the first Brillouin zone, which allows us to evaluate the dynamic stability of the suggested compounds. Figure 4 illustrates the phonon dispersion for the double perovskites Cs₂NaBiX₆ (with X = Cl, Br, I). The lack of negative frequencies indicates the dynamic stability of the materials being examined. Additionally, an analysis of the phonon dispersion curves reveals that as the X ion transitions from Br to I, the frequencies of the phonon modes decrease, suggesting that Cs₂NaBiBr₆ exhibits greater stability compared to Cs₂NaBiI₆.

Phonon dispersion spectra for perovskite of a Cs2NaBiI6, and b Cs2NaBiBr6,
3.2 Electronic properties
The study proceeds to investigate the electronic properties of the Cs2NaBiX6 (X = I and Br) compounds by calculating their band structures. The band structure for these double perovskite materials are plotted in Fig. 5. The results indicate that the Cs2NaBiX6 (X = I and Br) compounds possess indirect band gaps. In the case of Cs2NaBiBr6, the maximum of the valence band is located at the W point, while the minimum of the conduction band is at the L point. The calculated band gap of Cs2NaBiBr6 is 3.073 eV, which agrees well with previously reported results of 3.07, 3.06, and 3.15 eV (Zhao et al. 2018; Anbarasan et al. 2021; Ouhammou et al. 2024). For the Cs2NaBiI6 structure, the maximum of the valence band is at the W point, and the minimum of the conduction band is at the L point. The calculated band gap for this compound is 2.09 eV, which is found to be reasonably consistent with the previously reported results of 2.23, 2.1, and 1.95 eV (Zhao et al. 2018; Anbarasan et al. 2021; Ouhammou et al. 2024). The difference in the calculated band gaps is attributed to the use of different types of pseudo-potentials. While Zhao et al. (Zhao et al. 2018) employed Ultrasoft pseudo-potentials, the current study utilized the more accurate Projector-Augmented Wave pseudo-potentials to enhance the reliability of the electronic property calculations.

Band structure of a Cs2NaBiI6 and b Cs2NaBiBr6
The electronic structure of the perovskites under consideration can be described using the density of states (DOS) and partial density of states (PDOS). In Fig. 6, the DOS and PDOS for the considered materials are illustrated. It is evident, that substituting I with Br can lead to an increase in the number of electronic states in the valence band. Both the conduction band minimum (CBM) and valence band maximum (VBM) show the contribution of halogen atoms to the total DOS. Consequently, modifying the bandgap of the double perovskites can be achieved by replacing the halogen.

The calculated total DOS and PDOS for a Cs2NaBiI6, and b Cs2NaBiBr6
3.3 Optical properties
The study further examines the optical properties of the Cs2NaBiX6 (X = I and Br) compounds by calculating their dielectric functions and absorption spectra, as shown in Figs. 7 and 8. The imaginary part of the dielectric function for Cs2NaBiI6 exhibits two distinct peaks at energies of 3.58 and 5.4 eV. In contrast, the imaginary part of the dielectric function for Cs2NaBiBr6 presents a single peak at 4.6 eV. The real part of the dielectric function at zero energy, known as the static dielectric constant (SDC), is calculated to be 7.1 for Cs2NaBiI6 and 4.5 for Cs2NaBiBr6.

The real (ɛ1) part of the dielectric function of a Cs2NaBiI6 and c Cs2NaBiBr6, and imaginary (ɛ2) part of the dielectric function of b Cs2NaBiI6 and d Cs2NaBiBr6

a, b Absorption coefficient, c, d loss function, and e, f conductivity of Cs2NaBiI6 and Cs2NaBiBr6, respectively
The study highlights an interesting aspect related to the absorption coefficient of the Cs2NaBiX6 (X = I and Br) compounds, which is in the order of 105 cm−1. The absorption intensity of these double perovskites is lower compared to the well-known perovskite material, MAPbI3, which has a maximum absorption intensity of 14 × 105 cm−1 (Saadatmand et al. 2023b).
