
Abstract
In this work, theoretical investigations were performed for the structural, elastic, optoelectronic, thermodynamic, and thermoelectric characteristics of barium ceroplatinate Ba2CePtO6 double perovskite oxide. The direct band gaps 1.518 eV and 1.385 are calculated with TB-mBJ potential, and PBE-GGA approximation. The findings of elastic constants C11, C12 and C44, ensure the mechanical stability, ductile nature, anisotropic, Debye temperature, and ionic bonding. In visible and ultraviolet spectrums, Ba2CePtO6 exhibits a high absorption coefficient (> 105 cm−1), low optical reflectivity, low electron energy loss and excellent refractive index. Finally, the thermoelectric properties are computed using BoltzTraP code. At room temperature the figure of merit has been reported as 0.84 and 0.998. Therefore, the reported results are likely to serve as an inspiration for the forthcoming experimental and theoretical investigations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Recently, with the rapid development of humans and the improvement of living standards, the question of energy supply, and many critical ecological issues, have become progressively concerned. In this view, we are dealing with the inevitably depletion of conventional energy resources with adverse effects on the environment (Mostafaeipour et al. 2022; Ivanovski et al. 2021; Snaith 2013). Therefore, seeking out new sustainable energy resources, efficient and economically viable, and especially coping with global warming, are the most pressing current issues (Mantziaris et al. 2017; Bildirici and Gökmenoğlu 2017). In this regard, it is worth mentioning that about 50% of the energy generated from primary sources is discarded as squander heat energy. The use of photovoltaic (PV) and thermoelectric (TE) technologies offers the extremely reliable alternative methods to convert the heat generated from solar radiation and to harvest effectively the industrial waste heat and automotive exhausts for electricity production. Therefore, the primary challenging task in this research area is to substantially enhance the PV and TE conversion efficiencies as compared to the conventional techniques. This can be attained with the help of fundamental and promising materials for devices origin. The most intrinsic transport parameter used to estimate the TE conversion efficiency of a material is the dimensionless figure of merit, ZT = S2σT/κ. In this quantity S, σ, κ, T stand respectively to Seebeck coefficient, electrical conductivity, total thermal conductivity and the absolute working temperature. Therefore, a typical TE material possesses an excellent σ to reduce Joule heating effect, strong S for a maximum heat-to-electricity conversion, and low κ keep a heat at the junctions and retain a substantial temperature gradient. Clearly, the task is to propose a potential material with specific properties that satisfy the basic requirements of optoelectronic and TE technologies.
In recent paste, perovskite materials have attracted immense excitement and have been recognized as the material of choice for optoelectronics and TE applications due to their remarkable features (Wu et al. 2015; Soukiassian et al. 2008; Chen et al. 2010). There are many kinds of materials have been studied for optoelectronic and photovoltaic applications (Tarbi et al. 2022, 2023a; b, c). Interestingly, perovskite materials can display many intriguing structural and electro-optical properties which show their flexibility to accommodate many elements of the periodic table. The general stoichiometric formula of simple perovskites structures is ABX3, where A denotes a rare earth, alkaline earth or transition metal cation, B is the transition metal cation and X stands to an anion. In the current situation, considering the ABO3 primitive perovskites oxides, when the octahedral coordinated B cation is shared, with 1:1 proportion, by two distinct transitions metals cations B′ and B′′, the A2B′B′′O6 double perovskite oxide (DPO) is formed. A significant difference between the size of B′ and B′′ transition metal cations play an important role in stabilizing the ordering phenomenon (Lufaso et al. 2006; Anderson et al. 1993). Moreover, the charge balance is maintained by: 2QA + QB′ + QB′′ = 12. In the crystallographic point of view, the crystal symmetry and space group of DPOs depend mainly on the arrangement of both B′O6 and B′′O6 octahedral (Chen et al. 2005; Retuerto et al. 2004). Furthermore, the potential applications of DPOs as merited a great deal of consideration in many fundamental and applied areas (Tariq et al. 2020; Slavney et al. 2016; Faizan et al. 2016; Rammeh 2016; Bharti and Sinha 2011a) due to their versatile properties such as tunneling magneto-resistance, magneto-optic properties, ferromagnetism, metallicity, multi-ferocity magneto-dielectric properties, high dielectric constants and photo-catalytic activity and so on (Saha-Dasgupta 2020; Yin et al. 2019; Ray et al. 2017; Vasala and Karppinen 2015; Retuerto et al. 2006; Bairwa et al. 2023; Al-Qaisi et al. 2023; Mebed et al. 2023; Das et al. 2008; Kato et al. 2002).
As far as Ba-based DPOs are concerned, an extensive amount of work has already been published (Al-Qaisi et al. 2022a, b; Himanshu et al. 2022; Kockelmann et al. 2006; Korchagina and Shevchuk 2006; Fu and Ijdo 2005; Kazim et al. 2022; Reddy et al. 2018; Aczel et al. 2019; Dutta et al. 2016; Bharti and Sinha 2011b,c; Hatakeyama et al. 2010; Amador et al. 1992). In this regards, Al-Qaisi et al. (2022a) predicted that Ba2YBiO6 is an indirect p-type semiconductor (SC) with a high ZT which is suitable for the ultraviolet (UV) and visible (VIS) optoelectronic devices, and TE applications. The same group demonstrates that Ba2NaIO6 is a direct p-type SC. Their findings reveal that this compound is a good material for infrared optoelectronic and TE applications (Al-Qaisi et al. 2022b). Based on the synchrotron X-ray diffraction technique and the reflectance measurements in the UV–VIS range, Himanshu et al. (2022) report that Ba2ScTaO6 refined in an ordered cubic symmetry with a band gap of 4.7 eV. From the High-resolution neutron powder diffraction measurements, Kockelamann et al. show that at ambient temperatures Ba2PrIrO6 DOP is cubic space group Fm-3m. Also, they demonstrate that below 71 K the compound orders anti-ferromagnetically (Kockelmann et al. 2006). In the experimental work of Korchagina et al., the low-frequency dielectric and microwave features of Ba2LnTaO6 (Ln = La, Pr, Sm, Dy, Ce, Gd, Nd, Tm, Tb) compounds were evaluated. The reported results were conveniently correlated with the ionic radius of Ln cations (Korchagina and Shevchuk 2006). In addition, based on X-ray powder diffraction measurements at room temperature, the crystal symmetry of a series of Ba2LnSbO6 (Ln = La, Pr, Nd, Sm) DOPs have been explored by Fu and Ijdo. They demonstrate that the structure of Ba2LnSbO6 (Ln = La, Pr and Nd) are rhombohedral with the space group \(\overline{R }\) not monoclinic or cubic as were previously reported (Fu and Ijdo 2005).
Moreover, after a comprehensive literature review, we have noticed a few works on Ce- containing DOPs. In this respect, using the FP-LAPW method within DFT theory, Kazim et al. ensure that Ba2CeZrO6 is stable in cubic symmetry with band gap energy of 3.25 eV. The ZT of 0.65 was observed at 800 K. Therefore, the reported results demonstrated that this compound fits well the optoelectronic and TE applications (Kazim et al. 2022). Furthermore, in their work, Reddy et al. (2018) have prepared Ba2CeZrO6 DOPs for the first time using the sol–gel technique. Their results show that the sample is refined in cubic structure with space group Fm-3m with a semiconducting nature. Also, based on neutron powder diffraction results, Aczel et al. (2019) show that Ba2CeIrO6 crystallizes in the cubic space group Fm-3m. The structural and magnetic properties of Ba2CeSbO6 DPO have been synthesized by Dutta et al. Their Rietveld refinement results suggest the monoclinic ordered structure with an anti-ferromagnetic behavior. The XPS study reveals the existence of two oxidation states of Ce (Dutta et al. 2016). The barium cerium tantalate Ba2CeTaO6 has been synthesized and characterized by Bharti and Sinha. Based on XRD patterns, they show that at room temperature the sample crystallizes in a monoclinic symmetry. The frequency-dependent conductivity spectra show that at f = 110 Hz the conductivity varies from 3. 5 × 10−7 to 1.2 × 10−2 S.m−1 when the temperature increases from 303 to 673 K (Bharti and Sinha 2011b). Furthermore, Ba2CeNbO6 DPO was prepared and characterized for the first time by Bharti et al. The XRD data at ambient conditions confirms the monoclinic crystal structure (Bharti and Sinha 2011c). Finally, a series of Ba2RBiO6 (R = La, Ce, Nd, Sm, Eu, Gd, Dy) DOPs have been synthesized by Hatakeyama et al. (2010) The high observed photocatalytic activities under VIS irradiation have been interpreted within a first-principle calculation within FLAPW method. Meanwhile, the lack of Pt-containing DOP is observed. On the basis of refinement of the XR powder diffraction pattern, Amador et al. (1992) demonstrate that Ba2PrPtO6 is a cubic crystal with Fm-3m space group.
