
Abstract
The switch to sustainable power generation sources has gained substantial spotlight due to the essential need to decrease fossil fuels dependence, and alleviate climate change issues. This research aims to explore the first principle calculations for geometrical structure, enthalpy of formation, electronic structure behavior, optical spectra, and photocatalytic performance of the clean new Iodine-based triple perovskite compounds A3B2I9 (A = Rb or Cs, and B = Sb or Bi) using FP-LAPW method based on the density functional theory implemented in Wien2K software. The outcomes prove that these triple perovskite compounds can be formed spontaneously under standard conditions, because they are thermodynamically stable. Moreover, the electronic band structure shows an indirect semiconductor behavior of all studied compounds. Further, the optical spectra highlights that all studied compounds have high visible light absorption due to their suitable forbidden band values [1.8 eV–2.369 eV] which facilitate efficient electron–hole pairs generation maximizing the conversion of light into electrical energy. In addition, all studied compounds are satisfied the limits necessary to split water and then to produce hydrogen. Consequently, these compounds are most likely to be used as Lead-free perovskites in various optoelectronic applications, especially solar cells and hydrogen production from water splitting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Research on perovskite compounds began in the 1990s, nevertheless it was not until 2009 that perovskite solar cells were first reported. Since then, their power conversion efficiency has increased rapidly from 3.8% 2009 to 25.5% in 2023 (Green et al. 2021; Dahbi et al. 2022a). Metal perovskites have been studied and gained significant attention for over an extended period due to their unique properties (Qaid et al. 2023; Bawazir et al. 2022; Tahiri et al. 2021a, 2021b; Dahbi et al. 2021a). Lately, a novel category of perovskite compounds, the so-called triple perovskites, has become favorable options as compounds for numerous applications (Guo et al. 2020; Saparov et al. 2015a; Wang et al. 2021; Singh et al. 2020). The metal halide triple perovskites have the typical chemical formula of A3B2X9, where A is alkaline earth or rare-earth cations, B is transition metal cations, while X is a halide anion. The Lead-free metal halide triple perovskite materials have numerous benefits over conventional ones, they are more stable leading them to be less prone to degradation over time in sunlight which is the mean issues related to the organic cation of the conventional perovskites solar cells leading to reduce their power conversion efficiency, therefore hinding their commercialization (Dahbi et al. 2022a, b; Li et al. 2022; Ghrib et al. 2021; Liu et al. 2020). Moreover, they have strong defect tolerance, long carrier lifetimes and high carrier mobilities enhancing the efficiency of the solar energy conversion to electrical power which improve of photovoltaic performance (Dahbi et al. 2021b; Saparov et al. 2015b; Lehner et al. 2015a; Harikesh et al. 2016; Hoye et al. 2016; Pazoki et al. 2016; Hebig et al. 2016). The A3B2X9 compounds have crystallographic structures which can undergo a sequence phase transitions; for instance; Cs3Bi2I9, with the P63/mmc symmetry at room temperature was studied by Laue and four-circle neutron diffractometry from room temperature down to 50 K. At T = 220 K, the crystal undergoes a second-order proper ferroelastic phase transition to a polydomain structure with a nonprimitive monoclinic C12/m1 space group (Park et al. 2015). Luo et al. (2023) discovered that the transitioning A3Bi2I9 from a three-dimensional to a two-dimensional structure triggers significant property changes. In 2D, t A3Bi2I9 materials exhibit modified electronic, optical, and mechanical characteristics, along with shifts in thermal conductivity and chemical reactivity. These alterations offer potential for new applications but require careful exploration and understanding (Jorio et al. 2000). Furthermore, tunable electronic structure behavior and optical properties of triple perovskites make them promising compounds for optoelectronic and effective photocatalysts for water splitting for hydrogen production applications (Dahbi et al. 2021b; Saparov et al. 2015b; Lehner et al. 2015a; Harikesh et al. 2016; Hoye et al. 2016; Pazoki et al. 2016; Hebig et al. 2016; Park et al. 2015; Jorio et al. 2000; Luo, et al. 2023; Bai et al. 2022). For instance, antimony-based halide triple perovskites such cesium antimony triiodide (Cs3Sb2I9) has been found to exhibit a high efficient light emission, showing a potential candidate for use in light-emitting diode (LEDs) (Xie et al. 2023). Under a high pressure up to 14 GPa, the forbidden band of Cs3Sb2I9 is efficiently decreased from 2.05 eV to 1.36 eV (Geng et al. 2019). Further, at room temperature intense raman scattering was observed for Cs3Sb2I9, Rb3Sb2I9, Cs3Bi2I9 and Rb3Bi2I9 materials demonstrating strong electron − phonon coupling and high polarizability, high resistivity, and high photo-response at room temperature (Geng et al. 2020). Besides, high quality thin films derivative Cs3Sb2I9 perovskite exhibiting a forbidden band of 2.05 eV and enhancing its stability in ambient air compared to analogous CH3NH3PbI3 films (Saparov et al. 2015a). Experimentally by optical absorption and ultraviolet photoemission spectroscopy and computationally by DFT calculations, the optical properties and electronic structures of A3Bi2I9 (A = K, Rb, or Cs) are investigated, and it was observed that these compounds are easy to prepare, and A3Bi2I9 (A = K, Rb, or Cs) triple perovskites are more chemically stable than the related lead halides (Kyle et al. 2017). The optical measurements indicate these kinds of compounds are semiconductors with the forbidden band value in the [1.89 − 2.06 eV] range and therefore they exhibit high resistivities (1010–1011 Ω.cm) (Harikesh et al. 2016; Geng et al. 2020; Lehner et al. 2015b). Besides, photocatalytic hydrogen production from water splitting is an important area of research and development due to the increasing demand for clean and sustainable energy sources with no greenhouse gas emissions which can be utilized in various applications, including fuel cells for transportation fuel and power generation (Li et al. 2022; Liu et al. 2020). As far as we know, A3B2I9 (A = Rb or Cs; B = Sb or Bi) triple perovskites are insufficiently explored in literature, and there is few researches that studied the optical and electronic properties (Geng et al. 2020; Kyle et al. 2017), and no author studied the ability of the use of these triple perovskites as materials for hydrogen production from water splitting. Therefore, the aim of this work is to explore the first principle calculations of geometrical properties, thermodynamic stability electronic structure behavior, optical spectra, and photocatalytic performance for the clean triple halide perovskites compounds A3B2I9 (A = Rb or Cs, and B = Sb or Bi).
2 Computational details
In an effort to study the clean Iodine-based triple perovskites A3B2I9 (A = Rb or Cs, and B = Sb or Bi), we have used the FP-LAPW method based on the density functional theory (DFT) (Peresh et al. 2014), using ab initio calculations implemented in Wien2k software (Sham and Schlüter 1983), while the exchange–correlation function was determined by the Perdew-Burke-Ernzerhof generalized gradient approach (PBE-GGA) (Blaha et al. 2020) was used for computing the structural properties and the enthalpy of formation, while PBE-GGA coupled with the Tran Blaha-modified Becke Johnson (TB-mBJ) (Wu and Cohen 2006) were used for electronic, optical and photocatalytic performance in order to achieve the accurate results. To accomplish the optimal theoretical results, the Self-consistent calculations were carried out by taking the plane-wave cutoff value RMT × Max as 7.0. Whereas the Gmax presents the density Fourier-coefficient expansion is seted as 12 (a.u). The k-space meshes of 4 × 7 × 3 were used in the first Brillouin zone.
