
Abstract
The structural, optical, electronic, and transport properties of A2AgMoBr6 (A = K, Rb, Cs) have been examined using first-principles calculations in the present study. The measured value of Paugh’s ratio (B/G) in mechanical computations discloses the material’s ductility. The computed negative value of enthalpy of formation (ΔH) suggests that the examined materials are stable. The assessment of band structure reveals that the A2AgMoBr6 (A = K, Rb, Cs) compound has an indirect bandgap (L-X). The materials under examination have an incredible absorption coefficient (α(ω)) in the ultraviolet and visible regions of the light spectra, making them suitable for photovoltaic and optical equipment manufacturing. In addition, the transport characteristics have been projected using the BoltzTraP code at temperatures ranging from 100 to 1000 K. The estimated value of the figure of merit (zT) 0.89, 0.92, and 0.85 for K, Rb, and Cs respectively at room temperature suggests that A2AgMoBr6 (A = K, Rb, Cs) compounds are perfect contenders for thermoelectric device applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The quest for unconventional and non-fossil fuel energy sources has acquired more intense in recent years because of the drastic increment in the world's energy requirements. As a consequence, renewable energy sources are receiving greater emphasis globally (Elsheikh et al. 2014; Zheng et al. 2014). Thermoelectricity may be utilized to generate renewable energy that efficiently utilizes waste heat by directly converting thermal to electrical energy (Goldsmid 2017; Tritt and Subramanian 2006). The figure of merit (zT) specifies the efficiency of thermoelectric materials (TE), as zT = S2σT/κe + κl, where S is the Seebeck coefficient, σ is electrical conductivity, T is the absolute temperature, and κe and κl are electronic and lattice contributions to thermal conductivity. A large thermoelectric power factor (S2σ) and low thermal conductivity are prescribed for the prime zT (Mingo 2004; Dehkordi et al. 2015). Thus, novel and cutting-edge thermoelectric materials with excellent zT are emerging. The current research primarily focuses on finding low-cost, highly effective, and non-toxic materials. As per surveys, perovskite materials are regarded as promising candidates to accommodate some of these prerequisites. Semiconducting perovskite materials are intriguing candidates for thermoelectric and photovoltaic applications due to their potential to meet these requirements. On the contrary, Perovskite-based materials have recently attracted interest after being explored to have extraordinary zT. Numerous researchers and scientists are keen on these compounds because of their multiple physical, chemical, and catalytic properties (Andrianov et al. 2019; Houari et al. 2020; Nemtsev et al. 2018). Double perovskites and regular perovskites have mapped a lot of interest because of their incredible potential for advanced applications. The formula for the double perovskite is A2BB'X6, where X can be an oxide or a halide, the B and B' sites are possessed by transitional or non-transitional cations, and the A site is inhabited by a rare-earth metal or an alkaline-earth metal (Dahbi et al. 2021; Maiti et al. 2019). As a consequence, assessing the material characteristics of novel double perovskites may be beneficial in an array of applications. Some double perovskites have been investigated recently, and the outcomes in optoelectronics and transport properties are very significant. To generate lead-free A2+1B+1B3+X6−1 double perovskite compounds, monovalent (B+) and trivalent (B3+) ions can be used to replace Pb2+ ions in ABX3 (Bhamu et al. 2018; Yan et al. 2022; Gets et al. 2018). Recently, a diverse array of distinctive double perovskite compounds has been developed. In this framework, detailed descriptions of A as K, Cs, Rb, B+ cation as Cu, Ag, In, B3+ cation as Bi, Sb, and X as Cl, Br, I compounds were given, disclosing their optoelectronics and transport properties (Haque and Hossain 2018; Ravi et al. 2018; Noor et al. 2021; Murtaza et al. 2021; Zhou et al. 2018; Shi et al. 2020). Halide perovskites are perfectly robust and provide a larger variety of active absorbers.
From the literature survey, the Cs2AgFeCl6 (Radja et al. 2022) displays p-type ferromagnetic semiconducting properties. The magnetic moment of the 5μB is approximated to be massive, with essential participation from Fe atoms. The elastic properties of Cs2AgFeCl6 demonstrate its stability and ductility. The thermoelectric properties of Cs2AgFeCl6 are assessed using Boltzmann transport equations via DFT in the temperature range 100–900 K. Cs2AgFeCl6 is a worthy material for thermoelectric applications at both high and low temperatures, according to the literature. The associated characteristics of Cs2AgCrX6 (X = Cl, I) (Saeed et al. 2022) indicated that halogen replacement at the X-site would significantly shift the role of the onset of optical absorption towards the high- and low-energy infrared regions, respectively. The high-frequency dielectric constants, along with other factors like the indirect nature of bandgap transitions, the perceivable intensity of absorption coefficients and refractive indices, and others, indicated that cubic A2AgCrBr6 (A = Rb, Cs) and Cs2AgCrCl6 (Varadwaj 2020) may be a collection of optoelectronic materials for further experimental characterization. Also, observed that A2AgRhCl6 (A = K, Rb, and Cs) (Varadwaj and Marques 2020) is a suitable choice material for photovoltaic applications due to its immense refractive index and high absorption coefficient in the 1.0–4.5 eV range.