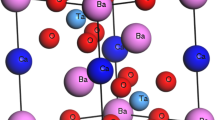
In the current work, we are focusing on barium ceroplatinate Ba2CePtO6 DPO for the motivated following reasons: To the best of our knowledge, except the below mentioned works, there are no reports on the properties of Ba2CePtO6 encountered in the literature. Furthermore, due to its nontoxic aspect it can be a potential material for useful applications. Additionally, owing to its reasonable direct semiconducting band gap, it can be selected as a plausible candidate for optoelectronic and TE and PV conversions. The formation of Ba2CePtO6 was first observed by Ouchetto et al. (1991), while working on the synthesis of barium procreates perovskite type (BaCeO3) in the platinum (Pt) crucibles in air using hydrothermal method. Accordingly, Ba2CePtO6 comes from the reaction between the synthesized BaCeO3 and Pt and air/O2. The structure determination was performed from the intensities of 15 diffraction peaks, up to dhkl = 0.1 nm, while, the space group was deduced from the systematic existence condition (hkl): h + k, k + l, h + l = 2n. As a result, Ouchetto et al. (1991) conveniently concluded that their DOP crystallizes in a cubic Fm3̅m space group (n◦0.225) with a primitive cell parameter a = 8.4088 Å. Furthermore, in the primitive unit cell the Wyckoff positions of Ba, Ce, Pt and O atoms are, respectively, 8c, 4b, 4a and 24e with coordinates (1/4, 1/4, 1/4), (1/2, 1/2, 1/2), (0, 0, 0) and (0.248, 0, 0) as displayed in Fig. 1. Moreover, Casapu et al. (2007) discovered Ba2CePtO6 DPO while dealing with a Pt/Ba/CeO2 catalyst and suggested a reaction in which a BaPtO3 compound is first formed and reacts with BaCeO3 to form the Ba2CePtO6 DPO.

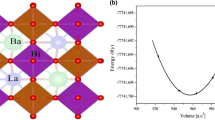
The crystal structure and the calculated total energy as a function of volume E(V) for the cubic Ba2CePtO6 optimized structure using PBE-GGA
In this light, the primary purpose of the current study is to show to what extent this newly predicted material can be used for optoelectronics devices and PV and TE conversion purposes. For this aim, we have convincing investigated the structural, elastic, optoelectronic, and TE properties of this proposed material. The rest of the present manuscript is structured as follows: in Sect. 2, the computational method details are described. The results and discussions are presented in Sect. 3 where a comparison is made, when possible, with similar compounds. Finally, in the last section we conclude this work with a summary of the reported results.
2 Computational method
To simulate the crystal primitive cell of Ba2CePtO6 we have adopted the previously mentioned experimental structural parameters measured by Ouchetto et al. (1991). Subsequently, the structural, elastic, optoelectronic, thermodynamic and TE properties were performed within the frame of density functional theory (DFT) using the full-potential linearized augmented plane wave plus local orbital (FP-LAPW + lo) method as implemented in the WIEN2K software package (Schwarz et al. 2002; Madsen et al. 2001; Blaha et al. 2001). In this method, a muffin-tin crystal potential is presupposed and the crystal primitive unit cell is partitioned into two regions: (i) non-overlapping muffin-tin spheres (MTS) with radii RMT centered at the atomic sites and separated by (ii) an interstitial space. In addition, these two regions were properly treated with different basis sets. Inside the non-overlapping MTS, the electron is extended in terms of radial functions times spherical harmonic basis functions in the interstitial and plane wave regions. Within the MT spheres, the basis set is adequately controlled with the maximum angular momentum lmax = 10. While in the interstitial space, a reasonable number of plane wave functions for Kmax(a.u)−1 = 7/(RMT)min is adopted, where Kmax denotes the highest k-vector magnitude, while (RMT)min is the lowest value amongst RMTs. In the present contribution, RMTs of 2.5, 2.17, 2.04, and 1.67 are chosen for the Ba, Ce, Pt, and O atoms, respectively. These values are adjusted in a fashion that no leakage of electrons occurs from the core into interstitial region and thus the convergence of the total energy is achieved with a minimal time calculation.
After simulating the crystal primitive unit cell, elastic and structural properties of the present material were optimized using the exchange–correlation functional to evaluate by generalized gradient approximation (GGA) in the scheme of Perdewe Burkee Ernzerhof (PBE) functional (Perdew et al. 1996). Subsequently, the computed ground states energies were added into the Birch-Murnaghan equation of state (EOS) to extract the lattice parameter (Murnaghan 1944). Similarly, for the electronic band structure computation, we have executed both PBE-GGA and Trans-Bhala modified Becke-Johnson (TB-mBJ) exchange–correlation potentials (Tran and Blaha 2009). Therefore, to overcome the failing of PBE-GGA to maintain reliable band gap, the outcomes of TB-mBJ approach were appropriately used in the computation of the optoelectronic properties for an incident photon energy range of 0–14 eV. As accessible in the WIEN2K software package (Schwarz et al. 2002; Madsen et al. 2001; Blaha et al. 2001), a dipole matrix element approach was adopted to conduct the optical features, with a Lorentzian broadening parameter of 0.1 eV. Additionally, the TE transport properties were investigated in the framework of the Boltzmann semi-classical transport theory under the constant relaxation time (τ) and rigid-band approximations. Subsequently, the TE transport equations were implanted in the BoltzTrap code with τ = 10−14 s (Madsen and Singh 2006). Moreover, the Integrations over the FBZ were performed using non-shifted k-points mesh of 3000 (14 × 14 × 14) for optoelectronic and 124,000 (49 × 49 × 49) for TE transport properties because they require dense k-mesh. Moreover, a self-consistency is reached when the system total energy convergence between two successive iterations is lower than 10−5 Ry.
3 Results and discussion
3.1 Structural properties
At the first stage and before starting the detailed computation, it is necessary to determine the most stable structure of Ba2CePtO6 in its ground state. For this purpose, an accurate optimization of the structural parameters of the interest compound and the relaxed atomic positions were initially conducted within the frame of PBE-GGA approximation by minimizing the system total energy against the primitive unit cell volume Etot(V). Therefore, the ground state properties of Ba2CePtO6 compound, namely equilibrium lattice parameter a0, equilibrium primitive unit cell volume V0, bulk modulus B0 and its first derivative B0′ have been obtained through the fitting of Etot(V) to the Birch-Murnaghan EOS (Murnaghan 1944):
On the Etot(V) optimization curve depicted in Fig. 1, the minimum point gives the ground state total energy. Since the perfectly cubic perovskite structure is concerned, the lattice parameter corresponding to the minimum energy is determined at the optimized volume. The obtained results are shown in Table 1. Interestingly, the optimized lattice parameter a0 reported in this work matches with the experimental value reported in Ouchetto et al. (1991). However, owing to the deficiency in the theoretical and experimental results regarding B0 and B0′, for the present compound, our results are predictions.
Furthermore, in this optimized structure the Ba–Ce, Pt–O, Ce–O and Ba–O chemical bond lengths were found to be 3.684, 2.038, 2.216 and 3.009 Å, respectively. These obtained values can directly employ to give the first idea on the nature of chemical bonds in Ba2CePtO6. Therefore, the distance Pt-Ce is about 4.254 Å, which is in the same order as a/2 parameter. Besides, the shortest distance between Ba and Ce is less than the sum of the two atomic radii. Furthermore, the distances between O atom and other atoms are lower than the sum of the corresponding atomic radii. All these indications ensure its strong bonding and preservation. Another remark that should be pointed out is that the stability of the material under focus depends mainly on the mutual suitability of Ba2+, Ce3+, Pt5+, and O2− ions in terms of their atomic radii characterized by the tolerance factor τ defined as follows (Bharti and Sinha 2010):
The perfectly cubic perovskite material is usually predictable when τ = 1. In the current work, the tolerance factor of 1 was obtained confirming that Ba2CePtO6 compound stabilizes in a perfect cubic perovskite structure. The accuracy of our results implies the reliability of to the current work and inspired us to investigate different properties of the Ba2CePtO6 DOP.
3.2 Elastic properties
To inspect the mechanical behavior of a material for specific application, mechanical properties play a primordial role because they are linked to different micro and macroscopic properties of materials. These mainly include the elastic constants (Cij), Shear (G), Bulk (B), and Young (Y) modulii. They allowed us to examine the possible features of a material in terms of its mechanical stability, brittleness, ductility, anisotropy, nature of the inter-atomic bonding, melting temperature, sound velocity, and Debye temperature and so on. Furthermore, in the present contribution, as the perfect cubic symmetry is concerned, the axial lengths are equal; therefore, only three independent elastic constants in the Cij tensor are required namely C11, C12 and C44. Our results yield values of C11 = 283.97, C12 = 78.98 and C44 = 59.3 GPa as listed on Table 2. It is obvious that the suggested compound is characterized by a large value of C11 contrasted to C12 and C44, implying that Ba2CePtO6 has more withstanding resistance to unidirectional compressive strength than shearing deformation. The elastic stability of the present material requires that the whole set of Cij must fulfill the Born elastic stability criterion: C11, C12, C44 > 0; C12 < B < C11; C11–C12 > 0 and C11 + 2C12 > 0 (Wang et al. 1993; Behera et al. 2023).