3 Results and discussion
3.1 Structural properties and enthalpy of formation
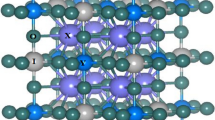
At room temperature, the iodine-based triple perovskites A3B2I9 (A = Rb and Cs, and B = Sb and Bi) crystalize in monoclinic structure (space group N° 14, P21/n), toms occupy 12 A atom, 8 B atom and 36 I atom, respectively, where the BI6 octahedra undergo a distortion toward a rhombohedral elongation (Geng et al. 2020; Camargo-Martínez and Baquero 2012) (See Fig. 1).

The unit cell of the monoclinic structure of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) triple perovskites
By optimizing the geometrical structure, we can discover the atoms equilibrium position in the crystal, which provide insight into the physical properties of a given compound, for instance; the geometrical structure affects the electronic structure behavior which is important for understanding the behavior of compounds. Therefore, by using X-ray diffraction, Rb3Sb2I9 unit cell crystal structures are a = 14.591(3) Å, b = 8.1879(16) Å, c = 20.584(4) Å and β = 90.36(3)° (Geng et al. 2020), while unit cell crystal structures are a = 14.537(7) Å, b = 8.385(4) Å, c = 21.11(1) Å and β = 90.09(4)° for Cs3Bi2I9 (Camargo-Martínez and Baquero 2012), and a = 14.6443(19) Å, b = 8.1787(9) Å, c = 20.885(2) Å and β = 90.421(7)° for Rb3Bi2I9 (Kyle et al. 2017). Table 1 the optimized lattice parameters and betta angle (β) of monoclinic structure of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) compounds are aligned with the experimental results (Geng et al. 2020; Kyle et al. 2017; Camargo-Martínez and Baquero 2012).
Moreover, to predict the chemical reactions and stability of a material, the enthalpy of formation of all studied compounds is simulated using the following equation (Lehner et al. 2015c; Dahbi et al. 2022c, 2022d):
where, Etotal (A12B8I36) shows the total energy each compound, while Etotal (A) is the total energy of Cs or Rb atoms, Etotal (B) is the total energy of Sb or Bi atoms, and Etotal (I) represents the total energy of I atoms. Although, Fig. 2 illustrate that the values of enthalpy formation of all studied structures have negative sign, proving that these structures can be formed spontaneously under standard conditions (Dahbi et al. 2022d, 2022e), and they release energy when formed because they are thermodynamically stable.

The calculated enthalpy of formation of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) compounds
4 Electronic behavior
The investigation of electronic properties is important due to several reasons because they provide valuable information about the electronic structure and properties at the atomic and molecule level of the material including; the energy levels of the electrons and the nature of the chemical bonds within the material by disclosing numerous solids applications like optoelectronic devices and solar cells. In this section, the Total (TDOS) and Partial Density Of States (PDOS) and the electronic band structure were calculated for A3B2I9 (A = Rb or Cs, and B = Sb or Bi) compounds along the high symmetry point of the Brillouin zone (A → Y → Γ → Z → A) are plotted in Fig. 3. It can be observed that the Fermi level of A3B2I9 compounds are lies above the valence Highest Occupied Molecular Orbital (HOMO), proving the p-type semiconductor behavior of A3B2I9 perovskites. On one hand, the Valence Band (VB) of all investigated compounds is principally composed by I-5p5 states with some admixture of Sb-5p3 and Sb-5s2 orbits for A3Sb2I9, Bi-6p3 and Bi-6s2 for A3Bi2I9 compounds, respectively. On the other hand, the Conduction Band (CB) is predominantly composed by Sb-5p3 and I-5p5 orbits, and by Bi-6p3 and I-5p5 orbits for A3Sb2I9 and A3Bi2I9, respectively. Additionally, the band structure of all studied compounds confirms that the highest occupied molecular orbital is positioned between Γ (0.0, 0.0, 0.0) and Z (0.5, 0.0, 0.0) high-symmetry point, while the lowest unoccupied Molecular Orbital (LUMO) is situated at Γ high-symmetry point, showing an indirect semiconductor behavior of all studied compounds. These outcomes are in complete agreement with the other simulated results (Kyle et al. 2017). Besides, the forbidden band values of these compounds are in the range of [1.8 eV–2.369 eV] which is aligned with the experimental data (Geng et al. 2020; Kyle et al. 2017) (Table 2).