3.4 Strain effects
Strain modifies the lattice parameters of the perovskite structure, leading to changes in bond lengths and angles. This distortion can influence the overlap of electronic wavefunctions, thereby affecting the electronic band structure. Under strain, the potential energy landscape experienced by the electrons changes. Compressive strain can increase the overlap between the valence and conduction bands, potentially decreasing the bandgap, whereas tensile strain may lead to a widening of the bandgap due to reduced overlap. Using density functional theory, the impact of triaxial strains ranging from − 3 to + 3% on the bandgaps and refractive indices of the proposed Cs2NaBiI6 and Cs2NaBiBr6 perovskites is investigated. The results, as shown in Fig. 9, indicate that by applying strain, the bandgaps of Cs2NaBiI6 and Cs2NaBiBr6 can be tuned within a range of 0.274 and 0.319 eV, respectively. Furthermore, the refractive indices of Cs2NaBiI6 and Cs2NaBiBr6 can be tuned within a range of 0.174 and 0.036, respectively. This strain, which can result from shrinkage and expansion due to temperature variations, will alter the properties of these materials. This finding demonstrates the ability to manipulate the bandgap of the double perovskites across a wide range by applying strain (Wu et al. 2023; Tang and Tang 2023b).

Strain effect on bandgaps and refractive indices of a Cs2NaBiI6 and b Cs2NaBiBr6
3.5 Elastic properties and mechanical parameters
After examining the electronic and optical properties of the Cs2NaBiX6 (X = I and Br) compounds through geometry optimization, the study proceeds to investigate their elastic properties and mechanical parameters. To calculate the elastic constants, the unit cells are subjected to three different types of deformation matrices (Jamal et al. 2014):
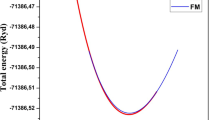
By applying strain (ɛ) in the range of − 2% to + 2% to the Cs2NaBiX6 (X = I and Br) compounds, the energy-strain curves for each of the deformation types are plotted, as shown in Fig. 10. To calculate the elastic coefficients, the study employed the second-order derivative of the energy-strain curve around the equilibrium state. Specifically, for the D2 deformation:

Energy-strain curves of Cs2NaBiX6 (X = I and Br) compounds under deformations D1, D2 and D3
And D1 deformation:
And also, for D3:
By calculating the second-order derivative of the energy-strain curves for the different deformation types (Eqs. 8–10), the independent elastic constants of the cubic perovskites (C11, C12, and C44) can be determined. In these equations, E and V0 represent the energy and volume of the equilibrium state, respectively.
The energy-strain curves for the Cs2NaBiX6 (X = I and Br) under the D1, D2, and D3 deformations are shown in Fig. 8. Using Eqs. 8–10, the elastic coefficients of the Cs2NaBiX6 (X = I and Br) compounds are calculated and listed in Table 3. The calculations show that the C11 value of Cs2NaBiBr6 (C11 = 32.3 GPa) is 3.7 GPa larger than that of Cs2NaBiI6 (C11 = 28.6 GPa).
For cubic crystals, the Born stability criteria (Born and Misra 1940), can be written as:\
The elastic constants of the analyzed compounds are found to be positive and satisfy the Born stability criteria for cubic crystals, thereby signifying the mechanical stability of these materials.
In the following, using the calculated elastic constant, the study proceeds to derive various mechanical parameters for the Cs2NaBiX6 (X = I and Br) compounds. The bulk modulus (B) is calculated based on the C11 and C12 constants, as shown in Eq. 12 (Babaei et al. 2022b; Saadatmand et al. 2023b):
The shear modulus (G) is then computed using the C11, C12, and C44 coefficients, as per Eq. 13 (Liu et al. 2018):
Finally, Young’s modulus (E) and Poisson’s ratio (ν) are derived from the bulk and shear modulus values (Saadatmand et al. 2024; Saadatmand 2024; Shokouhi et al. 2023):
The calculated mechanical parameters for the Cs2NaBiX6 (X = I and Br) compounds are listed in Table 4. The results show that the bulk modulus of Cs2NaBiBr6 (18.2 GPa) is 2.1 GPa higher than the bulk modulus of Cs2NaBiI6 (16.1 GPa). Similarly, Young's modulus of Cs2NaBiBr6 (25.1 GPa) is 0.5 GPa greater than that of Cs2NaBiI6 (24.6 GPa). To understand the reason behind the higher stiffness of Cs2NaBiBr6 compared to Cs2NaBiI6, the study investigated the bond dissociation energies (BDE) of the Na–Br and Na–I bonds. The Na–Br BDE (361.1 kJ/mol) is found to be 56.3 kJ/mol higher than the Na-I BDE (306.8 kJ/mol) (Luo 2007). This indicates that the Na–Br bond is stronger than the Na–I bond, which contributes to the enhanced mechanical properties of the Cs2NaBiBr6 compound.
