
Abstract
Thiosemicarbazones are considered as emerging scaffolds for various research domains due to their diverse applications. Herein, novel thiosemicarbazone based compounds 1–5 were derived from the condensation reaction between salicylaldehyde and differently substituted thiosemicarbazides with an excellent yield. The structures of yielded chromophores have been confirmed by UV–Vis, vibrational (IR) and NMR spectral techniques. Besides this, DFT study was also accomplished to elucidate the electronic properties of 1–5 chromophores at M06/6-311G(d,p) level. Different kind of investigation: natural bond orbital (NBO), natural population analysis (NPA), frontier molecular orbitals (FMOs), molecular electrostatic potential (MEP) and global reactivity parameters (GRPs) were determined to explore the NLO assets of chromophores. The NBO analysis elucidated that the intramolecular interactions, electron delocalization and hyper conjugative interactions played a significant role in stabilizing the compounds. The NPA and MEP analyses were performed for predicting the charge distributions for the atoms and chemical reactivity regions of studied compounds. The GRPs were calculated utilizing FMOs energies and compound 1 was found as soft, less stable, more reactive molecule from other compounds The non-linear optical (NLO) findings exploited that 1–5 compounds showed 40.6, 34.2, 34.6, 35.4 and 29.1 times greater hyperpolarizability than the urea and can be utilized as efficient optoelectronic materials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Thiosemicarbazones, also considered as Schiff bases related to a group of thiourea derivatives, are extensively studied compounds because of their broad applications in biological activities, industries and analytical determinations (Subhashree et al. 2017; Mysliwiec et al. 2009). The biological activities of thiosemicarbazones comprise antibacterial, antifungal (Kumar and Arunachalam 2009), antitumor (Silveira et al. 2008; Zhong et al. 2006), anticancer (Desai et al. 2001), antidiabetic, anti-inflammatory (Vančo et al. 2008), antiproliferative (Prajapati and Patel 2019), and herbicidal (Samadhiya and Halve 2001) activities. The analytical applications of thiosemicarbazones involve the sensitive and selective detection of metal ions in which thiosemicarbazone ligands act as chelating agents for metal ions and form colored complexes. Intended for the analytical recognition of metal ions, spectrophotometric (Reddy et al. 2003; Arroudj et al. 2016), potentiometric and stripping voltammetric techniques are mainly used (Mahajan et al. 2006). From the last few years, computational chemistry has been emerging as a significant implement for chemists and as a well-accepted companion with experimental chemistry. The DFT analysis has mainly been used for the theoretically detailed investigation of molecules (Shahab et al. 2017a, 2017b; Khalid et al. 2020, 2023a; Mahmood et al. 2023).
The NLO materials have attained noteworthy consideration due to their number of functions in laser technology, molecular switches, data storage, microfabrication, imaging, spectroscopic and electrochemical sensors (Forrest 1997; Achelle et al. 2013; Cinteza and Marinescu 2018; Mirershadi et al. 2016; Iliopoulos et al. 2012; Mehmood et al. 2023; Mahmood et al. 2015a). Organic materials possess a high degree of delocalization and exhibit potential NLO properties than inorganic materials (Kamanina and Plekhanov 2002; Likhomanova and Kamanina 2016; Kamanina et al. 2012; Kamanina 2001; Denisov 2009; Shafiq et al. 2023). A strong push–pull system in molecules along with donor‒π‒acceptor (D‐π‐A) conjugation improves the molecular polarizability which will further increase NLO responses (Palanisami and Moon 2014; Bullo et al. 2023; Khalid et al. 2021a, 2019a; Mahmood et al. 2015b).
The present report is based on the synthesis of different substituted novel salicylaldehyde-based thiosemicarbazone moieties and their characterization was performed via the use of diverse spectroscopic approaches. The DFT method is used for a thorough exploration of optoelectronic properties.
2 Materials and methods
The chemicals and solvents were purchased from Merck that was in pure form and no need to be purified before use. For the determination of melting points Stuart SMP 10 melting point apparatus was used. FT-IR spectrum was obtained from FT-IR spectrophotometer Tensor 27 using KBr pellets. The UV–Visible spectrophotometer BK D560 was used for electronic spectral analysis using ethanol solvent. The 1H and 13C NMR spectra were obtained by utilizing DMSO as solvent and TMS as internal standard on a Bruker AM 300 spectrophotometer at 300 MHz. Thin-layer chromatography was executed using aluminum sheets coated with silica gel. The mobile phase used for TLC was n-hexane and ethyl acetate in 3:1 quantity.
2.1 Synthesis of Thiosemicarbazones
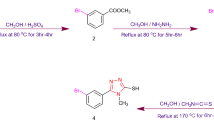
A condensation reaction of salicylaldehyde with different substituted thiosemicarbazides produced thiosemicarbazone compounds 1–5. The ethanolic solution (5 ml) of substituted thiosemicarbazide (0.01 mol) was mixed with the ethanolic solution (5 ml) of salicylaldehyde (0.01 mol) by adding few drops of glacial acetic acid as a catalyst. The mixture was heated and refluxed with continual stirring at 80–100 °C for four to five hours or until the formation of white color precipitates. The TLC was used to observe the reaction and check the purity of the product. The mixture was cooled and the resulting precipitates of products were filtered, washed, dried and then recrystallized from ethanol. The graphical synthesis representation of entitled compounds is illustrated in Scheme 1

Synthesis of compounds 1–5
3 Experimental
3.1 (E)-2-(2-hydroxybenzylidene)-N-(2-methylbenzyl)hydrazinecarbothioamide (1)
C16H17N3SO, White amorphous, Yield: 86%, M.p: 212–214 °C. UV–Vis (Ethanol) λmax: 344 nm. IR (KBr) ν (cm−1): 3005 (O–H), 3322 (N–H), 1539 (C=N), 1258 (C=S). 1H NMR (DMSO, 300 MHz) δ (ppm): 2.32 (s, 3H, C–H3), 4.79 (d, J = 4.8 Hz, 2H, Ph–CH2), 8.42 (s, 1H, O–H), 8.87 (t, J = 4.5 Hz, 1H, NH–CS), 9.95 (s, 1H, N=CH), 11.56 (s, 1H, NH–N), 6.82–7.95 (m, Ar–H). 13C NMR (DMSO, 300 MHz) δ (ppm): 21.3 (CH3), 46.4 (CH2), 158.7 (C=N), 177.7 (C=S), 114.0–140.0 (Ar–C).
3.2 (E)-2-(2-hydroxybenzylidene)-N-(3-methylbenzyl)hydrazinecarbothioamide (2)
C16H17N3SO, White amorphous, Yield: 82%, M.p: 214–215 °C. UV–Vis (Ethanol) λmax: 341 nm. IR (KBr) ν (cm−1): 3008 (O–H) 3323 (N–H), 1519 (C=N), 1229 (C=S). 1H NMR (DMSO, 300 MHz) δ (ppm): 2.50 (s, 3H, C–H3), 4.80 (d, J = 4.5 Hz, 2H, Ph–CH2), 8.40 (s, 1H, O–H), 8.97 (t, J = 4.8 Hz, 1H, NH–CS), 9.91 (s, 1H, N=CH), 11.51 (s, 1H, NH–N), 6.81–7.96 (m, Ar–H). 13C NMR (DMSO, 300 MHz) δ (ppm): 21.6 (CH3), 47.0 (CH2), 156.9(C=N), 177.7 (C=S), 116.5–140.0 (Ar–C).
3.3 (E)-2-(2-hydroxybenzylidene)-N-(4-methylbenzyl)hydrazinecarbothioamide (3)
C16H17N3SO, White amorphous, Yield: 84%, M.p: 206–208 °C. UV–Vis (Ethanol) λmax: 342 nm. IR (KBr) ν (cm−1): 3313 (O–H), 3398 (N–H), 1538 (C=N), 1296 (C=S). 1H NMR (DMSO, 300 MHz) δ (ppm): 2.50 (s, 3H, C–H3), 4.76 (d, J = 3.3 Hz, 2H, Ph–CH2), 8.40 (s, 1H, O–H), 8.93 (t, J = 4.5 Hz, 1H, NH–CS), 9.91 (s, 1H, N=CH), 11.50 (s, 1H, NH–N), 6.81–7.95 (m, Ar–H). 13C NMR (DMSO, 300 MHz) δ (ppm): 21.1 (CH3), 46.8 (CH2), 158.7 (C=N), 177.3 (C=S), 114.0–140.0 (Ar–C).