The calculated band structure and partial density of states of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) compounds
5 Optical spectra
The ability of a compound to efficiently absorb sunlight is essential for photocatalyts devices and for solar cells, because it design their performance (Lehner et al. 2015c; Dahbi et al. 2022c, 2022d, 2020; Adhikari et al. 2022; Mouhib et al. 2022). The absorption coefficient, reflectivity, and transmittance of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) perovskites in percentage are plotted in Fig. 4. It can be seen that all studied compounds have high visible light absorption due to their suitable forbidden band values [1.8 eV–2.369 eV] which determine the light wavelengths that can they absorb are facilitate efficient electron–hole pairs (excitons) generation. Further, Fig. 4 shows low transmittance and reflectivity of A3B2I9 triple perovskites contribute to efficient visible light harvesting that contribute to the electrical current generation, because maximizing a material’s absorption leads to minimize the transmittance through it, as well as minimize the incident light reflection off the materials surface. These advantages facilitate effective carrier transport, minimizing recombination and maximizing the conversion of light into electrical energy and maintain its performance and durability over an extended period. Thus, the optical properties advantages of A3B2I9 triple perovskites are desirable in various optoelectronic applications, including solar cells and photocatalytic.


The reflectivity, absorption and the transmittance of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) perovskites
6 Photocatalytic performance
Electrolysis, also known as water splitting, is a procedure that utilizes electricity to isolate dihydrogen monoxide molecules (H2O) into oxygen (O2) and hydrogen (H2) gases, respectively. Furthermore, the energy levels of HOMO and LUMO of all studied triple perovskites are determined using the following formula (Li et al. 2022; Liu et al. 2020; Lehner et al. 2015c):
Where \({{\varvec{E}}}_{{\varvec{L}}{\varvec{U}}{\varvec{M}}{\varvec{O}}}\) and \({{\varvec{E}}}_{{\varvec{H}}{\varvec{O}}{\varvec{M}}{\varvec{O}}}\) are the LUMO and HOMO energy level positions, respectively, χ represents the Mulliken electronegativity of A3B2I9 compounds, E0 is the free electron the energy in the hydrogen Normal Hydrogen Electrode (NHE) potential (− 4.5 eV), while Eg is its forbidden energy (Lehner et al. 2015c). Moreover, Fig. 5 highlights that the two fundamental conditions must be met to achieve the desired outcome are respected for all studies triple perovskites, because, the HOMO and LUMO of all A3B2I9 (A = Rb or Cs, and B = Sb or Bi) perovskites are satisfied the necessary conditions to split water, where HOMO are below the dihydrogen monoxide oxidation potential (1.23 eV) while HUMO are greater than the proton reduction potential (0 eV) (Li et al. 2022; Liu et al. 2020; Lehner et al. 2015c), as well as all studied compounds exhibit high visible light absorption which assess the viability materials for water splitting devices. Therefore, these compounds are most likely to be used for hydrogen production from mater splitting.