The ratio of bulk modulus to shear modulus (B/G), known as Pugh's ratio (Babaei et al. 2022b), is listed in Table 4 (Saadatmand et al. 2023b). If the B/G ratio is greater than 1.75, the material is considered ductile, while a B/G ratio less than 1.75 indicates a brittle material. According to this criterion, Cs2NaBiBr6 (B/G = 1.84) is classified as a ductile material, while Cs2NaBiI6 (B/G = 1.63) is considered brittle.
Then we examine the isotropic nature of the mechanical parameters for the Cs2NaBiX6 (X = I and Br) compounds using the ELATE software (Gaillac et al. 2016) to plot the three-dimensional (3D) shapes of the Young’s modulus, linear compressibility, and Poisson's ratio (Fig. 11).

3D diagram of Young's modulus, linear compressibility, and Poisson's ratio of a Cs2NaBiI6, and b Cs2NaBiBr6
According to the results, Young’s modulus, linear compressibility, and Poisson’s ratio of the Cs2NaBiBr6 structure are completely isotropic, indicating that the material exhibits the same behavior in all directions when subjected to external stresses. In contrast, while Young’s modulus and linear compressibility of the Cs2NaBiI6 structure are isotropic, its Poisson's ratio shows a 10% anisotropic behavior.
4 Conclusion
In this comprehensive study, we have conducted a thorough investigation of the Cs2NaBiX6 (X = I and Br) double perovskites using density functional theory (DFT). Our analysis has revealed several key findings. Firstly, we have examined the optical and electronic properties of the Cs2NaBiX6 (X = I and Br) double perovskites. Additionally, we have investigated the impact of strain (tensile and compressive) on their properties, providing valuable insights into the tunability of these materials. The negative formation energy values and the absence of negative frequencies in the phonon dispersion band structures provide evidence for the thermodynamic and dynamic stability of the compounds examined. By analyzing the electronic structure using the PBE method, the bandgap values for Cs₂NaBiBr₆ and Cs₂NaBiI₆ are found to be 2.1 and 3.07 eV, respectively. This study highlights the potential to modify the electronic bandgap of these materials by substituting the halogen atom, making them particularly suitable for optoelectronic applications. The results of our study indicate that Cs2NaBiBr6 emerges as the most thermodynamically and moisture-stable variant, as evidenced by its narrower energy fluctuation range, which suggests superior thermal stability. Furthermore, the DFT analysis has shown that the mechanical moduli of these materials increase as the halide changes from I to Br. The ratio of shear modulus to bulk modulus shows the ductility of considered compounds. Perovskite Cs2NaBiBr6 is relatively more flexible than perovskite Cs2NaBiI6 because the bond dissociation energy of Na–Br (361.1 kJ/mol) is higher than that of Na-I (306.8 kJ/mol). These findings highlight the promising potential of the Cs2NaBiX6 (X = I and Br) double perovskites for applications in optoelectronic devices. The ability to tune their properties through strain and the superior stability of the Cs2NaBiX6 (X = I and Br) variant make these materials attractive candidates for further exploration and development in the field of advanced materials and device engineering.
Data availability
No datasets were generated or analysed during the current study.