The evaluated Cij for Ba2CePtO6 are positive in the unit of GPa and adequately content with the mentioned stability conditions, implying that the current compound is mechanically stable. Thus, this result accurately allowed us to conduct the present investigation. A part from that, it is not useful to study a material that is not mechanically stable. Subsequently, the obtained Cij can be used to determine the macroscopic behavior of the material under focus. Hence, for industrial point of view applications, the key elastic coefficients namely shear (G), bulk (B), Young (Y) modulii, Cauchy pressure (CP), Pugh index (B/G), Poisson ratio (υ), anisotropic factor (A) are computed and listed in Table 2.
The G elastic modulus provides indications on the plastic deformation of a compound trough an external pressure. G is the arithmetic average of GV and GR which represent its upper and lower limiting values, respectively, obtained from G = (GR + GV)/2 where GV = (C11-C12 + 3C44)/5 and GR = 5(C11–C12)C44/(3(C11–C12) + 4C44) (Hill 1952). Therefore, by referring to Table 2, the recorded value of G = 73.95 GPa suggests that Ba2CePtO6 shows greater resistance plastic deformation compared to Ba2ZrCeO6 (Kazim et al. 2022). Also, the bulk modulus B = (C11 + 2C12)/3 (Hill 1952) is a proper quantity used to determine the resistance towards the volume change in response to external pressure. As a result, B (GPa) = 147.31 (~ 2G) indicates that Ba2CePtO6 is resistant to the volume deformations than to the shear stress. Furthermore, the Y modulus can be defined as the ratio of linear stress to strain. Also, Y is a proper quantity used to predict the stiffness of a material. Further, the Y modulus is directly evaluated in terms of the computed values of bulk B and shear G modulii using Y = 9BG/(3B + G) (Wang et al. 1993). In this contribution, the predicted value of Y = 190.05 GPa advocates that Ba2CePtO6 is relatively stiffer compared to Ba2CeNbO6 (Bharti and Sinha 2011c). This result is expected to be ascribed to the strong bond between adjacent atoms. Additionally, the reported moduli decrease in order of G < B < Y, implying that this compound reveals more shape deformation than volume deformation.
Also, B and G afford important indication regarding the malleability of materials trough Pugh index (B/G) (Pugh 1954). In this context, the amount value of the Pugh index is typically 1.75 for purely plastic materials. If B/G is smaller than 1.75, a brittle attitude is predicted, else the material is designated as ductile (Bougherara et al. 2022). Besides, for the compound of interest, the value of B recorded in Table 2 is two times larger than G suggesting the ductile behavior of Ba2CePtO6. This result is supported by the Cauchy pressure (CP = C12-C44) (Pettifor 1992). CP manifests as an effective way to identify the brittle and ductile behavior of a material in such a way that if CP > 0 the material is ductile. Otherwise, as for a brittle structure Cp < 0. Further, the positive CP observed for the Ba2CePtO6 DOP ensures that this material shows a ductile behavior. In the same way, the ductility nature of the present material can be confirmed using the Poisson ratio. The Poisson ratio, ν = (3B-Y)/6B, represents the volumetric change throughout uniaxial deformation (Li et al. 2012). Thus, for a completely plastic compound, a critical amount of υ is close to 0.26. However, if υ < 0.26, the compound shows a brittle nature, while, if υ > 0.26, the material affords ductile character. Based on these conditions, the present reported value of υ is about 0.285. Thus, the ductility of Ba2CePtO6 compound is accounted again. Accordingly, Ba2CePtO6 is an excellent candidate for device fabrication. Also, the good approval emphasizes the reliability and precision of the current study. In other hand, the chemical bonding character in a material can be outlined from the above mentioned ν. For covalent materials, ν ~ 0.1, whereas a material exhibits an ionic bonding if ν = 0.25. However, as for a metallic material ν = 0.33 (Rani et al. 2023). Accordingly, as reported in Table 2, the stability of the suggested material is maintained by ionic bonding with metallicity and hence showing ductile nature. In addition, the direction-dependent elastic properties of a material can be identified using the anisotropic factor A = 2C44/(C11-C12) evaluated from the elastic constants (Li et al. 2012). As a matter of fact, the elastic anisotropy originates basically from the difference in the longitudinal and shear elastic constants. For a purely isotropic material A = 1, otherwise the divergence from unity indicates the degree of elastic anisotropy. For the Ba2CePtO6 DOP, the recorded A = 0.58 inferred that the investigated compound present anisotropic character and can therefore exhibits different properties in various directions.
3.3 Thermodynamic properties
Likewise, another indispensable physical parameter, known as melting temperature (Tm), has been estimated under ambient conditions. The Tm is the critical temperature of a solid above which it starts melting and changes phase from solid to liquid. The value of Tm of a crystal is linked to the directional elastic constant C11 and can be predicted through the standard equation (Dar et al. 2019):
The present reported value of Tm = 2231 ± 300 K is consistent with what we concluded for Y modulus. Also, it confirms the strong bonding and retention of the studied material and provides a strong validation for its practical use for elevated temperatures applications. Furthermore, the average sound velocity vm, also known as average Debye velocity, is expressed in terms of transverse and longitudinal parts velocities as (Jasiukiewicz and Karpus 2003):
where vt and vl are the magnitudes of transverse and longitudinal sound velocities, respectively. By means of the computed value of, B and G, the νl and νt can be estimated through Navier equation of states vl = (G/ρ)0.5 and vt = (3B + 4G/3ρ)0.5 (Schreiber et al. 1975). Evidently, the sound velocities depend on the B and G, indicating that higher values of elastic moduli imply high values of sound velocities as well. Our results concerning vt, vl and vm for Ba2CePtO6 are given in Table 3. Thus, one can note that the vl is about 1.82 higher than vt. The Debye temperature (TD) is a material basic quantity directly linked to various physical properties namely lattice vibrations, thermal conductivity and so on. Moreover, the TD directly affects the specific heat capacity of a solid. Interestingly, TD is predicted from the computed Cij elastic constants through the average sound velocity vm mentioned above by the following expression (Jasiukiewicz and Karpus 2003):
where h is Planck’s constants, kB is Boltzmann’s constant, n stands to the number of atoms per unit cell, NA is Avogadro’s number, ρ is for the density, M represents the molecular mass per unit cell. Thus, Ba2CePtO6 has more withstanding to heat generated by lattice vibration. Also, this alloy can be considered as a low thermal conduction material which could be interesting for TE applications. The obtained values are recorded in Table 3. Due to the unavailability of both theoretical and experimental data of Tm, vm and TD of the current material, a comparison has been made with the data related to similar DOPs compounds.
3.4 Electronic properties
After checking the structural stability and formability of Ba2CePtO6 DPO, it is of crucial importance to conduct their substantial optoelectronic performances through an in-depth insight into its electronic band structure and the related total (TDOS) and partial density of states (PDOS). Indeed, knowing the band structure features of a material accurately provides valuable information for their synthesis and manufacturing.
3.4.1 Electronic band structure
The band structure calculated with PBE-GGA and TB-mBJ potentials along with the total density of states are illustrated in Fig. 2a–c. The Fermi level (EF) taken at zero is selected to be the energy reference. As depicted in Fig. 2a, b, the bands structure patterns obtained with PBE-GGA and TB-mBJ potentials conceptually simulate each other. Accordingly, it can be inferred that the compound under focus shows that the conduction band minimum (CBm) and the valence band maximum (VBM) are revealed at X-point of the FBZ, confirming the existence of a direct (X–X) band-gap (Eg). Furthermore, as regards to the magnitude of Eg, we have recorded the values of 1.385 eV and 1.518 eV within PBE-GGA and TB-mBJ approaches, respectively. However, it should be noted that the gap energy value is underestimated by about 9% within PBE-GGA potential. Therefore, the band gap value has been enhanced using TB-mBJ potential. Consequently, the present Eg agree reasonably with those of stable and direct compounds appropriate for PV conversion (1.35–2.4) eV. Moreover, a close examination of Fig. 2a, b shows that the group of CBs is slightly shifted to higher energies in the case of TB-mBJ potential inducing the enhanced Eg(X). However, in all circumstances, the EF level seems to do not cross the energy bands. Accordingly, the material under focus exhibits a semiconducting behavior.