The calculated HOMO and LUMO positons of A3B2I9 (A = Rb or Cs, and B = Sb or Bi) perovskites relative to the NHE potential
7 Conclusions
In summary, the first principle calculations are explored to study the geometrical structure, thermodynamic stability, electronic structure behavior, optical spectra and photocatalytic performance A3B2I9 (A = Rb or Cs, and B = Sb or Bi) were performed using ab-initio calculations. It was found that all studied structures are thermodynamically stable. Moreover, VB of A3B2I9 compounds is principally composed by I-5p5 states with some admixture of Sb-5p3 and Sb-5s2 orbits (Bi-6p3 and Bi-6s2 orbits) for A3Sb2I9 (A3Bi2I9) compounds. Besides, the calculated band structure of all studied compounds confirms that HOMO is positioned between Γ (0.0, 0.0, 0.0) and Z (0.5, 0.0, 0.0) high-symmetry point, while the LUMO)is situated at Γ high-symmetry point, confirming that A3B2I9 compounds are indirect semiconductor perovskites. Further, the optical spectra shows low transmittance and reflectivity of A3B2I9 triple perovskites contribute to efficient visible light harvesting that contribute to the electrical current generation. Finally, all studied compounds can split water to produce hydrogen. Thus, A3B2I9 triple perovskites can be used as potential candidate for photocatalytic and photovoltaic applications.
References
Adhikari, M., et al.: Luminescence from self-trapped excitons and energy transfers in vacancy-ordered hexagonal halide perovskite Cs2HfF6 doped with rare earths for radiation detection. Adv. Opt. Mater. 10, 2201374 (2022)
Bai, Z.J., et al.: Tuning photocatalytic performance of Cs3Bi2Br 9 perovskite by g-C3N4 for C(sp3)—H bond activation. Nano Res. (2022). https://doi.org/10.1007/s12274-022-4835-z
Bawazir, H.S., et al.: Phase state influence on photoluminescence of MAPb(BrxI1−x)3 perovskites towards optimized photonics applications. Photonics 10, 21 (2022)
Blaha, P., Schwarz, K., Tran, F., Laskowski, R., Madsen, G.K.H., Marks, L.D.: WIEN2k: an APW+lo program for calculating the properties of solids. J. Chem. Phys. 152, 074101 (2020)
Camargo-Martínez, J.A., Baquero, R.: Performance of the modified Becke-Johnson potential for semiconductors. Phys. Rev. B 86, 195106 (2012)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: The new eco-friendly lead-free zirconate perovskites doped with chalcogens for solar cells: Ab initio calculations. Opt. Mater. 109, 110442 (2020)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: Electronic, optical, and thermoelectric properties of perovskite BaTiO3 compound under the effect of compressive strain. Chem. Phys. 544, 111105 (2021a)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: Calcium hafnate perovskite from an insulator to a semiconductor for photovoltaic and photocatalytic hydrogen production from water splitting applications. Superlattices Microstruct. 160, 107058 (2021b)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: Importance of spin-orbit coupling on photovoltaic properties of Pb-free vacancy ordered double perovskites halides X2TeY6 (X= Cs, Rb, and Y= I, Br, Cl): first-principles calculations. Int. J. Energy Res. 46, 8433–8442 (2022a)
Dahbi, S., Mouhib, B., Tahiri, N., El Bounagui, O., Mounkachi, O., Ez-Zahraouy, H.: An oxygen vacancy defect and chalcogens impurities in Earth-Abundant Lead-Free calcium zirconate perovskite for non-toxic solar cells: first-principles calculation. Mater. Today Commun. 33, 104847 (2022b)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: Chalcogens’ impurities and a single F-center in SHO: Ab initio calculations. Mater. Sci. Semicond. Process. 138, 106271 (2022c)