References
Alidaei, M., et al.: Stability improvement of perovskite solar cell using photoswitchable and moisture resistant dual-function interfacial layer. J. Alloy. Compd. 903, 163891–16899 (2022)
Anbarasan, R., et al.: First principle insight on the structural, mechanical, electronic and optical properties of indirect band gap photovoltaic material Cs2NaBiX6 (X=Cl, Br, I). Comput. Condens. Matter 28, e00581–e00586 (2021)
Babaei, M., Hosseini, S., Kuchaki, S.: Improving gain and saturation output power in single-quantum-well semiconductor optical amplifiers by injection current. J. World Appl. Sci. 10(3), 342–347 (2010)
Babaei, M., Ahmadi, V., Darvish, G.: First-principles study of lead-free Ge-based 2D Ruddlesden–Popper hybrid perovskites for solar cell applications. Phys. Chem. Chem. Phys. 24(35), 21052–21060 (2022a)
Babaei, M., Ahmadi, V., Darvish, G.: Opto-electro-mechanical properties of lead-free hybrid double perovskites Cs2AgSbX6 (X=Cl, Br, I) for solar cells: a first-principles study. J. Phys. Chem. Solids 169, 110880–110890 (2022b)
Bairwa, J.K., et al.: Modeling and simulation of multifaceted properties of X2NaIO6 (X=Ca and Sr) double perovskite oxides for advanced technological applications. J. Mol. Model. 29(12), 379–386 (2023)
Bairwa, J.K., et al.: Highly efficient and stable Ra2LaNbO6 double perovskite for energy conversion device applications. Mater. Sci. Energy Technol. 7, 61–72 (2024)
Blöchl, P.E.: Projector augmented-wave method. Phys. Rev. B 50(24), 17953–17971(1994)
Born, M.: On the stability of crystal lattices. I. In: Mathematical Proceedings of the Cambridge Philosophical Society. Cambridge University Press 36(2), 160–172 (1940)
Chu, L., et al.: Lead-free halide double perovskite materials: a new superstar toward green and stable optoelectronic applications. Nano-Micro Lett. 11, 1–18 (2019)
Gaillac, R., Pullumbi, P., Coudert, F.-X.: ELATE: an open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 28(27), 275201–275211 (2016)
Giannozzi, P., et al.: QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21(39), 395502–395509 (2009)
Guo, X., Peng, Q., Shin, K., Zheng, Y., Tunmee, S., Zou, C., Zhou, X., Tang, Y.: Construction of a composite Sn-DLC artificial protective layer with hierarchical interfacial coupling based on gradient coating technology toward robust anodes for Zn metal batteries. Adv. Energy Mater. 24(4), 2402015–2402023 (2024). https://doi.org/10.1002/aenm.202402015
Han, Q., et al.: Single crystal formamidinium lead iodide (FAPbI3): insight into the structural, optical, and electrical properties. Adv. Mater. 28(11), 2253–2258 (2016)
Hu, D.-Y., et al.: Exploring the structural, electronic and optical properties of vacancy-ordered double perovskites Cs2TlAsX6 (X=I, Br, Cl) based on first-principles. Phys. Lett. A 427, 127917–127924 (2022)
Jamal, M., et al.: Elastic constants of cubic crystals. Comput. Mater. Sci. 95, 592–599 (2014)
Kim, H.-S., et al.: Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9%. Sci. Rep. 2(1), 591–597 (2012)
Laamari, M.E., et al.: Optimized opto-electronic and mechanical properties of orthorhombic methylamunium lead halides (MAPbX3)(X=I, Br and Cl) for photovoltaic applications. Sol. Energy 182, 9–15 (2019)
Lee, W., Choi, D., Kim, S.: Colloidal synthesis of shape-controlled Cs2NaBiX6 (X=Cl, Br) double perovskite nanocrystals: Discrete optical transition by non-bonding characters and energy transfer to Mn dopants. Chem. Mater. 32(16), 6864–6874 (2020)
Li, X., Aftab, S., Liu, H., Vikraman, D., Hussain, S., Al-Kahtani, A.A., Koyyada, G., Kang, J. and Akman, E.: Enhancing electron transport through metal oxide adjustments in perovskite solar cells and their suitability for X-ray detection. J. Mater. Chem. A 12, 22310–22324 (2024)