3.4 (E)-2-(2-hydroxybenzylidene)-N-(2,4-dichlorobenzyl)hydrazinecarbothioamide (4)
C15H14N3SOCl2, White amorphous, Yield: 79%, M.p: 191–192 °C. UV–Vis (Ethanol) λmax: 342 nm. IR (KBr) ν (cm−1): 3137 (O–H), 3394 (N–H), 1521 (C=N), 1228 (C=S). 1H NMR (DMSO, 300 MHz) δ (ppm): 4.70 (d, J = 4.2 Hz, 2H, Ph–CH2), 8.08 (s, 1H, O–H), 8.50 (t, J = 4.5, 1H, NH–CS), 10.05 (s, 1H, N=CH), 11.77 (s, 1H, NH–N), 6.83–7.58 (m, Ar–H). 13C NMR (DMSO, 300 MHz) δ (ppm): 47.1 (CH2), 157.1 (C=N), 176.2 (C=S), 116.5–140.6 (Ar–C).
3.5 (E)-2-(2-hydroxybenzylidene)-N-(3,4-dichlorobenzyl)hydrazinecarbothioamide (5)
C15H14N3SOCl2, White amorphous, Yield: 77%, M.p: 195–197 °C. UV–Vis (Ethanol) λmax: 342 nm. IR (KBr) ν (cm−1): 3135 (O–H), 3386 (N–H), 1515 (C=N), 1226 (C=S). 1H NMR (DMSO, 300 MHz) δ (ppm): 4.81 (d, J = 4.2 Hz, 2H, Ph–CH2), 8.42 (s, 1H, O–H), 8.53 (t, J = 4.5, 1H, NH–CS), 9.94 (s, 1H, N=CH), 11.62 (s, 1H, NH–N), 6.81–7.98 (m, Ar–H). 13C NMR (DMSO, 300 MHz) δ (ppm): 46.0 (CH2), 156.9(C=N), 177.9 (C=S), 116.5–141.4 (Ar–C).
3.6 Computational methodology
The complete theoretical calculations for compounds 1–5 were accompanied utilizing Gaussian 09 program package (Gaussian 2009). The DFT (Braga et al. 2006, 2005; García-Melchor et al. 2013; Guest et al. 2015) study was executed using M06 functional with 6-311G (d, p) basis set. At first, global minimum geometries (Figure S22) were obtained through optimization at above-mentioned functional then these structures were utilized for further investigations. The NBO (Reed et al. 1988) analysis was carried out with the embedded package of the NBO 3.1 program. TD-DFT study was used at M06/6-311G (d, p) level for the UV–Vis and FMO analysis. The global reactivity descriptors were examined by utilizing the HOMO and LUMO energy values. The NLO (Muhammad et al. 2018, 2017, 2016) analysis was attained at the same level of theory. The GaussView 5.0 (Dennington et al. 2009) was used to organize the input files. The interpretation of output files was carried out through Avogadro (Cerqueira 2016), GaussView 5.0 (Frisch et al. 2000), Gauss sum (N.M. O'boyle et al. 2008) and the Chemcraft program (Zhurko 2014). The 3D representation of entitled chromophores along xyz-axes was illustrated in Figure S21.
4 Result and discussion
The synthesized compounds 1–5 were solid and soluble in ethanol and dimethyl sulfoxide solvent and the percentage yield varies from 77 to 86%. The structure of the newly prepared compounds was investigated by the inspection of their spectral data.
UV–Vis spectra exhibited intense absorption bands at 344, 341, 342, 342 and 342 nm for the entitled compounds. FT-IR spectra showed the O–H vibrational frequencies in the region of 3005–3313 cm−1 as characteristic intense bands due to the phenolic group. The spectra exhibited bands at 3322–3398 cm−1 were allocated to the N–H group. The vanishing of carbonyl (C=O) band in their characteristics frequency region and appearance of new vibrational band at 1515–1539 cm−1 accredited to the imine (C=N) group confirmed the formation of thiosemicarbazone compounds. The band corresponding to the thiocarbonyl (C=S) group vibrations were noticed at the 1226–1296 cm−1 region.
A singlet peak in the shielded region of 1H NMR spectrum appeared at 2.322–2.498 ppm is assigned to methyl (C–H3) protons. One proton singlet at the region of 8.078–8.422 ppm, ascribed as the phenolic (O–H) proton. The spectrum shows triplet in the region 8.495–8.966 ppm due to the NH–CS proton. The singlet of imine (N=CH) proton is displayed at region of 8.966–10.052 ppm. The NH–N proton is observed as a singlet at 11.503–11.766 ppm region. The multiplet of aromatic protons has appeared in the expected region of 6.809–7.979 ppm has displayed peak of methyl (CH3) carbon at 21.171–21.568 ppm region. The peak in the region of 45.993–47.125 ppm is related to the methylene (CH2) carbon. The aromatic carbons emerged in the expected range of 114.029–141.421 ppm. The spectra showed the peak of imine (C=N) and thiocarbonyl (C=S) carbons at 156.871–158.663 and 176.196–177.917 ppm, correspondingly. 1H NMR and 13C NMR spectrum of compounds 1–5 are displayed in Figure S11-20.
4.1 Natural bond orbital (NBO) analysis
The NBO analysis is an efficacious way for studying the interactions between bonds in both filled and empty orbitals and also offers a useful basis for exploring the conjugative interactions in molecular structures (Snehalatha et al. 2009). The outcome of an interaction is an occupancy loss from the localized NBO (idealized Lewis structure) to a vacant NBO (non-Lewis structure). The second-order perturbation interaction energy E(2) is used to clearly express bonding (donor) and anti-bonding (acceptor) interaction in NBO approaches. Each type of NBO donor (i) and acceptor (j) interactions and their stabilization energies related to electron delocalization between donor and acceptor is predicted from the second-order Fock-matrix approach as given below.
where εi and εj denotes as diagonal elements (orbital energies), qi symbolizes the donor orbital occupancy and Fi,j is the off-diagonal Fock matrix element among the NBOs (Inkaya et al. 2013). The higher value of E(2) demonstrates that the interaction is more intensive among donor and acceptor orbitals. The more affinity of electrons donation from donor to acceptor increases the degree of conjugation in a complete molecule (Weinhold and Landis 2001).
NBO analysis has been executed for studied compounds 1–5 at the DFT/M06/6-311G(d,p) level to determine the interactions between orbitals and the stabilization energies of molecules and the results are tabularized in Tables S1-S5.
The significant interactions such as \(\sigma \to {\sigma }^{*}\), \({\pi \to \pi }^{*}\), \(\mathrm{LP}\to {\sigma }^{*}\), \(\mathrm{LP}\to {\pi }^{*}\) are investigated for each titled compound. Among these four types of transitions, the most noticeable transition is \({\pi \to \pi }^{*}\) due to its higher stabilization energy values than other transitions and causes the extended conjugation in the molecular system. The \(\mathrm{LP}\to {\sigma }^{*}\), \(\mathrm{LP}\to {\pi }^{*}\) transitions are moderate dominant and \(\sigma \to {\sigma }^{*}\) transitions are slightly dominant transitions.