The computed electronic band structure for Ba2CePtO6 using a PBE-GGA and b TB-mBJ approaches. c represents the corresponding total density of states (TDOS)
The proposed DPO material not only exhibits a semiconducting direct Eg but also it shows substantial flatter bands along the (X–W–K) path. This results in an enhancement of the probability of electron transition from VBM states to CBm states associated with a photon absorption. Interestingly, the lowest CB is almost flat in the whole BZ, whereas the highest VB shows a notable dispersion. The observed flatness in the first CBs indicates that the individual electrons are weakly affected by each other. Also, this feature lead to a small hole’s effective masse with extremely high mobility allowing a p-type conductivity character. Moreover, the presence of p-type semiconducting nature is shown by the closeness of EF level to VB. In the absence of both theoretical and experimental findings to be contrasted with our data regarding the magnitude of Eg of Ba2CePtO6 DPO, and owing to the fact the present findings are obtained from the TB-mBJ framework, we expect that the present results are more accurate. Also, since the present compound is characterized by moderate and direct band gap, it can be believed as a potential candidate for PV and TE conversion applications.
3.4.2 Density of states
As means to provide sufficient explanation about the electronic properties and to inspect the provenance of electronic states in the computed band structure, the partial (PDOS) and total (TDOS) density of states as a function of energy were computed in the TB-mBJ potential framework. The obtained results, projected between − 6 and 6 eV, are shown in Fig. 3a–c. Clearly, one can observe that diverse peaks with high TDOS dominate both valence and conduction regions suggesting the availability of a large number of unoccupied electronic states. A close inspection of these figures reveals that in the energy range of − 6 to − 4 eV of the lowest VBs, the TDOS is typically originates from the Pt-5d, O-2p and Ba-p bonding orbitals, where the predominant contribution of the Pt-5d is larger than that of O-2p and Ba-p. Furthermore, in the energy range extending from − 4 to − 1 eV, the significant contributions are provided by the O-2p orbital and the p and f-type orbitals of Ce atom. Moreover, the CB as a whole is mainly constituted by the unoccupied Ce-4f, Pt-5d, and p-type states of O and Ce atoms. Therefore, one may say that the main contributions are due to the Ce-4f and Pt-5d with a little involvement from the O-2p and Ce-2p orbitals. A further consideration of these results specifies that the edge associated with the VBM has a contribution from Pt and O atoms, while CBm is mainly obtained from Ce atom with a slight contribution from Pt and O atoms. Besides, the band gap energy is characterized by direct transitions from the dominating Pt-5d metal states in VB to the dominating Ce-4f states in CB.

The partial (PDOS) and total (TDOS) density of states for Ba2CePtO6 computed within the frame of TB-mBJ approximation
It is a known fact that the optoelectronic features of SC compounds are tightly related to the distribution of states around the EF level. For the DPO under focus, these results highlight that for the upper VBs near the EF level, the most important contribution originates basically from the 5d electrons of Pt atom with a few contributions of O-2p states. This indicates that the Pt-5d electrons have an interesting role in the conductivity of the present material. Besides, in the valence region, a small value of TDOS at the EF level explains the p-type semiconducting nature for this material. While in CB regions the TDOS vanishes at the EF level. Furthermore, the value gap of 1.52 eV closes to the EF level in TDOS confirms the SC character of our material.
3.5 Optical properties
The optical characteristics exhibit the light transmission, transparency and polarization ability of materials in different energy ranges, which can be regarded as important indexes for the study of optoelectronic materials. Actually, the key function which can precisely describe the optical behavior of a material owing to an external electromagnetic (EM) radiation is the frequency-dependent complex dielectric function ε(ω). From the theoretical point of view, ε(ω) can be split into two parts as given by Ehrenreich and Cohen as ε(ω) = ε1(ω) + iε2(ω) (Hilal et al. 2016), where ε1(ω) and ε2(ω) stand to the real and the imaginary components of ε(ω), correspondingly. Mostly, ε is a tensor with nine different direction-depending components. However, with regard to the crystallographic symmetry, only some of these components may be independent. As Ba2CePtO6 compound crystallizes in cubic phase, the three principal x, y, and z directions are equivalent. Accordingly, the principal tensor components are equal: εxx = εyy = εzz. Therefore, it is crucial to compute only one component. The imaginary component ε2(ω) of the dielectric function can be achieved by summing up, over the first Brillouin zone (FBZ), all possible transitions of the electrons from the occupied electronic states at the VB to the empty states at the CB. Considering the appropriate transition dipole matrix elements, ε2(ω) can be stated in the following form (Al-Qaisi et al. 2021):
where e stands to the elementary charge, m is the electron mass, M denotes the dipole matrix element, < i| and |j > represent the initial and final electronic states, respectively, Ei,k presents the energy level of the electron in the ith state, fi stands to the Fermi distribution function taken for the ith state. Indeed, ε1(ω) and ε2(ω) are interlinked by the Kramers–Kronig relation (Bobrov et al. 2010):
where P represents the principal value of the Cauchy integral.
Thereafter, all other optical response functions namely the refractive index n(ω), the optical absorption coefficient α(ω), the optical conductivity σ(ω), the extension coefficient κ(ω), the optical reflectivity R(ω) and the electron energy loss L(ω) can be derived from ε1(ω) and ε2(ω) (Gajdoš et al. 2006).
3.5.1 Dielectric function
The dispersion of light energy has been illustrated in Fig. 4a by ε1(ω) from which it is obvious that in the low photon energy side, the highest dispersion of light is 7.42 near the energy 1.73 eV corresponding to the resonance frequency in the visible band (VIS). The highest value of ε1(ω) spectra in the VIS windows indicates that the materials under load can be efficiently used for PV conversion and for other optoelectronics devices. Afterwards, ε1(ω) shows a set of minor peaks essentially attributed to the CB and VB inter-band optical transition. A slight enhancement in the photon energy declines these peaks drastically to their lowest values in the ultraviolet (UV) region. Furthermore, the photons in the energy range from 8.53 to 9.86 eV, and ε1(0) show that the longitudinal waves are possible. Thereafter, ε1(ω) value becomes negative in energy regions from 8.53 to 10.95 eV. Moreover, for the positive values of ε1(ω), the photons propagate through the compound under focus, whereas the negative values describe the damping of the incident photons corresponding to the maximum reflection clearly shown by the maximum of reflectivity. The zero energy limit of ε(ω) or static dielectric constant ε(ω → 0) is the value of ε(0) = ε1(0) estimated for zero frequency with neglecting the phonon contribution. As depicted on Table 4, the extracted value of ε1(0) = 5.11 for Ba2CePtO6 is higher than ε1(0) = 4.20 reported by Kazim et al. (2022) for Ba2ZrCeO6 DPO. Moreover, this relatively larger value of ε(0) dictates the relatively higher polarizability of Ba2CePtO6 material which causes a better optical absorption, and therefore, improves low-level of electronic defects and decreases the radiative recombination. This result is expected owing to the fact that Pt atom has an extra polarization compared to Zr due to its bigger atomic radius. On the other hand, it has also been remarked that the larger ε1(0) are attributed to the small band gap SCs in consistent with the mathematical equation of the Penn model: ε1(0) = 1 + (ħωp/Eg)2 (Penn 1962).

Computed results of a real ε1(ω) and imaginary ε2(ω) part of the complex dielectric function b refraction index and Extinction coefficient c electronic energy loss and optical reflectivity d absorption coefficient and e optical conductivity of Ba2CePtO6 compound
By referring to the computed ε2(ω) spectra depicted in Fig. 4a, one can see that ε2(ω) remains zero for an incident photon energy less than that of the first optical critical point A0 at which the energy referred to as the fundamental absorption threshold. Thus, the optical Eg can be extracted by the threshold energy of ε2(ω) at about 1.52 eV in consistent with the Eg obtained directly from band structure. In Fig. 4a a set of peaks are observed at 1.97, 3.41, 3.85, 4.83, 6.51, 7.74 and 8.07 eV. Moreover, these peaks come chiefly from the most probable direct electronic transferences from occupied electronic states in VBs to the empty states in the CBs. By means of DOS diagram and band structure, it can be realized that A1 peak is mainly related to transition of the Pt-5d electrons to Ce-4f states. Whereas, the peaks A2, A3, and A4 are mainly connected to the optical transitions of electron from 5d orbital under the EF to the 5d orbital above the EF of Pt atom. Additionally, the peak A5 is attributed to the transition of Pt-5d electrons to O-2p states above EF. These peaks are followed by other minors peaks related to inter-band transitions of electrons between O-2p states below and above EF.
3.5.2 Refraction index and extinction coefficient
The absorption and optical transparency of materials are basically measured by the extinction coefficient κ(ω) and refractive index n(ω). In this regard, the n(ω) index is regarded as the parameter by which the radiation speed and wavelength are reduced compared to their vacuum values. In other words, it describes the transparency degree of a material. In addition, the ionic polarization and local field inside the material is connected to n(ω) index. Moreover, κ(ω) appears due to the consequence of the propagating EM radiation in the same material medium. Also, the attenuation of EM waves in a material is directly described by the κ(ω) coefficient.