Liu, C., et al.: All-inorganic CsPbI2Br perovskite solar cells with high efficiency exceeding 13%. J. Am. Chem. Soc. 140(11), 3825–3828 (2018)
Luo, Y.R.: Comprehensive handbook of chemical bond energies. CRC press, 1312–1345 (2007)
McClure, E.T., et al.: Cs2AgBiX6 (X=Br, Cl): new visible light absorbing, lead-free halide perovskite semiconductors. Chem. Mater. 28(5), 1348–1354 (2016)
Monkhorst, H.J., Pack, J.D.: Special points for Brillouin-zone integrations. Phys. Rev. B 13(12), 5188–5197 (1976)
Ouhammou, A., Fazouan, N., Es-Smairi, A., Khuili, M., Atmani, E.H.: DFT analysis of Cs2NaBiCl6, Cs2NaBiBr6, and Cs2NaBiI6 perovskites for optoelectronic and thermoelectric applications. Comput. Theor. Chem. 1238, 114673–114684 (2024)
Pandit, N., Singh, R., Kumar, A., Joshi, T.K., et al.: Physical properties and power conversion efficiency of SrZrX3 (X= S and Se) chalcogenide perovskite solar cell. Modern Phys. Lett. B, 2450345–2450353 (2024a). https://doi.org/10.1142/S0217984924503457
Pandit, N., Dubey, A., Joshi, T.K., Shukla, A., Rani, U., Kamlesh, P.K., Gupta, R., Kumar, T., Kaur, K., Verma, A.S.: Effect of anion (S− 2 & Se− 2) replacement on photovoltaic properties in transition metal (Ba-Barium) chalcogenide perovskites. Int. J. Modern Phys. b, 2550059–2550067 (2024b). https://doi.org/10.1142/S0217979225500596
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77(18), 3865–3882 (1996)
Qi, F., et al.: Exploring high-performance all-inorganic perovskite materials for next-generation photovoltaic applications: a theoretical study on Cs2TlBiX6 (X=Cl, Br, I). Comput. Theor. Chem. 1233, 114500–114508 (2024)
Qiu, L., et al.: Progress of surface science studies on ABX3-based metal halide perovskite solar cells. Adv. Energy Mater. 10(13), 1902726–1902735 (2020)
Ramdane, O., Labidi, M., Labidi, S., Masrour, R.: A comparative study on the structural, electronic, and magnetic properties of the cubic Sr-based perovskite SrXO3 (X=Mn, Sn, Cr): DFT calculation. J. Korean Ceram. Soc., 1–13 (2024). https://doi.org/10.1007/s43207-024-00397-7
Rani, U., et al.: Emerging study on lead-free hybrid double perovskite (CH3NH3) 2AgInBr6: potential material for energy conversion between heat and electricity. Energ. Technol. 10(9), 2200002–2200008 (2022)
Rani, U., et al.: Exploring properties of organometallic double perovskite (CH3NH3) 2AgInCl6: a novel material for energy conversion devices. Mod. Phys. Lett. B 38(18), 2450144–2450153 (2024)
Saadatmand, S.B., et al.: Design and analysis of highly sensitive plasmonic sensor based on 2-D inorganic Ti-MXene and SrTiO3 interlayer. IEEE Sens. J. 23(12), 12727–12735 (2023a)
Saadatmand, S.B., et al.: Plasmonic heterostructure biosensor based on perovskite/two dimensional materials. Optik 290, 171328–171339 (2023b)
Saadatmand, S.B., et al.: Design and analysis of a flexible Ruddlesden–Popper 2D perovskite metastructure based on symmetry-protected THz-bound states in the continuum. Sci. Rep. 13(1), 22411–22427 (2023c)
Saadatmand, S.B., et al.: Metastructure engineering with Ruddlesden–popper 2D perovskites: stability, flexibility, and quality factor trade-Offs. ACS Omega, 24925–24932 (2024)
Saadatmand, S.B., Shokouhi, S. and Ahmadi, V.: Investigation of 2D Ruddlesden–Popper BZA2PbX4 (X= I, Br, and Cl) and mixed‐halides BZA2PbBrxCl4‐x perovskites: Opto‐electro‐mechanical, thermodynamic properties, moisture and strain effects. Advanced Engineering Materials, Early access (2024)
Shi, H., Du, M.-H.: Discrete electronic bands in semiconductors and insulators: potential high-light-yield scintillators. Phys. Rev. Appl. 3(5), 054005–054013 (2015)
Shokouhi, S., Saadatmand, S.B., Ahmadi, V., Arabpour, R.F.: Comprehensive study on optical, electrical, and stability properties of BA2PbBr4-xClx (x=0, 2, and 4) Ruddlesden Popper perovskites for high-performance PeLEDs. AUT J. Electr. Eng. 56(3), 389–398 (2024)