The \({\pi \to \pi }^{*}\) interactions are the most substantial interactions due to their greater values of stabilization energies causes extended conjugation in the molecular system and stabilized the entire molecule. The significant interactions noticed in compounds 1–5 are \(\pi\)(C31-C33)\(\to {\pi }^{*}\)(C27-C29), \(\pi\)(C25-C27)\(\to {\pi }^{*}\)(C21-C23),), \(\pi\)(C23-C25)\(\to {\pi }^{*}\)(C19-C21), \(\pi\)(C22-C24)\(\to\) \({\pi }^{*}\)(C18-C20) and \(\pi\)(C22-C24)\(\to\) \({\pi }^{*}\)(C18-C20) with the stabilization energy values 25.95, 26.12, 26.10, 26.08 and 26.54 kcal/mol, respectively. Further, some other remarkable \({\pi \to \pi }^{*}\) interactions observed in compounds 1–5 are \(\pi\)(C27-C29)\(\to {\pi }^{*}\)(C28-C30), \(\pi\)(C3-C33)\(\to\) \({\pi }^{*}\)(C4-C5), \(\pi\)(C19-C21)\(\to\) \({\pi }^{*}\)(C20-C22),), \(\pi\)(C19-C21)\(\to\) \({\pi }^{*}\)(C22-C24) and \(\pi\)(C19-C21)\(\to\) \({\pi }^{*}\)(C22-C24) with stabilization energy values 24.78, 24.87, 24.60, 25.51 and 25.87 kcal/mol, correspondingly. In contrast, the interactions originate from \(\sigma\) (donor) \(\to {\sigma }^{*}\) (acceptor) are weak interactions such as \(\sigma\)(N18-H19)\(\to {\sigma }^{*}\)(C20-N22), \(\sigma\)(N12-H13)\(\to\) \({\sigma }^{*}\)(C14-N16), \(\sigma\)(C12-S13)\(\to\) \({\sigma }^{*}\)(C12-S13), \(\sigma\)(C11-S12)\(\to {\sigma }^{*}\)(C11-S12) and \(\sigma\)(C11-S12)\(\to {\sigma }^{*}\)(C11-S12) having the least values of stabilization energy 7.03, 6.52, 5.73, 6.92 and 7.43 kcal/mol, respectively in compounds 1–5. Similarly, the resonance energy values in compounds 1–5 are 32.26, 33.38, 33.23, 32.15 and 32.62 kcal/mol are exhibited by the interactions LP1(N22)\(\to {\pi }^{*}\)(N24-C25), LP2(O31)\(\to {\pi }^{*}\)(C22-C24), LP2(O29)\(\to {\pi }^{*}\)(C20-C22), LP2(O28)\(\to {\pi }^{*}\)(C19-C21) and LP2(O28)\(\to {\pi }^{*}\)(C19-C21), respectively. Moreover, LP1(N18)\(\to {\sigma }^{*}\)(C20-S21), LP1(N12)\(\to\) \({\sigma }^{*}\)(C14-S15), LP1(N10)\(\to {\sigma }^{*}\)(C12-S13), LP1(N9)\(\to {\sigma }^{*}\)(C11-S12) and LP1(N13)\(\to {\sigma }^{*}\)(C11-S12) are also observed in compounds 1–5 with stabilization energy values 81.30, 77.51, 78.26, 66.15 and 41.00 kcal/mol, respectively (Tables S1-S5).
The ICT results in stabilization of the studied molecules due to the interactions formed by the overlapping of the donor and the acceptor orbitals. The transfer of electronic cloud from donor towards acceptor orbital causes ICT which increases the polarizability of molecule that is accountable for the NLO properties of a molecule. So the entitled compounds might show good NLO applications.
4.2 Natural population analysis (NPA)
The NPA is the most useful population analysis method. NPA gives information regarding the charge distribution for each atom of the molecule and is also significant to recognize the molecular electrostatic interactions and the chemical reactivity of an extensive range of chemical systems (Demircioğlu et al. 2019; Khalid et al. 2021b). The effective atomic calculations also play a substantial part in the chemical analysis of the molecular framework as charges of atoms influence the electronic structure, polarizability, dipole moment, and many other assets of molecules (Sıdır et al. 2010; Khalid et al. 2021c).
The values of natural atomic charges are used to describe the electronic distribution among atoms of the molecule. They help to find that which atom loses an electron and has a positive charge and which atom accepts electrons and has a negative charge. The NPA has been performed to reveal the electrophilic and nucleophilic nature of the titled compounds. The NPA analysis of the studied compounds 1–5 shows that all hydrogen atoms possess positive atomic charges and nitrogen, oxygen, sulfur, chlorine atoms possess negative atomic charges. The carbon atoms that are attached to hydrogen atoms have negative charges and those that are attached to oxygen, sulphur and nitrogen atoms have positive atomic charges as presented in Fig. 1.

Natural population analysis depiction of compounds 1–5
4.3 UV–Vis analysis
UV–Visible analysis of the entitled compounds has been examined by theoretical calculations along with the experimental studies. The major contributions of each maximum wavelength are beneficial to a deep insight into the study of the molecule. The experimental and simulated data of the entitled compounds have been described in Table S6 and UV–Vis spectra are presented in Figures S1-S5.
The simulated maximum absorption band (\(\lambda\)max) of compound 1 is observed at 335 nm (E = 3.702 eV, f = 0.892) with the contributions H → L (93%), H-2 → L (2%) which is attributed to methyl group at ortho position of benzene ring that shows hyper conjugative effect. The calculated maximum absorption band appears at 334 nm (E = 3.708 eV, f = 0.879) with the H → L (94%) transition for compound 2. The computed band of maximum absorption is occurred at 334 nm (E = 3.711 eV, f = 0.885) with the H → L (94%) excitation in compound 3. The reduction in \(\lambda\)max value to 334 nm in compound 2 and 3 is because of methyl group at meta and para positions, respectively. Further, the \(\lambda\)max value is observed to be increased to 338 nm (E = 3.668 eV, f = 0.767) in compound 4 owing to the manifestation of chloro group at ortho and para positions of benzene ring. The maximum absorption for compound 5 is observed at 341 nm (E = 3.636 eV, f = 0.676) related to the H → L (90%), H-1 → L + 3 (2%), H → L + 3 (2%) transitions. The highest value of \(\lambda\)max of compound 5 is accredited to the presence of dichloro substituent at 3 and 4 positions of benzene ring. The calculated maximum absorption wavelength values are in good harmony with experimental values: 344, 341, 342, 342 and 342 nm respectively, for studied compounds 1–5.
4.4 Vibrational analysis
The vibrational spectral analysis is a significant method used for the identification of functional groups of a molecule through their fundamental vibrational modes. The vibrational modes of the organic compound are relying on the planes of the functional groups of the entire molecule. The realistic vibrational modes were allocated rendering to the distinctive region of functional group frequencies. Theoretical harmonic frequencies, experimental fundamental frequencies and vibrational modes of studied compounds 1–5 calculated at DFT/M06/6-311G(d,p) level are described in Tables S7-S11 and the corresponding FT-IR spectrum has been depicted in Figures S6-S10.
4.4.1 C=S vibrations
The C=S stretching vibrations normally occurred in the frequency of 1570–1395 cm−1 (Moorthy et al. 2016a, 2016b). In entitled compounds 1–5, the C=S bond stretching vibrational frequencies are positioned at 1258, 1229, 1296, 1228 and 1226 cm−1 correspondingly in the FT-IR spectrum. The computed C=S bond stretching frequencies have occurred at 1257, 1282, 1274, 1201 and 1196 cm−1 respectively.
4.4.2 N–H vibrations
The high-frequency region above 3000 cm−1 is the representative region of N–H stretching vibrations (Silverstein and Bassler 1962). The N–H in-plane bending vibrations are usually assigned in the region of 1640–1080 cm−1 in thioamides which are slightly affected through the inductive effect of a more electronegative group (Roeges and Baas 1994; Ohno et al. 1992; Singh et al. 2012). The N–H out-of-plane bending vibrations in thioamides generally give bands in the region 710–580 cm−1 (M. susai Boobalan et al. 1072). In the entitled compounds 1–5, the bands appeared at 3322, 3323, 3398, 3394 and 3386 respectively cm−1 in the experimental spectra and calculated at 3581, 3602, 3605, 3580 and 3587 cm−1 respectively computationally belong to stretching vibrations of N–H. Additionally, the N–H in-plane bending vibrational bands were noticed at 3322, 3323, 3398, 3394 and 3386 cm−1 respectively and calculated at 3605–3580 cm−1. The bands at 747, 668, 750, 693 and 751 cm−1 respectively and their corresponding computed values in the region 668–627 cm−1 ensured the presence of N–H out-of-plane bending vibrations.
4.4.3 C=N and C–N vibrations
The heteronuclear bond (dipole bond) C-N and C = N stretching vibrational modes predicted in the range of 1600–1490 cm−1 (Silverstein et al. 2005). In the entitled compounds 1–5, the C=N stretching vibration bands were detected at 1539, 1519, 1538, 1521 and 1515 cm−1 respectively in FT-IR spectra and calculated at 1690, 1690, 1689, 1692 and 1687 cm−1 respectively. The bands that appeared in the region 1508, 1539, 1520, 1539 and 1536 cm−1 respectively of FT-IR spectra and happened theoretically at 1528, 1529, 1529, 1516 and 1517 cm−1 respectively are attributed to the C–N stretching modes.