In this work, the computed absorption and transparency of light in terms of n(ω) and κ(ω) are plotted in Fig. 4b. The trend of these two spectrums is alike to εi(ω). This can be made clearer by looking at the link between these parameters through the relations: 2nκ(ω) = ε2(ω) and n2(ω) − κ2(ω) = ε1(ω). Further, at ω → 0 limit, the static n(ω → 0) of the present material is recorded as n(0) = 2.26. The spectra of n(ω) exhibits a set of appreciable peaks principally ascribed to the excitonic transitions. Moreover, these peaks are more intense at low photon energy and this effect is diminished with increasing photon energy. In addition, we can clearly observe that n(ω) is considerable for a weak incident photon energy. However, it decreases speedily at higher energies in the shape of fluctuations to 14 eV. Its values in the VIS spectrum n(ω) has peak value 2.75 at 1.76 eV. Thus, the material under focus possesses high n(ω) within VIS region which is significantly suitable for optoelectronic devices. The situation is extremely quite different for Ba2ZrCeO6 which exhibits a pronounced principal peak at around 4 eV situated in UV region (Kazim et al. 2022). Also, the n(ω) index is less than unity beyond 10.7 eV, implying that Ba2CePtO6 material is more refractive. Accordingly, the optical transparency of this material extends over a large range of solar spectrum from UV to IR. Similarly, Fig. 4b displays the κ(ω) coefficient of the Ba2CePtO6 DPO. One can note that the κ(ω) values are zero till the fundamental Eg, indicating that the photons will be fully transmitted without any attenuation. Moreover, the κ(ω) coefficient of Ba2CePtO6 have many distinct peaks located in both VIS and UV regions, indicating the maximum penetration of light in this region with a strong extinction effect in the UV region. Also, we can see clearly that the peaks κ(ω) are consistent with that of ε2(ω). Therefore, one expects that Ba2CePtO6 compound is an excellent candidate for optoelectronic applications.
3.5.3 Absorption coefficient
The absorption coefficient α(ω) characterizes the distance traveled by the incident photons inside a material before absorption. In other word, it deals with the change in the intensity of light at a given wavelength per unit length. With regard to the design and fabrication of optoelectronic and PV devices, α(ω) is the most compelling and sensitive criteria for checking the ability and usefulness of SC materials. For this purpose, α(ω) has been computed for Ba2CePtO6 DPO in the incoming photon energy range of 0–14 eV. The obtained spectrum is illustrated in Fig. 4d. The noticed peaks are consistent with that of ε2(ω) and κ(ω). Additionally, it is noteworthy that the wider peaks are resulted from the transitions in very close bands. Nevertheless, at high energies, the sharp peaks may be accordance with inter-band optical transitions between valance and CBs that can be considerably far from each other.
The onset of absorption edge in Ba2CePtO6 is recorded at around 1.52 eV which is the same point from where the ε2(ω) rises from zero, in agreement with computed band gap energy. Near the threshold point, the absorption process comes from the dominant bands of the optical transition from Pt-5d orbital at VBM to the Ce-4f CB orbital. Afterwards, the α(ω) rate increases with the photon energy toward the UV spectrum. This is due to the augmentation of the number of electrons that have an energy allowing them to interact with photons. Also, the titled compound demonstrates substantial optical absorption over a wide band of solar spectrum involving both VIS and UV bands with a strong wider peak noticed at about 5.2 eV. Thus, the compound understudy can be suggested as a potential material having possible applications in optoelectronics within VIS and UV regions. We note that in the VIS region, the absorption of Ba2CePtO6 is higher than that of Ba2ZrCeO6 reported by Kazim et al. (2022). As a result, Ba2CePtO6 is the most suitable contender for such uses because of its higher α(ω) values. Since the elevated α(ω) enhances the PV conversion efficiency, the observed high α(ω) (> 105 cm−1) with a broad absorption band in the VIS range suggests that Ba2CePtO6 material is a potential candidate for PV absorber.
3.5.4 Optical conductivity
The conduction of electrons, with respect to particular incident photon, is controlled by σ(ω). In the present work, the σ(ω) has been conducted for Ba2CePtO6 compound material. Our results are depicted in Fig. 4e. However, at lower energies, the σ(ω) spectra show a zero-conductivity correlated with the zero absorption α(ω). Likewise, the highest values of σ(ω) occur at nearly the same position of the maximum α(ω). Additionally, we can clearly see that when the incident photon energy is less than Eg, σ(ω) is zero, meaning that the incident photons do not interact much with the electrons of Ba2CePtO6 within that photon energy range. Afterwards, a sudden increase to the first extreme peak of about 883 (Ω.cm)−1 is observed in the VIS range at 2 eV. Furthermore, a multiple intense peaks of σ(ω) spectra indicate that Ba2CePtO6 is optically conductive in a wide range from VIS to UV and may have broad applications in this range. However, a maximum optical conductivity of 4658.24 (Ω.cm)−1 noticed at 8.12 eV denotes that the material is more photon absorbent in the UV range.
3.5.5 Optical reflectivity
The reflectivity spectra against photon energy, R(ω), computed for the Ba2CePtO6 is exposed in Fig. 4c. The zero frequency limit R(0) = R(ω → 0) is recorder as 15%. Then, the R(ω) increases with photon energy to extent a maximum value at UV region. The maximum reflectivity is found to occur when ε1(ω) goes below zero. Additionally, the most appreciable peaks values of the present R(ω) are noted in the energy ranging from 1.78 eV to afterwards. In the VIS spectrum, the material shows an insignificant maximum reflectivity of only 22% as its first reflectivity peak appear at around 1.78 eV indicating that the Ba2CePtO6 has a relatively high transparency in the VIS range and ensure the reliability of this material for optoelectronics devices applications.
3.5.6 Electron energy loss
The electron energy loss spectra L(ω) is also an indispensable optical parameter to be investigated. Usually this parameter is used to illustrate how fast electron travelling throughout a given homogeneous material loses energy in terms of scattering of light and heating of materials. Moreover, the interactions causing the loss of energy include mainly the intra-band and inter-band transitions together with both plasmon and phonon excitations. We note that, plasmons are collective oscillations of free electrons which cause sharp peaks in L(ω) spectrum. However, peaks in L(ω) diagram can also attributed to the inter-band transitions. Figure 4c shows L(ω) in the photon energy scope of 0–14 eV for the Ba2CePtO6 DPO. One can see that L(ω) spectra shows a set of peaks at different energy regions. Furthermore, the most appreciable peaks have been observed in the UV range. These peaks are derived from inter-band transitions whose widths are attributed to the phonon–phonon scattering. In the VIS range, the prominent peaks are located at around 2.28 eV. A nominal optical L(ω) in low energy side proves the usefulness of the present material for optoelectronics applications.
3.6 Thermoelectric transport properties
The current and promising applications of TE materials require the study of its heat-to-electricity conversion efficiency. Hence, understanding TE transport properties is of crucial importance in designing and improving the TE performances of materials. These can be evaluated by determining its TE parameters such as Seebeck coefficient (S), thermal conductivity (κ), electrical conductivity (σ), power factor (PF = S2σ) and TE figure of merit (ZT = σS2T/κ) at varying temperatures. The interdependency of TE transport properties of a material can be pursued from a number of factors related to the electronic band profile near the EF level. The key factor affecting the value of S is the occurrence of flat bands at the VBM which confirm the larger carrier’s effective mass. While, the σ is crucially enhanced by the existence of a wider dispersive band at the VBM implying lighter effective mass and improved mobility. Furthermore, the observed flat and wider dispersive bands at the VBM, a moderate semiconducting Eg and adequate amount of TDOS at the vicinity of EF confirm that the investigated compound is a promising candidate for TE applications. Consequently, these relevant results inspired us to explore its TE transport properties. As all these characteristics depend mainly on the carrier’s concentration and thus, they cannot be controlled independently. However, to clearly see the doping contribution on these parameters, TE parameters are investigated against chemical potential in the range of − 2 to 2 eV for three fixed temperatures, namely, 300 K, 600 K, and 900K. In this respect, we note that, the chemical potential describes the requisite quantity of energy for moving carriers against the faced Coulomb potential. Moreover, the positive value of chemical potential indicates n-type conductivity with an upward shift of the EF level, whereas the negative values are associated with the p-type conductivity with a downward shift of EF.
The chemical potential and the temperature based σ(T, µ), S(T, µ) and the electronic thermal conductivity κe(T, µ) are estimated by integrating the transport distribution function as follows (Ziman 2001; Allen 1996; Hurd 1972):
where μ, ε, T, e, V, f, α and β, correspond respectively to the chemical potential, the band energy, the absolute temperature, the electron charge, the volume of the primitive unit cell under investigation, the Fermi–Dirac function, and tensor independent indices. The coefficient of electrical conductivity σαβ(ε) is reported as (Madsen and Singh 2006):
where τi,k and v(i, k) refer respectively to the relaxation time and the group velocity.