Dahbi, S., Tahiri, N., Bounaguiand, O.E., Ez-Zahraouy, H.: Earth Abundant Nontoxic ternary calcium nitrides inverse perovskites for single-junction solar cells; ab-initio simulations. Mater. Sci. Semiconduct. Process. 150, 106959 (2022d)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H., Benyoussef, A.: Effects of oxygen group elements on thermodynamic stability, electronics tructures and optical properties of the pure and pressed BaTiO3 perovskite. Comput. Condens. Matter 32, e00728 (2022e)
Geng, T., et al.: Bandgap engineering in two-dimensional halide perovskite Cs3Sb2I9 nanocrystals under pressure. Nanoscale 12, 1425–1431 (2020)
Ghrib, T., Rached, A., Algrafy, E., et al.: A new lead free double perovskites K2Ti(Cl/Br)6; a promising materials for optoelectronic and transport properties; probed by DFT. Mater. Chem. Phys. 264, 124435 (2021)
Green, M.A., Dunlop, E.D., Hohl-Ebinger, J., Yoshita, M., Kopidakis, N., Hao, X.: Solar cell efficiency tables (version 58). Prog. Photovolt. Res. Appl. 29, 657–667 (2021)
Guo, W.-H., et al.: Two-dimensional 111-Type In-based halide perovskite Cs3In2X9 (X = Cl, Br, I) with optimal band gap for photovoltaics and defect insensitive blue emission. Phys. Preview Appl. 13, 024031 (2020)
Harikesh, P.C., et al.: Rb as an alternative cation for templating inorganic lead-free perovskites for solution processed photovoltaics. Chem. Mater. 28, 7496–7504 (2016)
Hebig, J.-C., Kühn, I., Flohre, J., Kirchartz, T.: Optoelectronic properties of (CH3NH3)3Sb2I9 thin films for photovoltaic applications. ACS Energy Lett. 1(1), 309–314 (2016). https://doi.org/10.1021/acsenergylett.6b00170
Hoye, R.L.Z., et al.: Methylammonium bismuth iodide as a lead-free, stable hybrid organic−inorganic solar absorber. Chem. - Eur. J. 22, 2605–2610 (2016)
Jorio, A., et al.: Ferroelastic phase transition in Cs3Bi2I9: a neutron diffraction study. Phys. Rev. B 61, 3857 (2000)
Lehner, A.J., et al.: Crystal and Electronic Structures of Complex Bismuth Iodides A3Bi2I9 (A = K, Rb, Cs) Related to Perovskite: Aiding the Rational Design of Photovoltaics. Chem. Mater. 27, 7137–7148 (2015a)
Lehner, A.J., et al.: Crystal and electronic structures of complex bismuth iodides A3Bi2I9 (A = K, Rb, Cs) related to perovskite: aiding the rational design of photovoltaics. Chem. Mater. 27(20), 7137–7148 (2015b)
Lehner, A.J., Fabini, D.H., Evans, H.A., Hébert, C.-A., Smock, S.R., Hu, J., Wang, H., Zwanziger, J.W., Chabinyc, M.L., Seshadri, R.: Crystal and electronic structures of complex bismuth iodides A3Bi2I9 (A = K, Rb, Cs) related to perovskite: aiding the rational design of photovoltaics. Chem. Mater. 27, 7137–7148 (2015c)
Li, J., et al.: Two-dimensional Cs3Sb2I9−xClx film with (201) preferred orientation for efficient perovskite solar cells. Materials 15, 2883 (2022)
Liu, D., Zha, W., RushengYuan, J.C., Sa, R.: First-principles study on the optoelectronic properties of mixed-halide double perovskites Cs2TiI6-xBrx. New J. Chem. 44, 13613–13618 (2020)
Luo, Q., et al.: Computational screening of 2D all-inorganic lead-free halide perovskites A3B2X9 for photovoltaic and photocatalytic applications. Adv. Theory Simulat. (2023). https://doi.org/10.1002/adts.2023009885
McCall, K.M., et al.: Strong Electron−Phonon Coupling and Self-Trapped Excitons in the Defect Halide Perovskites A3M2I9 (A = Cs, Rb; M = Bi, Sb). Chem. Mater. 29(9), 4129–4145 (2017)