4.4.4 O–H vibrations
The O–H stretching vibrations are normally lie in the region around 3500 cm−1. The in-plane phenolic O–H bending vibration, commonly appeared in the region 1250–1150 cm−1 and O–H out of plane usually appeared at 950–900 cm−1 (Michalska et al. 1996). In the entitled compounds 1–5, the O–H stretching vibrations were examined in the region 3872, 3853, 3854, 3876 and 3863 cm−1 respectively in terms of theoretical calculations and at 3005, 3008, 3313,3137 and 3135 cm−1 respectively in experimental FT-IR spectra. The in-plane O–H bending vibration band was obtained at 1209, 1261, 1234, 1262 and 1295 cm−1 respectively in FT-IR spectrum and calculated at region 1368–1114 cm−1. The O–H out of plane bending wavenumbers were noticed at 815, 948, 766, 947 and 751 cm−1 respectively (experimentally) and calculated at 961–858 cm−1 (theoretically).
4.4.5 C–C vibrations
The absorption bands for C–C stretching vibrations of the aromatic ring are located between the frequency at 1666, 1665, 1665, 1619 and 1640 cm−1 respectively (Muthu and Paulraj 2012; Govindarajan et al. 2011). In the entitled compounds 1–5, the bands appeared at 1585, 1573, 1520, 1605 and 1618 cm−1 respectively in the FT-IR spectrum and theoretically happened at 1692–1251 cm−1.
4.4.6 N–N vibrations
The homonuclear bond N–N is a significant core bond of the azine group in thiosemicarbazones and acts as a bridge point for the NLO properties of molecules. The N–N bond stretching vibrations are found around 1080 cm−1 (Dhandapani et al. 2014). In the entitled compounds 1–5, the vibrations observed at 1095, 1034, 966, 1042 and 1195 cm−1 respectively in the FT-IR spectrum, and theoretically calculated in the region 1039, 1058, 114, 1045 and 1058 cm−1 respectively have been allocated to N–N stretching vibrations. The peaks of azine group obtained correspond with those in the literature which confirmed their presence and revealed the conducting properties of the entitled compounds.
4.4.7 C–H vibrations
The C–H stretching vibrations of the aromatic ring are seemed in the region 3100–3000 cm−1 that is the specific region of C–H stretching vibration (Arivazhagan 2011; Erdogdu 2013). The region of in-plane bending vibrations of C–H bonds is 1300–1000 cm−1 (Sundaraganesan et al. 2007). The out-of-plane bending C–H bond vibrations are assigned in the range of 1000–780 cm−1 (Arjunan et al. 2013). In the entitled compounds 1–5 the C–H stretching bands are located at 3322, 3385, 3001, 3394 and 2997 cm−1, correspondingly in the experimental FT-IR spectrum. In addition, theoretical results appeared in the region between 3156, 3159, 3142, 3164 and 3137 cm−1 correspondingly. The C-H in-plane bending vibrational modes were appeared at 1666–1045 cm−1 (theoretically) and noticed at 1575, 1543, 1520, 1539 and 1536 cm−1 correspondingly (experimentally). The C-H out-of-plane bending modes were detected at 972, 968, 966, 962 and 1195 cm−1 respectively in the FT-IR spectrum and theoretically computed at 964–753 cm−1.
4.4.8 C–H3 vibrations
The stretching vibrational modes of C-H3 are commonly noticed in the region of 2990–2950 cm−1 (Lin-Vien et al. 1991). In the entitled compounds 1–3, the C–H stretching mode was predicted at 3015, 3013 and 3102 cm−1 respectively computationally and perceived at 3005, 3008 and 3313 cm−1 respectively in experimental FT-IR spectra. The compounds theoretically exhibit symmetric stretching at 3015, 3013 and 3011 cm−1 respectively, asymmetric stretching at 3102 and 3081 cm−1 and mixed stretching vibrations (symmetric and asymmetric) at 3108, 3106 and 3079 cm−1 respectively.
4.4.9 C–Cl vibrations
The C–Cl absorption bands are detected in the range between 850 and 550 cm−1 (Arjunan et al. 2011; Govindarajan et al. 2010). In the entitled compounds 4–5, the calculated frequency of C–Cl stretching vibrations has appeared at 862 and 881 cm−1 theoretically while determined at 781 and 751 cm−1 in the experimental FT-IR spectrum.
4.5 Frontier molecular orbital (FMO) analysis
The FMO analysis plays a significant role in quantum chemistry, UV–Visible spectra and optoelectronic properties (Khalid et al. 2023b, 2021d; Arshad et al. 2023). The HOMO is the higher energy orbital having electrons and behaves as an electron donor and LUMO is the lowest-lying energy orbital that can accept electrons and act as an electron acceptor (Khalid et al. 2022a, 2019b). The energy difference between the energies of HOMO (EHOMO) and LUMO (ELUMO) is called the energy gap that significantly describes the charge transfer interaction, kinetic stability and reactivity of the molecule from the quantum chemical perspective (Saka et al. 2016; Khalid et al. 2022b). The small value of energy gap (∆E) displays that the molecule is more reactive and less stable and the large energy gap value exhibits that the molecule is less reactive and more stable. The computed values of HOMO, LUMO, HOMO-1, LUMO + 1, HOMO−2 and LUMO + 2 at TD-DFT/M06/6-311G(d, p) level for compounds 1–5 are described in Table S12 and their pictorial representations is shown in Fig. 2.

Frontier molecular orbitals representation of compounds 1–5
The HOMO and LUMO energy gap describes the ICT inside the molecule. The HOMO, LUMO energy gaps are found to be 4.223, 4.344, 4.342, 4.277 and 4.276 eV, respectively, for compounds 1–5 which shows that compound 1 is more reactive and less stable due to its small value of energy gap and compound 2 is less reactive, more stable because of high value of the energy gap. The least ∆E value (4.223 eV) of compound 1 might be due to the hyper conjugative effect of methyl substituent. The slight increase in ∆E value of 4.276 and 4.277 eV is viewed in compound 5 and 4 owing to the chloro substituent at the 3, 4 and 2, 4 positions of position of benzene ring, respectively. The further escalation in ∆E value of 4.342 eV is spotted in compound 3 owing to the manifestation of –CH3 group at ortho position. The highest value of ∆E (4.344 eV) is viewed in compound 2 which might be because of CH3 group at meta position of benzene ring. The decreasing order of reactivity is 1 > 5 > 4 > 3 > 2 between compounds 1–5. The small value of the band gap between FMO enables intramolecular charge transference (ICT) that makes the material act as NLO active. The energy gap values of the entitled compounds 1–5 are smaller than that of the urea (ΔE = 6.7063 eV) (Uludağ and Serdaroğlu 2018) so, they exhibit more NLO active material property than that of the urea. The positive and negative phases have been signified by blue and red color respectively. The HOMO discloses that parts of the molecule having ability to donate electrons to the corresponding vacant LUMO of an acceptor moiety. The orbitals figure of studied compounds show that the electronic cloud of HOMO is mainly presides over hydrazinecarbothioamide moiety whereas the LUMO region of the molecule is presides over (E)-2-(2-hydroxybenzylidene) hydrazinecarbothioamide.
4.6 Global reactivity parameters (GRP) analysis
The quantum chemical reactivity parameters are significant parameters that provide beneficial information associated with the physical and physicochemical characteristics of the molecule (Khan et al. 2021). The EHOMO and ELUMO are used in the calculation of global reactivity parameters such as chemical potential (µ), ionization potential (I), electronegativity (X), electron affinity (\({\rm E}{\rm A}\)), global hardness (η) electrophilicity (ω) and chemical softness (σ) (Demircioğlu et al. 2017; Khalid et al. 2023c). The computed global reactivity descriptors value of studied compounds 1–5 are tabularized in Table S13.