Within the constant time relaxation (τ) approximation adopted here, S coefficient can be evaluated independently of any constraint. While, σ and κe is associated with the carrier relaxation time τ and therefore must be computed with respect to τ.
3.6.1 Seebeck coefficient
The S coefficient is a key measure of TE power of a material, or its ability to generate an electric potential due to the temperature difference across two contact materials. Moreover, a good TE material provides a large potential gradient induced by a small temperature variation between the two contacts. Furthermore, S can be referred to describe the carrier’s contribution in electronic transport. A positive magnitude of S exhibits p-type conduction due to the holes as dominant charge carriers; nevertheless S < 0 defines n-type conduction due to electrons. Figure 5a clearly shows the contribution of carriers to the S above (μ > μF) and below (μ < μF) the EF level at three chosen temperatures, namely, 300, 600 and 900 K. It is clear that S shows a variation between − 0.27 and 1.75 eV, while in the outside of this energy range, S has low and almost zero values. Moreover, the observed prominent peaks in the n-type side are consistent with the fact that in this energy range, the bands are less dispersive, which is a direct consequence of large effective mass, with the forbidden region in the vicinity of EF, in that way fewer charge carriers are present. For the considered temperatures, the magnitude of peaks reduces remarkably as the temperature rises and the highest possible value of |S|sup has been noticed at 300 K. This behavior demonstrates that the bound electrons are excited by acquiring a sufficient amount of thermal energy given rise to the electron–hole pairs. Moreover, the observed extreme values of the present S at different temperatures are listed in Table 5. As can be seen, the reported extreme values correspond to the narrow range of µ, thus they can be reached by a weak doping concentration which is useful in practice. While for higher µ, its value decreases gradually. In addition, from Table 5, we can notice that at the same temperature, the positive value of S is higher than the corresponding negative magnitude, signaling that Ba2CePtO6 compound reveals p-type conductivity.

Computed results of a seebeck coefficient (S) b electrical conductivity (σ/τ) c electronic thermal conductivity (κe/τ) d TE power factor (PF) and e TE figure of merit (ZT) of Ba2CePtO6 against μ–μF for three selected temperatures
3.6.2 Electrical conductivity
As seen in the Fig. 5b, for the selected temperatures, the increasing magnitude of µ for p-and n-type doped regions, σ/τ shows alternative peaks and valleys. Moreover, in the chemical potential range of 0.36–1.30 eV near the EF level it appears to have no conductivity as there are no energy bands and this region is devoid of charge carriers. However, outside this range, the Ba2CePtO6 reveals a high σ/τ. Furthermore, the chemical potential dependence of σ/τ is similar at different temperatures. In contrast to the S coefficient, the peak values of σ/τ do not vary greatly with temperature. Consequently, the temperature has an insignificant effect on σ/τ in the investigated chemical potential window. Moreover, because the σ/τ is inversely correlated to the carrier’s effective mass, the p-type doped region exhibits a relatively higher σ/τ than the n-type side owing to the observed light holes effective masse in the VB contrasted to electrons in the CB. Accordingly, the values of µ where σ/τ reaches its maximum value lie in a short range.
3.6.3 Electronic thermal conductivity
Another important parameter that influences the figure of merit is the thermal conductivity, κ, which needs to be reduced. This parameter corresponds to the rate of heat transferred trough unit area of material under the temperature gradient of 1 K per meter. Instinctively, a good κ prevent a large temperature gradient developing. It is well known that the flow in the compounds is mainly attributed to the carrier’s motion and lattice vibrations (phonons). Therefore, the net κ is predominately consisted of two main contributions: the electronic contribution, κe, caused by the carrier’s motion, and the lattice contribution due to the lattice phonons κL which induces elastic waves that trigger the carriers (κ = κe + κL). Therefore, in the present contribution we have ignored the phonon component while the electronic is only accountable. For this reason, we have adopted the BoltzTrap ((Boltzmann Transport Properties) code based on semi-classical Boltzmann transport theory, which computes only the electronic part, to synthetically investigate the electronic transport properties. As seen in Fig. 5c, for p-type doping region we remark that at a particular temperature, with increasing µ, κe/τ increases quickly with temperature contrasted to σ/τ. Also, with tuning chemical potential the κe/τ increases substantially with temperature, ensuring that the κe/τ is mainly attributable to the free carriers. Furthermore, the p-type doped side exhibits higher κe/τ than the n-type side. Moreover, with the same trend as for electrical conductivity, we can clearly see that κe/τ is almost zero in a certain range of positive chemical potential with a negligible variation for varying temperatures which forecast that the studied perovskite has a highly potential for TE devices.
3.6.4 Thermoelectric power factor
The TE response efficiency of materials is evaluated without incorporation of κe/τ by the TE power factor (PF = S2σ/τ). Therefore, a remarkable S and σ/τ are essential in a possible TE material with a superior power factor, though their coexistence is unusual. It is clear from Fig. 5d that, because of its high σ/τ, Ba2CePtO6 exhibits the highest PF in p-type doping region revealing that the electrons contribution in electronic transport is insignificant compared to the holes. Further, the observed pronounced peaks are directly connected to the doping level, indicating that the TE performance can be improved under appropriate doping levels. The predicted peak power factors values for p- and n-type regions are listed in Table 5. Both Fig. 5 and Table 5 demonstrate that in all circumstances, the PF shows the highest values at 900 K and it gradually decreases with temperature. The PF improvement with temperature is known to contribute positively to enhancing TE performances. Hence, the DPO under focus may find practicality in TE devices thanks to its higher PF values.
3.6.5 Figure of merit
The working efficiency of a TE material is specified in terms of the dimensionless quantity called the figure of merit (ZT = σS2T/κ). Further, a material is considered to be a relevant TE material if its ZT ≥ 1. On the basis of the above conferred electron transport properties, we have computed the figure of merit of Ba6CePtO6 DPO at different temperatures under p-and n-type doping levels. The obtained results are depicted in Fig. 5e. In n-type doping region, Fig. 5e indicates that the present compound exhibits a significant ZT over a wide range of chemical potential. Furthermore, ZT reaches its enhanced value of around unity noticed in the chemical potential windows where S becomes higher which is appreciable for TE applications. Similar to the PF, in the whole considered chemical potential range, all temperatures show similar trends. Moreover, owing to the enhancement of σ, two apparent peaks are present for the hole-doped material. Thus, ZT values exceed 0.841 and 0.998 at 300 K under an optimized p-type and n-type doping, respectively. Although, the relatively poor TE performance shown in p-type doping region, is mainly ascribed to the high thermal conductivity. Also, at the EF level, the obtained ZT value is approximately equal to 0.741 which is found to be temperature independent. The highest values of |ZT|sup in both p-and n-type doping regions of the proposed material have been recorded in Table 5. Accordingly, these findings suggest that for Ba2CePtO6 DPO, the improvement of ZT is expected with a proper n-type doping. Consequently, Ba2CePtO6 emphasizes a great potential as a high-temperature TE material.
4 Conclusion
Double perovskite oxides are promising materials for optoelectronic and thermoelectric applications. In this paper, the structural, optoelectronic, and thermoelectric (TE) features of SC Ba2CePtO6 are explored systematically. The studied material is a stable cubic structure, stiffer, ductile with ionic bonding character and anisotropic. Also, the reported melting and Debye temperatures ensure its strong bonding and retention even for elevated temperatures. The broad band of solar spectrum, Ba2CePtO6 is found to be optically conductive with substantial optical absorption, low reflectivity, low energy loss and high refractive index. Furthermore, the TE properties show a significant value of figure of merit (ZT). Accordingly, the reported results ensure the reliability of Ba2CePtO6 for optoelectronics and thermoelectric applications even at higher temperatures. However, owing to the deficiency in theoretical and experimental data, these findings offer new pathway for TE and optoelectronic technologies and are expected to serve as a stimulated reference for the innovative investigations of Ba2CePtO6.