Mouhib, B., Dahbi, S., Douayar, A., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: Theoretical investigations of electronic structure and optical properties of S, Se or Te doped perovskite ATiO3 (A=Ca, Ba, and Sr) materials for eco-friendly solar cells. Superlattices Microstruct. Micro Nanostruct. 163, 107124 (2022)
Park, B.-W., Philippe, B., Zhang, X., Rensmo, H., Boschloo, G., Johansson, E.M.J.: Bismuth based hybrid perovskites A3Bi2I9 (A: Methylammonium or Cesium) for solar cell application. Adv. Mater. 27, 6806–6813 (2015)
Pazoki, M., Johansson, M.B., Zhu, H., Broqvist, P., Edvinsson, T., Boschloo, G., Johansson, E.M.J.: Bismuth Iodide perovskite materials for solar cell applications: electronic structure, optical transitions, and directional charge transport. J. Phys. Chem. C 120, 29039–29046 (2016)
Peresh, E.Y., Sidei, V.I., Gaborets, N.I., Zubaka, O.V., Stercho, I.P., Barchii, I.E.: Influence of the average atomic number of the A2TeC6 and A3B2C9 (A = K, Rb, Cs, Tl(I); B = Sb, Bi; C = Br, I) compounds on their melting point and band gap. Inorg. Mater. 50, 101–106 (2014)
Qaid, S., Ghaithan, H., Bawazir, H.S., Aldwayyan, A.: Surface passivation for promotes Bi-excitonic amplified spontaneous emission in CsPb(Br/Cl)3 perovskite at room temperature. Polymers 15, 1978 (2023)
Saparov, B., et al.: Thin-film preparation and characterization of Cs3Sb2I9: a leadfree layered perovskite semiconductor. Chem. Mater. 27, 5622–5632 (2015a)
Saparov, B., et al.: Thin-film preparation, and characterization of Cs3Sb2I9: a lead-free layered perovskite semiconductor. Chem. Mater. 27, 5622–5632 (2015b)
Sham, L.J., Schlüter, M.: Density-functional theory of the energy gap. Phys. Rev. Lett. 51, 1888–1891 (1983)
Singh, A., et al.: Modulating performance and stability of inorganic lead-free perovskite solar cells via lewis-pair mediation. ACS Appl. Mater. Interfaces 12, 32649–32657 (2020)
Tahiri, N., Dahbi, S., Dani, I., El Bounagui, O., Ez-Zahraouy, H.: Magnetic, magnetocaloric and thermoelectric investigations of perovskite LaFeO3 compound: first principles and Monte Carlo calculations. Comput. Theor. Chem. 1204, 113421 (2021a)
Tahiri, N., Dahbi, S., Dani, I., El Bounagui, O., Ez-Zahraouy, H.: Magnetocaloric and thermoelectric properties of the perovskite LaMnO3 material: a DFT study and Monte Carlo technique. Phase Trans. 94, 826–834 (2021b)
Wang, Y., Zhou, Q., Zhu, Y., Xu, D.: High efficiency reduction of CO2 to CO and CH4 via photothermal synergistic catalysis of lead-free perovskite Cs3Sb2I9. Appl. Catal. B Environ. 294, 120236 (2021)
Wu, Z., Cohen, R.E.: More accurate generalized gradient approximation for solids. Phys. Rev. B 73, 235116 (2006)
Wu, L., et al.: High-pressure band-gap engineering and metallization in the perovskite derivative Cs3Sb2I9. Chemsuschem 12, 3971–3976 (2019)
Xie, B., Chen, D., Li, N., Xu, Q., Li, H., Lu, J.: Lead-Free Cs3Bi2Br 9 perovskite In-situ growth on 3D Flower-like g-C3N4 microspheres to improve photocatalytic performance. Chem. Eng. J. 452, 139662 (2023)
Acknowledgements
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project no. (IFKSUOR3–396–1)
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
SD and SMHQ wrote the main manuscript text, HMG, and AAAA prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have not disclosed any conflict interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dahbi, S., Qaid, S.M.H., Ghaithan, H.M. et al. Iodine-based triple halide perovskites for photovoltaic and photocatalytic applications: a ab-initio study. Opt Quant Electron 56, 702 (2024). https://doi.org/10.1007/s11082-024-06343-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-024-06343-5