The value of the ionization potential of a molecule is associated with the energy value needed to donate an electron. Electronegativity provides an insight into the capability of a molecule to attract electrons towards itself and the electron affinity is associated to the ability of the molecule to accept specifically one electron from a donor. The I and EA values of the titled compounds show the following trend: 5 > 1 > 4 > 2 > 3 which clears that compound 5 loses the highest amount of energy when added to its LUMO, leading to a more stabilizing interaction. The chemical potential calculations are a good measure to describe the tendency of electrons escaping from an equilibrium system and exhibit the order of decreasing for compounds 1–5: 3 > 2 > 1 > 4 > 5. The value of the electrophilicity index is helpful to measures the susceptibility of the electrophilic influence of a molecule. The high values of the electrophilicity index suggest that the molecule is a good electrophile while its low value suggests that the molecule is a good nucleophile. The electrophilicity index values of titled compounds exhibited the decreasing order: 5 > 1 > 4 > 2 > 3 which shows that compound 5 has a more electrophilic nature and can accept the electrons. The global hardness and softness measure the extent of chemical reactivity and are inverse to each other. The following trend: 2 > 3 > 4 > 5 > 1 from hardness to softness values of title compounds shows that compound 2 is a hard, more stable, less reactive molecule and compound 1 is a soft, less stable, more reactive molecule from other compounds.
The computed values of global reactivity parameters revealed that compound 1 has more proficient NLO properties than other entitled compounds.
4.7 Non-linear optical (NLO) properties analysis
The NLO materials have gained considerable attention in scientific aspects as they offer potential applications in frequency shifting and optical memory in emerging optoelectronic technologies, signal processing and telecommunications (Muthu and Maheswari 2012; Govindarasu and Kavitha 2014; Govindarasu et al. 2014; Bouchouit et al. 2016). The organic molecules increase the movement of charges by \(\pi\)-electron delocalization are important NLO materials because they possess synthetic resilience and higher hyperpolarizability in comparison to inorganic materials (Uludağ and Serdaroğlu 2018; Guezguez et al. 2014). The electric \(\mu\), \(\alpha\), and \(\beta\) values are significant to predict NLO properties of the material are calculated via x, y, z constituents from the equations given below (Rajamani and Muthu 2013; Mydlova et al. 2018).
For dipole moment calculations.
For polarizability calculations.
For hyperpolarizability calculations.
DFT study is an effective method in the investigation of the organic NLO material. The NLO properties of the examined compounds have been computed at M06, B3LYP and M06-2 × along with /6-311G(d, p) level. The values of polarizability, hyperpolarizability and dipole moment of the studied compounds 1–5 are described in Tables S14–S16.
The greater values of polarizability, hyperpolarizability and dipole moment are significant for NLO properties. The tensors of polarizability (αxx, αyy, αzz) and hyperpolarizability (βxxx, βxxy, βxyy, βyyy, βxxz, βyyz, βxzz, βyzz, βzzz) are used to calculate the total values. The calculated polarizability values of compounds 1–5 are 151.802, 239.598, 241.346, 261.350 and 256.288 a.u. respectively at M06, 247.018, 240.426, 241.980, 261.411 and 256.355 a.u., respectively at B3LYP whereas 237.422, 231.666, 233.109, 251.258 and 246.841 a.u., at M06-2x. Among the other tensors, αxx is dominant and exhibits remarkably larger values than other tensor components. The values obtained for hyperpolarizability (βtot) are found in range of 1472.182–1522.007 a.u. at M06, 1814.907–2148.175 a.u. at B3LYP and 957.44–1247.69 a.u. at M06-2 × for compounds 1–5. The βxxx depicted the largest values among the individual components that entails the better intramolecular charge transference along x-axis. Significant results for NLO are examined at B3LYP functional. Further, the dipole moment values of compounds 1–5 are appeared to be 6.811, 6.159, 6.475, 6.311 and 5.6395 D, respectively at M06, 7.108, 6.466, 6.776, 6.605, 5.906 at B3LYP whereas 7.302, 6.641, 6.956, 6.755 and 6.041 D, respectively for compound 1–5. Urea is an accepted prototypical molecule to use as a reference in the examination of NLO properties of molecules. The dipole moment of titled compounds are 4.10, 4.5, 4.7, 4.6 and 4.1 times greater than urea (1.3732 D) and the hyperpolarizability of titled compounds are 40.6, 34.2, 34.6, 35.4 and 29.1 times larger from the value of urea (βtotal = 43 a.u.) at M06 leel (Prasad and Williams 1991).
It is concluded from the comparative study of entitled compounds with urea that all the investigated compounds have excellent NLO properties but compound 1 may be proved a powerful molecule in the future for the study of NLO features and in the development of NLO materials.
4.8 Molecular electrostatic potential (MEP) analysis
The MEP analysis is associated with the electron density and is a precise method to describe the sites of nucleophilic and electrophilic reactivity and also designate hydrogen-bonding interactions (Güveli et al. 2010). MEP is an actual physical term that can be calculated through computational methods or experimentally via diffraction techniques (Truhlar 1981). The various regions of MEP surface diagram are distinguished on the basis of color distribution. The red color is used to signify the negative regions (electrophilic region), the blue color represents the positive regions (nucleophilic region) and the green color for the zero potential regions as shown for studied compounds 1–5 in Fig. 3.

Molecular electrostatic potential Surfaces of compounds 1–5
It is clear from the MEP map of the entitled compounds that negative part is over the azomethine nitrogen (N) atom, thiolate sulfur (S) atom and phenolate oxygen (O) atom, so it has three feasible sites for the electrophilic attack. The positive region has covered the hydrogen (H) atoms, which is a probable position for the nucleophilic attack. These outcomes provide evidence regarding the regions of intermolecular interaction and metallic bonding.
5 Conclusion
In this work, new thiosemicarbazone compounds 1–5 have been synthesized in an excellent yield 77% to 86% by refluxing the salicylaldehyde with different substituted thiosemicarbazides in the presence of acetic acid. The synthesized products were examined by UV–Vis, IR, 1H NMR and 13C NMR spectroscopic methods. Quantum chemical computations have been used to perform detailed investigations of NLO properties. The outcomes of NBO analysis revealed that the compounds transitions have the highest stabilization energy values that lead to the stability of the compounds. The NPA findings show that all the hydrogen atoms possess positive atomic charges and nitrogen, oxygen, sulfur, chlorine and mostly carbon have negative atomic charges. The FMO results disclosed that energy gap of compound 1–5 is found to be 4.344- 4.223 eV. Also, GRP results exhibited the softness values in the range 0.230–0.237 eV for the entitled compounds. Additionally, the λmax is noticed in UV region from 341 to 344 nm with lesser excitation energies (3.702–3.711 eV). The study MEP maps describe that the azomethine nitrogen, thiolate sulfur and phenolate oxygen atoms are negative regions (red color) having three feasible sites for the electrophilic attack and the amino nitrogen atom (blue region) is the positive region and probable site for nucleophilic attack. The NLO properties of the synthesized compounds have been examined and results show that compounds 1–5 are 40.6, 34.2, 34.6, 35.4 and 29.1 times greater than the urea. Among the studied compounds, that compound 1 is found to be more reactive, soft and less stable molecule and exhibit excellent NLO properties. Thus it can be utilized as favorable candidates for different applications in photonic and electronic devices.