References
Aczel, A.A., Clancy, J.P., Chen, Q., Zhou, H.D., Reig-i-Plessis, D., MacDougall, G.J., Ruff, J.P.C., Upton, M.H., Islam, Z., Williams, T.J., Calder, S., Yan, J.-Q.: Revisiting the Kitaev material candidacy of Ir4+ double perovskite iridates. Phys. Rev. B 99, 134417–134428 (2019)
Allen, P.B.: Boltzmann theory and resistivity of metals. In: Chelikowsky, J.R., Louie, S.G. (eds.) Quantum Theory of Real Materials, pp. 219–250. Klüwer, Boston (1996)
Al-Qaisi, S., Rai, D.P., Alshahrani, T., Ahmed, R., Haq, B.U., Tahir, S.A., Khuili, M., Mahmood, Q.: Structural, elastic, thermodynamic, electronic, optical and thermoelectric properties of MgLu2X4 (X= S, Se) spinel compounds from ab-initio calculations. Mater. Sci. Semicond. 128, 105766–105777 (2021)
Al-Qaisi, S., Mushtaq, M., Alzahrani, J.S., Alkhaldi, H., Alrowaili, Z.A., Rached, H., Bakhtiar, U.H., Mahmood, Q., Al-Buriahi, M.S., Morsi, M.: First-principles calculations to investigate electronic, structural, optical, and thermoelectric properties of semiconducting double perovskite Ba2YBiO6. Micro. Nano 170, 207397–207407 (2022a)
Al-Qaisi, S., Malak, A.A., Tahani, A.A., Tuan, V.V., Morsi, M., Bakhtiar, U.H., Ahmed, R., Mahmood, Q., Afzal Tahir, S.: First-principles investigations of Ba2NaIO6 double perovskite semiconductor: material for low-cost energy technologies. Mater. Chem. Phys. 275, 125237–125247 (2022b)
Al-Qaisi, S., Rached, H., Ali, M.A., Abbas, Z., Alrebdi, T.A., Hussein, K.I., Khuili, M., Rahman, N., Verma, A.S., Ezzeldien, M., Morsi, M.: A comprehensive first-principles study on the physical properties of Sr2ScBiO6 for low-cost energy technologies. Opt. Quantum Electron. 55, 1015–1033 (2023)
Amador, U., Hetherington, C.J.D., Moran, E., Alario-Franco, M.A.: Ba2PrPtO6: a novel double perovskite. J. Solid State Chem. 96, 132–140 (1992)
Anderson, M.T., Greenwood, K.B., Taylor, G.A., Poeppelmeier, K.R.: B-cation arrangements in double perovskites. Prog. Solid State Chem. 22, 197–233 (1993)
Bairwa, J.K., Rani, M., Kamlesh, P.K., Singh, R., Rani, U., Al-Qaisi, S., Kumar, T., Kumari, S., Verma, A.S.: Modeling and simulation of multifaceted properties of X2NaIO6 (X = Ca and Sr) double perovskite oxides for advanced technological applications. J. Mol. Model. 29, 379–391 (2023)
Behera, D., Al-Qaisi, S., Manzoor, M., Sharma, R., Srivastava, V., mana Al-Anazy, M., El Shiekh, E., Mukherjee, S.K.: First principles studies on optoelectronics and transport properties of KSrY (Y = Sb, Bi) for renewable energy application. Mater. Sci. Eng. B 297, 116765–116776 (2023)
Bharti, C., Sinha, T.P.: Dielectric properties of rare earth double perovskite oxide Sr2CeSbO6. Solid State Sci. 12, 498–502 (2010)
Bharti, C., Sinha, T.P.: Electrical analysis of a newly synthesized rare earth double perovskite oxide: Sr2CeNbO6. Phys. b: Condens. 406, 624–627 (2011a)
Bharti, C., Sinha, T.P.: Synthesis, structure and dielectric properties of a rare earth double perovskite oxide Ba2CeTaO6. Mater. Res. Bull. 46, 1431–1436 (2011b)
Bharti, C., Sinha, T.P.: Structural and ac electrical properties of a newly synthesized single phase rare earth double perovskite oxide: Ba2CeNbO6. Phys. b: Condens. 406, 1827–1832 (2011c)
Bildirici, M.E., Gökmenoğlu, S.M.: Environmental pollution, hydropower energy consumption and economic growth: evidence from G7 countries. Renew. Sust. Energy Rev. 75, 68–85 (2017)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2K: An Augmented Plane Wave and Local Orbitals Program for Calculating Crystal Properties. Vienna University of Technology, Austria (2001)
Bobrov, V.B., Trigger, S.A., Van Heijst, G.J.F., Schram, P.P.J.M.: Kramers–Kronig relations for the dielectric function and the static conductivity of Coulomb systems. EPL 90, 10003–10007 (2010)
Bougherara, K., Al-Qaisi, S., Laref, A., Vu, T.V., Rai, D.P.: Ab initio insight of the electronic, structural, mechanical and optical properties of X3P2 (X= Mg, Ca) from GGA and hybrid functional (HSE06). J. Supercond. Nov. Mag. 35, 79–86 (2022)
Casapu, M., Grunwaldt, J.D., Maciejewski, M., Baiker, A., Eckhoff, S., Göbel, U., Wittrock, M.: The fate of platinum in Pt/Ba/CeO2 and Pt/Ba/Al2O3 catalysts during thermal aging. J. Catal. 251, 28–38 (2007)
Chen, L., Yuan, C., Xue, J., Wang, J.: B-site ordering and magnetic behaviours in Ni doped double perovskite Sr2FeMoO6. J. Phys. D Appl. Phys. 38, 4003–4008 (2005)
Chen, P., Podraza, N.J., Xu, X.S., Melville, A., Vlahos, E., Gopalan, V., Ramesh, R., Schlom, D.G., Musfeldt, J.L.: Optical properties of quasi-tetragonal BiFeO3 thin films. Appl. Phys. Lett. 96, 131907–131909 (2010)
Dar, S.A., Sharma, R., Srivastava, V., Sakalle, U.K.: Investigation on the electronic structure, optical, elastic, mechanical, thermodynamic and thermoelectric properties of wide band gap semiconductor double perovskite Ba2InTaO6. RSC Adv. 9, 9522–9532 (2019)
Das, H., De Raychaudhury, M., Saha-Dasgupta, T.J.A.P.L.: Moderate to large magneto-optical signals in high Tc double perovskites. Appl. Phys. Lett. 92, 201912–201914 (2008)
Dutta, A., Mukhopadhyay, P.K., Sinha, T.P., Dipankar, D., Shannigrahi, S.: Structural and magnetic properties of double perovskite oxide Ba2CeSbO6. Solid State Sci. 58, 64–69 (2016)
Faizan, M., Murtaza, G., Khan, S.H., Khan, A., Mehmood, A., Khenata, R., Hussain, S.: First-principles study of the double perovskites Sr2XOsO6 (X= Li, Na, Ca) for spintronics applications. Bull. Mater. Sci. 39, 1419–1425 (2016)
Fu, W.T., Ijdo, D.J.W.: X-ray and neutron powder diffraction study of the double perovskites Ba2LnSbO6 (Ln= La, Pr, Nd and Sm). J. Solid State Chem. 178, 2363–2367 (2005)
Gajdoš, M., Hummer, K., Kresse, G., Furthmüller, J., Bechstedt, F.: Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 73, 045112–045120 (2006)
Hatakeyama, T., Takeda, S., Ishikawa, F., Ohmura, A., Nakayama, A., Yamada, Y., Matsushita, A., Yea, J.: Photocatalytic activities of Ba2RBiO6 (R = La, Ce, Nd, Sm, Eu, Gd, Dy) under visible light irradiation. J. Ceram. Soc. Jpn. 118, 91–95 (2010)
Hilal, M., Rashid, B., Khan, S.H., Khan, A.: Investigation of electro-optical properties of InSb under the influence of spin-orbit interaction at room temperature. Mater. Chem. Phys. 184, 41–48 (2016)
Hill, R.: The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. a. 65, 349–354 (1952)
Himanshu, A.K., Kumar, S., Dey, U., Ray, R.: Multiple low-energy excitons and optical response of d0 double perovskite Ba2ScTaO6. Phys. b: Condens. 637, 413856–413863 (2022)
Hurd, C.M.: The Hall Effect in Metals and Alloys. Plenum Press, New York-London (1972)
Ivanovski, K., Hailemariam, A., Smyth, R.: The effect of renewable and non-renewable energy consumption on economic growth: non-parametric evidence. J. Clean. Prod. 286, 124956–124970 (2021)
Jasiukiewicz, C., Karpus, V.: Debye temperature of cubic crystals. Solid State Commun. 128, 167–169 (2003)
Kato, H., Okuda, T., Okimoto, Y., Tomioka, Y., Takenoya, Y., Ohkubo, A., Kawasaki, M., Tokura, Y.: Metallic ordered double-perovskite Sr2CrReO6 with maximal Curie temperature of 635 K. Appl. Phys. Lett. 81, 328–330 (2002)
Kazim, M.Z., Yassen, M., Shatha, A.A., Mudassir, I., Mubashar, N., Zahid, M., Neffati, R.: DFT study of optoelectronic and thermoelectric properties of cubic Ba2ZrMO6 (M=Ce, Ti) double perovskites. J. Solid State Chem. 315, 123419–123427 (2022)
Kockelmann, W., Adroja, D.T., Hillier, A.D., Wakeshima, M., Izumiyama, Y., Hinatsu, Y., Knight, K.S., Visser, D., Rainford, B.D.: Neutron diffraction and inelastic neutron scattering investigations of the ordered double perovskite Ba2PrIrO6. Phys. b: Condens. 378, 543–545 (2006)