Shokouhi, S., Saadatmand, S.B., and Ahmadi, V.: First principles study of optical and electrical properties for mixed-halide 2D BA2PbBr4-xClx (x=0, 2, and 4) as an active layer of perovskite light emitting diode. In: 2023 5th Iranian International Conference on Microelectronics (IICM). IEEE, 219–221 (2023)
Tang, T.-Y., Tang, Y.-L.: Physical and optoelectronic properties of double halide perovskites A2CuSbX6 (A=Cs, Rb, K; X=Cl, Br, I) based on first principles calculations. Chem. Phys. 570, 111897–111907 (2023a)
Tang, T., Tang, Y.: Electronic structure, mechanical properties and optical properties of three cobalt-based double halide perovskites: a first-principles study. J. Phys. Chem. Solids 179, 111415–111422 (2023b)
Tang, T., et al.: First-principles study on the mechanical, electronic and optical properties of double halide perovskite Cs2TlSbX6 (X=Cl, Br, I). Phys. Scr. 97(12), 125821–125828 (2022)
Volonakis, G., et al.: Cs2InAgCl6: a new lead-free halide double perovskite with direct band gap. J. Phys. Chem. Lett. 8(4), 772–778 (2017)
Wang, M., et al.: Reversible calcium alloying enables a practical room-temperature rechargeable calcium-ion battery with a high discharge voltage. Nat. Chem. 10(6), 667–672 (2018)
Wu, Y., et al.: The effect of uniaxial strain on electronic and optical properties of halide double perovskites Cs2AgXCl6 (X=Sb, Bi): a DFT approach. J. Alloy. Compd. 961, 170995–171002 (2023)
Xiao, Z., et al.: Intrinsic instability of Cs2In (I) M (III) X6 (M=Bi, Sb; X=halogen) double perovskites: a combined density functional theory and experimental study. J. Am. Chem. Soc. 139(17), 6054–6057 (2017)
Yaseen, M., Aldaghfag, S.A., Zahid, M.: Physical characteristics of X2NaMoBr6 (X=K, Rb): a DFT study. Mater. Sci. Semicond. Process. 147, 106760–106766 (2022)
Yin, H., et al.: Structurally stabilizing and environment friendly triggers: double-metallic lead-free perovskites. Solar Rrl 3(9), 1900148–1900155 (2019)
Zhang, X., et al.: A novel aluminum–graphite dual-ion battery. Adv. Energy Mater. 6(11), 1502588–1502596 (2016)
Zhang, C., et al.: Design of a novel and highly stable lead-free Cs2NaBiI6 double perovskite for photovoltaic application. Sustain. Energy Fuels 2(11), 2419–2428 (2018)
Zhang, Y., et al.: Achieving reproducible and high-efficiency (> 21%) perovskite solar cells with a presynthesized FAPbI3 powder. ACS Energy Lett. 5(2), 360–366 (2019)
Zhao, S., et al.: First-principles study of electronic and optical properties of lead-free double perovskites Cs2NaBX6 (B=Sb, Bi; X= Cl, Br, I). J. Phys. Chem. Solids 117, 117–121 (2018)
Zheng, L., Wang, J., Xuan, Y., Yan, M., Yu, X., Peng, Y., Cheng, Y.B.: A perovskite/silicon hybrid system with a solar-to-electric power conversion efficiency of 25.5%. J. Mater. Chem. a. 7(46), 26479–26489 (2019)
Zhu, C., et al.: Optimizing solar-driven multi-generation systems: a cascade heat recovery approach for power, cooling, and freshwater production. Appl. Therm. Eng. 240, 122214–122223 (2024a)
Zhu, C., et al.: Analytical optical solutions to the nonlinear Zakharov system via logarithmic transformation. Res. Phys. 56, 107298–107306 (2024b)
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
M.B.: Initiated the idea, conceptualization, validation, investigation, supervission, discussed the content, review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Babaei, M. Investigation of the properties of lead-free Cs2NaBiX6 (X = I and Br) double perovskites using density functional theory (DFT). Opt Quant Electron 56, 1449 (2024). https://doi.org/10.1007/s11082-024-07387-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-024-07387-3