References
Achelle, S., Baudequin, C., Plé, N.: Luminescent materials incorporating pyrazine or quinoxaline moieties. Dyes Pigm. 98, 575–600 (2013)
Arivazhagan, M.: Vibrational analysis of 4-amino pyrazolo (3, 4-d) pyrimidine a joint FTIR, laser Raman and scaled quantum mechanical studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 82, 228–234 (2011)
Arjunan, V., Balamourougane, P., Mythili, C., Mohan, S.: Experimental spectroscopic (FTIR, FT-Raman, FT-NMR, UV–Visible) and DFT studies of 2-amino-5-chlorobenzoxazole. J. Mol. Struct. 1003, 92–102 (2011)
Arjunan, V., Sakiladevi, S., Marchewka, M., Mohan, S.: FTIR, FT-Raman, FT-NMR and quantum chemical investigations of 3-acetylcoumarin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 109, 79–89 (2013)
Arroudj, S., Bouchouit, M., Bouchouit, K., Bouraiou, A., Messaadia, L., Kulyk, B., Figa, V., Bouacida, S., Sofiani, Z., Taboukhat, S.: Synthesis, spectral, optical properties and theoretical calculations on schiff bases ligands containing o-tolidine. Opt. Mater. 56, 116–120 (2016)
Arshad, M.N., Shafiq, I., Khalid, M., Asad, M., Asiri, A.M., Alotaibi, M.M., Braga, A.A., Khan, A., Alamry, K.A.: Enhancing the photovoltaic properties via incorporation of selenophene units in organic chromophores with A2-π2-A1-π1-A2 configuration: a DFT-based exploration. Polymers 15, 1508 (2023)
Bouchouit, M., Elkouari, Y., Messaadia, L., Bouraiou, A., Arroudj, S., Bouacida, S., Taboukhat, S., Bouchouit, K.: Synthesis, spectral, theoretical calculations and optical properties performance of substituted-azobenzene dyes. Opt. Quant. Electron. 48, 1–11 (2016)
Braga, A.A., Morgon, N.H., Ujaque, G., Maseras, F.: Computational characterization of the role of the base in the Suzuki−Miyaura cross-coupling reaction. J. Am. Chem. Soc. 127, 9298–9307 (2005)
Braga, A.A., Ujaque, G., Maseras, F.: A DFT study of the full catalytic cycle of the Suzuki−Miyaura cross-coupling on a model system. Organometallics 25, 3647–3658 (2006)
Bullo, S., Jawaria, R., Faiz, I., Shafiq, I., Khalid, M., Asghar, M.A., Perveen, S.: Efficient synthesis, spectroscopic characterization, and nonlinear optical properties of novel salicylaldehyde-based thiosemicarbazones: experimental and theoretical studies. ACS Omega 8(15), 13982–13992 (2023)
Cerqueira, N.: Avogadro. Revista De Ciência Elementar 4(2), 017 (2016)
Cinteza, L.-O., Marinescu, M.: Synthesis and nonlinear studies on selected organic compounds in nanostructured thin films. Advanced surface engineering research. London: IntechOpen (2018), pp. 1-23
Demircioğlu, Z., Kaştaş, Ç.A., Büyükgüngör, O.: X-ray structural, spectroscopic and computational approach (NBO, MEP, NLO, NPA, fukui function analyses) of (E)-2-((4-bromophenylimino) methyl)-3-methoxyphenol. Mol. Cryst. Liq. Cryst. 656, 169–184 (2017)
Demircioğlu, Z., Kaştaş, G., Kaştaş, Ç.A., Frank, R.: Spectroscopic, XRD, hirshfeld surface and DFT approach (chemical activity, ECT, NBO, FFA, NLO, MEP, NPA& MPA) of (E)-4-bromo-2-[(4-bromophenylimino) methyl]-6-ethoxyphenol. J. Mol. Struct. 1191, 129–137 (2019)
Denisov, A.: Soluble fullerene derivative in liquid crystal: polymer composites and their impact on photorefractive grating efficiency and resolution. Appl. Opt. 48, 1926–1931 (2009)
Dennington, R., Keith, T., Millam, J.: GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009, Received 23 May (2013)
Desai, S.Β, Desai, P., Desai, K.: Synthesis of some Schiff bases, thiazolidinones and azetidinones derived from 2, 6-diaminobenzo [1, 2-d: 4, 5-d’] bisthiazole and their anticancer activities. Heterocycl. Commun. 7, 83–90 (2001)
Dhandapani, A., Manivarman, S., Subashchandrabose, S., Saleem, H.: Molecular structure and vibrational analysis on (E)-1-(3-methyl-2, 6-diphenyl piperidin-4-ylidene) semicarbazide. J. Mol. Struct. 1058, 41–50 (2014)
Erdogdu, Y.: Investigations of FT-IR, FT-Raman, FT-NMR spectra and quantum chemical computations of Esculetin molecule. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 106, 25–33 (2013)
Forrest, S.R.: Ultrathin organic films grown by organic molecular beam deposition and related techniques. Chem. Rev. 97, 1793–1896 (1997)
Frisch, A., Nielson, A., Holder, A.: Gaussview user manual, Gaussian Inc., Pittsburgh, PA, 556 (2000)
García-Melchor, M., Braga, A.A., Lledós, A., Ujaque, G., Maseras, F.: Computational perspective on Pd-catalyzed C-C cross-coupling reaction mechanisms. Acc. Chem. Res. 46, 2626–2634 (2013)
Gaussian, R.A.: 09, 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al., gaussian, Inc., Wallingford CT, 121 (2009), pp. 150–166
Govindarajan, M., Ganasan, K., Periandy, S., Karabacak, M.: Experimental (FT-IR and FT-Raman), electronic structure and DFT studies on 1-methoxynaphthalene. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 79, 646–653 (2011)
Govindarajan, M., Ganasan, K., Periandy, S., Karabacak, M., Mohan, S.: Vibrational spectroscopic analysis of 2-chlorotoluene and 2-bromotoluene: a combined experimental and theoretical study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 77, 1005–1013 (2010)
Govindarasu, K., Kavitha, E.: Vibrational spectra, molecular structure, NBO, UV, NMR, first order hyperpolarizability, analysis of 4-methoxy-4′-nitrobiphenyl by density functional theory. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 122, 130–141 (2014)
Govindarasu, K., Kavitha, E., Sundaraganesan, N.: Synthesis, structural, spectral (FTIR, FT-Raman, UV, NMR), NBO and first order hyperpolarizability analysis of N-phenylbenzenesulfonamide by density functional theory. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 133, 417–431 (2014)
Guest, D., Menezes da Silva, V.H., de Lima Batista, A.P., Roe, S.M., Braga, A.A., Navarro, O.: (N-heterocyclic carbene)-palladate complexes in anionic mizoroki-heck coupling cycles: a combined experimental and computational study. Organometallics 34, 2463–2470 (2015)
Guezguez, I., Ayadi, A., Ordon, K., Iliopoulos, K., Branzea, D.G., Migalska-Zalas, A., Makowska-Janusik, M., El-Ghayoury, A., Sahraoui, B.: Zinc induced a dramatic enhancement of the nonlinear optical properties of an azo-based iminopyridine ligand. J. Phys. Chem. C 118, 7545–7553 (2014)
Güveli, Ş, Özdemir, N., Bal-Demirci, T., Ülküseven, B., Dinçer, M., Andaç, Ö.: Quantum-chemical, spectroscopic and X-ray diffraction studies on nickel complex of 2-hydroxyacetophenone thiosemicarbazone with triphenylphospine. Polyhedron 29, 2393–2403 (2010)
Iliopoulos, K., El-Ghayoury, A., Derkowska, B., Ranganathan, A., Batail, P., Gindre, D., Sahraoui, B.: Effect of the counter cation on the third order nonlinearity in anionic Au dithiolene complexes. Appl. Phys. Lett. 101, 261105 (2012)
Inkaya, E., Dincer, M., Şahan, E., Yıldırım, İ: Synthesis, spectroscopic and structural characterization of 5-benzoyl-4-phenyl-2-methylthio-1H-pyrimidine with theoretical calculations using density functional theory. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 114, 92–100 (2013)
Kamanina, N.V.: Peculiarities of optical limiting effect in π-conjugated organic systems based on 2-cyclooctylamino-5-nitropyridine doped with C70. J. Opt. A Pure Appl. Opt. 3, 321 (2001)
Kamanina, N., Plekhanov, A.: Mechanisms of optical limiting in fullerene-doped π-conjugated organic structures demonstrated with polyimide and COANP molecules. Opt. Spectrosc. 93, 408–415 (2002)
Kamanina, N.V., Serov, S., Shurpo, N., Likhomanova, S., Timonin, D., Kuzhakov, P., Rozhkova, N., Kityk, I., Plucinski, K., Uskokovic, D.: Polyimide-fullerene nanostructured materials for nonlinear optics and solar energy applications. J. Mater. Sci. Mater. Electron. 23, 1538–1542 (2012)