Korchagina, S.K., Shevchuk, Y.A.: Low-frequency and microwave dielectric properties of Ba2LnTaO6 (Ln= La, Pr, Sm, Dy, Ce, Gd, Nd, Tm, Tb) ceramics. Inorg. Mater. 42, 64–67 (2006)
Li, X.Q., Zhao, J.J., Xu, J.C.: Mechanical properties of bcc Fe–Cr alloys by first-principles simulations. Front. Phys. 7, 360–365 (2012)
Lufaso, M.W., Barnes, P.W., Woodward, P.M.: Structure prediction of ordered and disordered multiple octahedral cation perovskites using SPuDS. Acta Crystallogr. b: Struct. Sci. Cryst. Eng. Mater. 62, 397–410 (2006)
Madsen, G.K., Singh, D.J.: BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 175, 67–71 (2006)
Madsen, G.K., Blaha, P., Schwarz, K., Sjöstedt, E., Nordström, L.: Efficient linearization of the augmented plane-wave method. Phys. Rev. B 64, 195134–195142 (2001)
Mantziaris, S., Iliopoulos, C., Theodorakopoulou, I., Petropoulou, E.: Perennial energy crops vs. durum wheat in low input lands: economic analysis of a Greek case study. Renew. Sust. Energy Rev. 80, 789–800 (2017)
Mebed, A., Mushtaq, M., Muhammad, I., Lone, I.U.N., Samah, A.Q., Algethami, N., EL-Shamy, E.F., Laref, A., AL-Hosiny, N.M.: Structure, half-metallic and magnetic properties of bulk and (001) surface of Rb2XMoO6 (X = Cr, Sc) double perovskites: a DFT + U study. Phys. Scr. 98, 015807–015820 (2023)
Mostafaeipour, A., Bidokhti, A., Fakhrzad, M.B., Sadegheih, A., Mehrjerdi, Y.Z.: A new model for the use of renewable electricity to reduce carbon dioxide emissions. Energy 238, 121602 (2022)
Murnaghan, F.D.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. u.s.a. 30, 244–247 (1944)
Ouchetto, K., Archaimbault, F., Pineau, A., Choisnet, J.: Chemical and structural characterization of a new barium ceroplatinate: Ba2CePtO6 a double perovskite mixed oxide. J. Mater. Sci. 10, 1277–1279 (1991)
Penn, D.R.: Wave-number-dependent dielectric function of semiconductors. Phys. Rev. 128, 2093–2097 (1962)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
Pettifor, D.: Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 8, 345–349 (1992)
Pugh, S.: XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 45, 823–843 (1954)
Rammeh, N.: Crystal structure, electronic and magnetic properties of double perovskite Ba2FeWO6: a combined experimental–theoretical study. Phys. b: Condens. 481, 217–223 (2016)
Rani, U., Kamlesh, P.K., Joshi, T.K., Sharma, S., Gupta, R., Al-Qaisi, S., Verma, A.S.: Alkaline earth based antiperovskite AsPX3 (X= Mg, Ca, and Sr) materials for energy conversion efficient and thermoelectric applications. Phys. Scr. 98, 075902–075920 (2023)
Ray, R., Himanshu, A.K., Sen, P., Kumar, U., Richter, M., Sinha, T.P.: Effects of octahedral tilting on the electronic structure and optical properties of d0 double perovskites A2ScSbO6 (A= Sr, Ca). J. Alloys Compd. 705, 497–506 (2017)
Reddy, Y., Suresh, Y., Markandeya, B., Appa, R., Bhikshamaiah, G.: Characterization and impedance study of Ba2CeZrO6 double perovskite. J. Mater. Sci. Mater. Electron. 29, 2966–2973 (2018)
Retuerto, M., Alonso, J.A., Martínez-Lope, M.J., Martínez, J.L., García-Hernández, M.: Record saturation magnetization, Curie temperature, and magnetoresistance in Sr2FeMoO6 double perovskite synthesized by wet-chemistry techniques. Appl. Phys. Lett. 85, 266–268 (2004)
Retuerto, M., Alonso, J.A., García-Hernández, M., Martínez-Lope, M.J.: Synthesis, structure and magnetic properties of the new double perovskite Ca2CrSbO6. Solid State Commun. 139, 19–22 (2006)
Saha-Dasgupta, T.: Double perovskites with 3d and 4d/5d transition metals: compounds with promises. Mater. Res. Express. 7, 014003–014015 (2020)
Schreiber, E., Anderson, O.L., Soga, N., Bell, J.F.: Elastic constants and their measurement. J. Appl. Mech. 42, 747–748 (1975)
Schwarz, K., Blaha, P., Madsen, G.K.: Electronic structure calculations of solids using the WIEN2k package for material sciences. Comput. Phys. Commun. 147, 71–76 (2002)
Slavney, A.H., Hu, T., Lindenberg, A.M., Karunadasa, H.I.: A bismuth-halide double perovskite with long carrier recombination lifetime for photovoltaic applications. J. Am. Chem. Soc. 138, 2138–2141 (2016)
Snaith, H.J.: Perovskites: the emergence of a new era for low-cost, high-efficiency solar cells. J. Phys. Chem. Lett. 4, 3623–3630 (2013)
Soukiassian, A., Tian, W., Vaithyanathan, V., Haeni, J.H., Chen, L.Q., Xi, X.X., Schlom, D.G., Tenne, D.A., Sun, H.P., Pan, X.Q., Choi, K.J.: Growth of nanoscale BaTiO3/SrTiO3 superlattices by molecular-beam epitaxy. J. Mater. Res. 23, 1417–1432 (2008)
Tarbi, A., Chtouki, T., Bouich, A., Elkouari, Y., Erguig, H., Migalska-Zalas, A., Aissat, A.: InP/InGaAsP thin films based solar cells: lattice mismatch impact on efficiency. Opt. Mater. 131, 12704–12711 (2022)
Tarbi, A., Chtouki, T., Sellam, M.A., Elkouari, Y., Erguig, H., Migalska-Zalas, A.: Predicting the bandgap energy of distorted GaSbxAs1-x and InSbxAs1-x using design of experiment (DoE) and artificial intelligence (AI): a comparative study. J. Phys. Chem. Solids 175, 111180–111186 (2023a)
Tarbi, A., Chtouki, T., Erguig, H., Migalska-Zalas, A., Aissat, A.: Modeling and optimization of Sb and N resonance states effect on the band structure of mismatched III-NV alloys using artificial neural networks. Mater. Sci. Eng. B 290, 116312–116321 (2023b)
Tarbi, A., Chtouki, T., Sellam, M.A., Benahmed, A., El Kouari, Y., Erguig, H., Migalska-Zalas, A., Goncharova, I., Taboukhat, S.: Optimization of ultra-thin CIGS-based solar cells by strained In1−xGaxAs absorption layer: 1D SCAPS modeling. J. Comput. Electron. 22, 1–8 (2023c)
Tariq, M., Ali, M.A., Laref, A., Murtaza, G.: Anion replacement effect on the physical properties of metal halide double perovskites Cs2AgInX6 (X= F, Cl, Br, I). Solid State Commun. 314, 113929–113936 (2020)
Tran, F., Blaha, P.: Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401–226405 (2009)
Vasala, S., Karppinen, M.: A2B′B″O6 perovskites: a review. Prog. Solid State Chem. 43, 1–36 (2015)
Wang, J., Yip, S., Phillpot, S.R., Wolf, D.: Crystal instabilities at finite strain. Phys. Rev. Lett. 71, 4182–4185 (1993)
Wu, M., Fang, L., Liu, L., Li, G., Elouadi, B.: Dielectric and ferroelectric properties of (1–x) BiFeO3-xBi0.5Na0.5TiO3 solid solution. Ferroelectrics 478, 18–25 (2015)
Yin, W.J., Weng, B., Ge, J., Sun, Q., Li, Z., Yan, Y.: Oxide perovskites, double perovskites and derivatives for electrocatalysis, photocatalysis, and photovoltaics. Energy Environ. Sci. 12, 442–462 (2019)
Ziman, J.M.: Electrons and Phonons: The Theory of Transport Phenomena in Solids. Oxford University Press, London (2001)
Acknowledgements
The author (Tahani A. Alrebdi) extends her sincere appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R71), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through research groups program under grant number (RGP.2/583/44).
Funding
No funding received.
Author information
Authors and Affiliations
Contributions
Abderrazak Boutramine, Samah Al-Qaisi, Malak Azmat Ali, Tahani A. Alrebdi, wrote the main manuscript. Afaf Khadr Alqorashi, Ajay Singh Verma, Zeesham Abbas prepared figures. El Sayed Yousef, Ramesh Sharma, Muhammad Mushtaq reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Boutramine, A., Al-Qaisi, S., Ali, M.A. et al. A theoretical investigation of the Ba2CePtO6 double perovskite for optoelectronic and thermoelectric applications. Opt Quant Electron 56, 395 (2024). https://doi.org/10.1007/s11082-023-06113-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-06113-9