Khalid, M., Ahmed, R., Shafiq, I., Arshad, M., Asghar, M.A., Munawar, K.S., Imran, M., Braga, A.A.: First theoretical framework for highly efficient photovoltaic parameters by structural modification with benzothiophene-incorporated acceptors in dithiophene based chromophores. Sci. Rep. 12, 20148 (2022b)
Khalid, M., Ali, A., Asim, S., Tahir, M.N., Khan, M.U., Vieira, L.C.C., de la Torre, A.F., Usman, M.: Persistent prevalence of supramolecular architectures of novel ultrasonically synthesized hydrazones due to hydrogen bonding [X–H⋯ O; X= N]: experimental and density functional theory analyses. J. Phys. Chem. Solids 148, 109679 (2021a)
Khalid, M., Ali, A., Jawaria, R., Asghar, M.A., Asim, S., Khan, M.U., Hussain, R., Ur Rehman, M.F., Ennis, C.J., Akram, M.S.: First principles study of electronic and nonlinear optical properties of A-D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives. RSC Adv. 10, 22273–22283 (2020)
Khalid, M., Hussain, R., Hussain, A., Ali, B., Jaleel, F., Imran, M., Assiri, M.A., Usman Khan, M., Ahmed, S., Abid, S.: Electron donor and acceptor influence on the nonlinear optical response of diacetylene-functionalized organic materials (DFOMs): density functional theory calculations. Molecules 24, 2096 (2019a)
Khalid, M., Khan, M.U., Shafiq, I., Hussain, R., Mahmood, K., Hussain, A., Jawaria, R., Hussain, A., Imran, M., Assiri, M.A.: NLO potential exploration for D–π–A heterocyclic organic compounds by incorporation of various π-linkers and acceptor units. Arab. J. Chem. 14, 103295 (2021b)
Khalid, M., Khan, M., Shafiq, I., Mahmood, K., Akhtar, M.N., Iqbal, J., Assiri, M.A., Imran, M., Braga, A.A.C.: Role of donors in triggering second order non-linear optical properties of non-fullerene FCO-2FR1 based derivatives: a theoretical perspective. Heliyon 9, e13033 (2023c)
Khalid, M., Lodhi, H.M., Khan, M.U., Imran, M.: Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: a DFT approach. RSC Adv. 11, 14237–14250 (2021c)
Khalid, M., Naseer, S., Tahir, M.S., Shafiq, I., Munawar, K.S., de Alcântara Morais, S.F., Braga, A.A.: A theoretical approach towards designing of banana shaped non-fullerene chromophores using efficient acceptors moieties: exploration of their NLO response properties. Opt. Quant. Electron. 55, 258 (2023b)
Khalid, M., Shafiq, I., Mahmood, K., Hussain, R., Ur Rehman, M.F., Assiri, M.A., Imran, M., Akram, M.S.: Effect of different end-capped donor moieties on non-fullerenes based non-covalently fused-ring derivatives for achieving high-performance NLO properties. Sci. Rep. 13, 1395 (2023a)
Khalid, M., Shafiq, I., Zhu, M., Khan, M.U., Shafiq, Z., Iqbal, J., Alam, M.M., Braga, A.A.C., Imran, M.J.J.O.S.C.S.: Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc. 25, 101305 (2021d)
Khalid, M., Ullah, M.A., Adeel, M., Khan, M.U., Tahir, M.N., Braga, A.A.C.: Synthesis, crystal structure analysis, spectral IR, UV–Vis, NMR assessments, electronic and nonlinear optical properties of potent quinoline based derivatives: interplay of experimental and DFT study. J. Saudi Chem. Soc. 23, 546–560 (2019b)
Khalid, M., Zafar, M., Hussain, S., Asghar, M.A., Khera, R.A., Imran, M., Abookleesh, F.L., Akram, M.Y., Ullah, A.: Influence of end-capped modifications in the nonlinear optical amplitude of nonfullerene-based chromophores with a D− π–A architecture: a DFT/TDDFT study. ACS Omega 7, 23532–23548 (2022a)
Khan, I., Khalid, M., Adeel, M., Niaz, S.I., Shafiq, I., Muhammad, S., Braga, A.A.C.: Palladium-catalyzed synthesis of 5-(arylated) pyrimidines, their characterization, electronic communication, and non-linear optical evaluations. J. Mol. Struct. 1237, 130408 (2021)
Kumar, R.S., Arunachalam, S.: DNA binding and antimicrobial studies of polymer–copper (II) complexes containing 1, 10-phenanthroline and L-phenylalanine ligands. Eur. J. Med. Chem. 44, 1878–1883 (2009)
Likhomanova, S., Kamanina, N.: COANP-fullerenes system for optical modulation, in: Journal of Physics: Conference Series, IOP Publishing (2016), pp. 012146
Lin-Vien, D., Colthup, N.B., Fateley, W.G., Grasselli, J.G.: The handbook of infrared and Raman characteristic frequencies of organic molecules, Elsevier (1991)
Mahajan, R.K., Walia, T., Lobana, T.: Cyclopentanone thiosemicarbazone, a new complexing agent for copper determination in biological samples by adsorptive stripping voltammetry. Anal. Sci. 22, 389–392 (2006)
Mahmood, A., Abdullah, M.I., Khan, S.U.-D.: Enhancement of nonlinear optical (NLO) properties of indigo through modification of auxiliary donor, donor and acceptor. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 139, 425–430 (2015a)
Mahmood, A., Khan, S.U.D., Rana, U.A., Janjua, M.R.S.A., Tahir, M.H., Nazar, M.F., Song, Y.: Effect of thiophene rings on UV/visible spectra and non-linear optical (NLO) properties of triphenylamine based dyes: a quantum chemical perspective. J. Phys. Org. Chem. 28, 418–422 (2015b)
Mahmood, A., Sandali, Y., Wang, J.-L.: Easy and fast prediction of green solvents for small molecule donor-based organic solar cells through machine learning. Phys. Chem. Chem. Phys. 25, 10417–10426 (2023)
Mehmood, H., Akhtar, T., Haroon, M., Khalid, M., Woodward, S., Shafiq, I., Braga, A.A.C., Alhokbany, N.: Facile synthesis, spectroscopic and nonlinear optical insights of hydrazinyl-based functional materials: experimental and DFT approach. Polycycl. Aromat. Cmpd. (2023). https://doi.org/10.1080/10406638.2023.2235871
Michalska, D., Bieńko, D., Abkowicz-Bieńko, A., Latajka, Z.: Density functional, Hartree− Fock, and MP2 studies on the vibrational spectrum of phenol. J. Phys. Chem. 100, 17786–17790 (1996)
Mirershadi, S., Ahmadi-Kandjani, S., Zawadzka, A., Rouhbakhsh, H., Sahraoui, B.: Third order nonlinear optical properties of organometal halide perovskite by means of the Z-scan technique. Chem. Phys. Lett. 647, 7–13 (2016)
Moorthy, N., Prabakar, P.J., Ramalingam, S., Govindarajan, M., Gnanamuthu, S.J., Pandian, G.: Spectroscopic analysis, AIM, NLO and VCD investigations of acetaldehyde thiosemicarbazone using quantum mechanical simulations. J. Phys. Chem. Solids 95, 74–88 (2016b)
Moorthy, N., Prabakar, P.J., Ramalingam, S., Pandian, G., Anbusrinivasan, P.: Vibrational, NMR and UV–visible spectroscopic investigation and NLO studies on benzaldehyde thiosemicarbazone using computational calculations. J. Phys. Chem. Solids 91, 55–68 (2016a)
Muhammad, S., Al-Sehemi, A.G., Irfan, A., Algarni, H., Qiu, Y., Xu, H., Su, Z., Iqbal, J.: The substitution effect of heterocyclic rings to tune the optical and nonlinear optical properties of hybrid chalcones: a comparative study. J. Mol. Graph. Model. 81, 25–31 (2018)
Muhammad, S., Al-Sehemi, A.G., Irfan, A., Chaudhry, A.R.: Tuning the push–pull configuration for efficient second-order nonlinear optical properties in some chalcone derivatives. J. Mol. Graph. Model. 68, 95–105 (2016)
Muhammad, S., Chaudhry, A.R., Al-Sehemi, A.G.: A comparative analysis of the optical and nonlinear optical properties of cross-shaped chromophores: quantum chemical approach. Optik 147, 439–445 (2017)
Muthu, S., Maheswari, J.U.: Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H, UV) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 4-[(4-aminobenzene) sulfonyl] aniline by ab initio HF and density functional method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 92, 154–163 (2012)
Muthu, S., Paulraj, E.I.: Molecular structure, vibrational spectra, first order hyper polarizability, NBO and HOMO–LUMO analysis of 4-amino-3 (4-chlorophenyl) butanoic acid. Solid State Sci. 14, 476–487 (2012)
Mydlova, L., Taboukhat, S., Ayadi, A., Migalska-Zalas, A., El-Ghayoury, A., Zawadzka, A., Makowska-Janusik, M., Sahraoui, B.: Theoretical and experimental investigation of multifunctional highly conjugated organic push-pull ligands for NLO applications. Opt. Mater. 86, 304–310 (2018)
Mysliwiec, J., Sznitko, L., Bartkiewicz, S., Miniewicz, A., Essaidi, Z., Kajzar, F., Sahraoui, B.: Amplified spontaneous emission in the spiropyran-biopolymer based system. Appl. Phys. Lett. 94, 241106 (2009)
Oboyle, N.M., Tenderholt, A.L., Langner, K.M.: Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845 (2008)
Ohno, K., Mandai, Y., Matsuura, H.: Vibrational spectra and molecular conformation of taurine and its related compounds. J. Mol. Struct. 268, 41–50 (1992)
Palanisami, N., Moon, I.S.: Synthesis, characterization, and nonlinear optical properties of transition metal complexes bearing donor-acceptor ferrocenyl schiff bases. Sci. Adv. Mater. 6, 2378–2383 (2014)
Prajapati, N.P., Patel, H.D.: Novel thiosemicarbazone derivatives and their metal complexes: recent development. Synth. Commun. 49, 2767–2804 (2019)
Prasad, P.N., Williams, D.J.: Introduction to nonlinear optical effects in molecules and polymers, A Wiley-Interscience Publication (1991)
Rajamani, T., Muthu, S.: Vibrational spectra, first order hyperpolarizability, NBO, fukui function and HOMO–LUMO analysis of 2-[4-(1, 3-benzodioxol-5-ylmethyl)-1-piperazinyl] pyrimidine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 115, 654–666 (2013)
Reddy, B., Kumar, J., Reddy, K., Sarma, L., Reddy, A.: A rapid and sensitive extractive spectrophotometric determination of copper (II) in pharmaceutical and environmental samples using benzildithiosemicarbazone. Anal. Sci. 19, 423–428 (2003)
Reed, A.E., Curtiss, L.A., Weinhold, F.: Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899–926 (1988)
Roeges, N.P., Baas, J.: A guide to the complete interpretation of infrared spectra of organic structures. Wiley, New York (1994)
Saka, E.T., Uzun, S., Çağlar, Y.: Synthesis, structural characterization, catalytic activity on aerobic oxidation of novel Co (II) and Fe (II) phthalocyanines and computational studies of 4-[2-(2, 3-dichlorophenoxy) ethoxy] phthalonitrile. J. Organomet. Chem. 810, 25–32 (2016)
Samadhiya, S., Halve, A.: Synthetic utility of Schiff bases as potential herbicidal agents. Orient. J. Chem. 17, 119–122 (2001)
Shafiq, I., Amanat, I., Khalid, M., Asghar, M.A., Baby, R., Ahmed, S., Alshehri, S.M.: Influence of azo-based donor modifications on nonlinear optical amplitude of D-π-A based organic chromophores: a DFT/TD-DFT exploration. Synth. Met. 297, 117410 (2023)
Shahab, S., Filippovich, L., Sheikhi, M., Kumar, R., Dikusar, E., Yahyaei, H., Muravsky, A.: Polarization, excited states, trans-cis properties and anisotropy of thermal and electrical conductivity of the 4-(phenyldiazenyl) aniline in PVA matrix. J. Mol. Struct. 1141, 703–709 (2017b)
Shahab, S., Filippovich, L., Sheikhi, M., Yahyaei, H., Aharodnikova, M., Kumar, R., Khaleghian, M.: Spectroscopic (polarization, excitedstate, FT-IR, UV/Vis and 1H NMR) and thermophysical investigations of new synthesized azo dye and its application in polarizing film. Am. J. Mater. Synth. Process 5, 17–23 (2017a)
da Silveira, V.C., Luz, J.S., Oliveira, C.C., Graziani, I., Ciriolo, M.R., da Costa Ferreira, A.M.: Double-strand DNA cleavage induced by oxindole-Schiff base copper (II) complexes with potential antitumor activity. J. Inorgan. Biochem. 102, 1090–1103 (2008)
Silverstein, R.M., Bassler, G.C.: Spectrometric identification of organic compounds. J. Chem. Educ. 39, 546 (1962)
Silverstein, R., Webster, F., Kiemle, D.: Silverstein-Spectrometric Identification of Organic Compounds 7th ed. pdf, in, State University of New York, College of Environmental Science and Forestry (2005)
Singh, R., Kumar, A., Tiwari, R., Rawat, P., Verma, D., Baboo, V.: Synthesis, molecular structure and spectral analysis of ethyl 4-formyl-3, 5-dimethyl-1H-pyrrole-2-carboxylate thiosemicarbazone: a combined DFT and AIM approach. J. Mol. Struct. 1016, 97–108 (2012)
Snehalatha, M., Ravikumar, C., Joe, I.H., Sekar, N., Jayakumar, V.: Spectroscopic analysis and DFT calculations of a food additive carmoisine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 72, 654–662 (2009)
Subhashree, G., Haribabu, J., Saranya, S., Yuvaraj, P., Krishnan, D.A., Karvembu, R., Gayathri, D.: In vitro antioxidant, antiinflammatory and in silico molecular docking studies of thiosemicarbazones. J. Mol. Struct. 1145, 160–169 (2017)
Sundaraganesan, N., Ilakiamani, S., Joshua, B.D.: FT-Raman and FT-IR spectra, ab initio and density functional studies of 2-amino-4, 5-difluorobenzoic acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 67, 287–297 (2007)
Susai Boobalan, M., Ramalingam, S., Amaladasan, M., Tamilvendan, D., Prabhu, G.V., Bououdina, M.: A computational perspective on equilibrium geometry, vibrational spectra and electronic structure of antioxidant active Mannich base 1-[(Pyridin-2-yl amino) methyl] pyrrolidine-2, 5-dione. J. Mol. Struct. 1072, 153–172 (2014)
Sıdır, İ, Sıdır, Y.G., Kumalar, M., Taşal, E.: Ab initio Hartree-Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 7-acetoxy-6-(2, 3-dibromopropyl)-4, 8-dimethylcoumarin molecule. J. Mol. Struct. 964, 134–151 (2010)
Truhlar, D.G.: Effective potentials for intermediate-energy electron scattering: testing theoretical models, in: Chemical applications of atomic and molecular electrostatic potentials, Springer (1981), pp. 123–172
Uludağ, N., Serdaroğlu, G.: An improved synthesis, spectroscopic (FT-IR, NMR) study and DFT computational analysis (IR, NMR, UV–Vis, MEP diagrams, NBO, NLO, FMO) of the 1, 5-methanoazocino [4, 3-b] indole core structure. J. Mol. Struct. 1155, 548–560 (2018)
Vančo, J., Marek, J., Trávníček, Z., Račanská, E., Muselík, J., Švajlenová, O.G.: Synthesis, structural characterization, antiradical and antidiabetic activities of copper (II) and zinc (II) Schiff base complexes derived from salicylaldehyde and β-alanine. J. Inorgan. Biochem. 102, 595–605 (2008)
Weinhold, F., Landis, C.R.: Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 2, 91–104 (2001)
Zhong, X., Yi, J., Sun, J., Wei, H.-L., Liu, W.-S., Yu, K.-B.: Synthesis and crystal structure of some transition metal complexes with a novel bis-Schiff base ligand and their antitumor activities. Eur. J. Med. Chem. 41, 1090–1092 (2006)
Zhurko, G.: Chemcraft. http://www.chemcraftprog.com, Received 22 Oct (2014)
Acknowledgements
The authors extend their appreciation to the Ministry of Education in KSA for funding this research work through the project number KKU- IFP2-P-7.
Funding
The authors extend their appreciation to the Ministry of Education in KSA for funding this research work through the project number KKU- IFP2-P-7.
Author information
Authors and Affiliations
Contributions
RJ: Conceptualization; methodology; software. FF: Conceptualization; methodology. IS: Data curation; formal analysis. U-e-H: Conceptualization; resources. NK: Data curation; formal analysis; validation. MI: Data curation; formal analysis. NR: methodology; software editing and rewriting.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Our work is not related to human and/ or animal studies therefore this section is not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jawaria, R., Fareed, F., Shafiq, I. et al. Exploration of key electronic and optical nonlinearity properties of synthesized salicylaldehyde based compounds via quantum chemical approach. Opt Quant Electron 55, 1175 (2023). https://doi.org/10.1007/s11082-023-05494-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05494-1