Inspiring by the fascinating features of the above halide double perovskites we look for a deep study in analyzing the amazing features of semiconductor halide double perovskites A2AgMoBr6 which has got a figure of merit zT 0.96, 0.98, and 0.93 for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 respectively at 50 K which is very efficient for converting waste heat into useful energy that makes the environment eco-friendlier. There is a profound urge to find out more about perovskite materials because of the ever-growing demand for optoelectronic and thermal technologies. HDPs have been the subject of numerous studies, but A2AgMoBr6 (A = K, Rb, Cs) compounds still require theoretical and experimental investigation. This inspired us to look into the various characteristics of A2AgMoBr6 (A = K, Rb, Cs) of HDPs. Utilizing the DFT method within the WIEN2K code, the TE, structural, elastic, optical, and electronic properties of the stated compounds were explored.
2 Methods of calculation
The WIEN2K software premised on the FP-LAPW method has been implemented to investigate the properties of A2AgMoBr6 (A = K, Rb, Cs) double perovskites (DPs) (Blaha et al. 2001; Schwarz et al. 2002; Petersen et al. 2000). The PBE-GGA technique has been employed to optimize the electrical structures (Perdew et al. 1996). The PBE-GGA effectively identifies the structural type and ground state features such as lattice constant, bulk modulus, and energy, yet undervalues electronic properties and band gaps. To get the requisite band gap, the TB-mBJ potential was combined with the PBE-GGA (Tran and Blaha 2009). The mBJ potential proves to be the most adaptable, precise, and simple to operate. Within the core, the result appears as spherical harmonic waves and plane waves. Execute the programming for the structure utilizing fundamental rearing parameters of angular momentum lmax = 10 and vector product KmaxRMT = 10. The significant wave vector in the reciprocal lattice is Kmax, and the radius of the muffin tin is RMT. The k-mesh for charge/energy conversion was enhanced to the order of 10 × 10 × 10 by initiating to iterate. Up to 0.01 mRy, the energy convergence has indeed been examined. Thermoelectric characteristics have been identified utilizing the BoltzTraP2 code.
2.1 Structural parameters
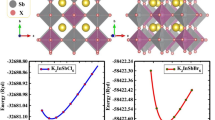
The lattice parameter has been used to recognize A2AgMoBr6 (A = K, Rb, Cs) structural parameters, and the crystal's total energy was reduced by applying Murnaghan's equation of states for unit cell volume (Birch 1947). Fm-3 m space group crystallization has been seen in A2AgMoBr6 (A = K, Rb, Cs). The unit cell's A atom is situated in the Wyckoff location 8c (0.25, 0.25, 0.25), while the Ag and Mo atoms were located at 4a (0, 0, 0) and (0.5, 0.5, 0.5), and the Br atom was located at 24e (0, 0, 0.25). Table 1 lists the ground structure findings for lattice parameters such as the bulk modulus (B), the lattice constant (a), and the pressure derivative of the bulk modulus (B′). In the optimization curve, as seen in Fig. 1, the E–V curves exhibit low energy under the Ferromagnetic (FM) phase. As a consequence, the Ferromagnetic (FM) condition of A2AgMoBr6 remained stable.

Optimization plot of investigated double perovskites A2AgMoBr6 (A = K, Rb, Cs)
The enthalpy of formation (ΔH) has been calculated to assess the material's stability. ΔH values explored for A2AgMoBr6 (A = K, Rb, Cs) are − 1.99, − 1.98, and − 1.94, respectively. The negative sign of ΔH reflects the endothermic phase indicating structural stability (Mousa et al. 2020; Chettri et al. 2016).
where Etotal indicates the aggregate energy of the materials and a, b, c, and d define the number of atoms. The tolerance factor (τ) is employed to quantify the distortion and integrity of a crystal, and the stable perovskites have a range of 0.8–1.4 (Haq et al. 2021).
where rX and rA are the ionic radii of X and A, respectively, and rBB' is the average of B and B` ionic radii. Our predicted tolerance factors for A2AgMoBr6 (A = K, Rb, Cs) are 0.90, 0.91, and 0.94, respectively, yielding A2AgMoBr6 (A = K, Rb, Cs) stability in a cubic lattice.
Cohesive energy has been estimated to comprehend the durability of interatomic bonds and, consequently, stability in a cubic structure. The cohesive energy refers to the energy that is needed to remove the compounds' attractive interactions and release the constituents. K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 have yielded cohesive energies of 2.71, 2.70, and 2.64 eV, respectively. The positive value of cohesive energy indicates structural stability.
2.2 Elastic properties
The mechanical and anisotropic response of solid materials can be explored by employing elastic characteristic assessment. Consequently, the elastic constants of double perovskites have been deduced to explore the mechanical properties of materials as well as their flexible and brittle behavior. To assess the mechanical stability of these cubic perovskite structures, just three independent elastic constants C11, C12, and C44 are sufficient. According to the Born mechanical stability criterion (Nabi et al. 2019), C11–C12 > 0, C11 + 2C12 > 0, and C44 > 0, all of the compounds tested meet mechanical stability. This confirmed the stability of these materials' elastic properties under deforming force. Furthermore, elastic constants are utilized for estimating the important mechanical characteristics of the materials under consideration using conventional relations (Shafiq et al. 2015, 2014) as shown in Table 2. The G shear modulus is the arithmetic mean of the GR and GH, and it quantifies the material's plastic deformation in response to external stress. Rb2AgMoBr6 has a higher GH and Young modulus Y, indicating that it is more resistant to plastic deformation and thus tougher than K2AgMoBr6 and Cs2AgMoBr6. Pughs ratio (B/G) (Pugh 1954) monitors the material's brittleness and ductility. Table 2 reveals that the B/G ratio for both materials is greater than the critical value of 1.75, signifying that these materials are ductile in nature. The brittle/ductile tendency of materials is also illustrated by the negative/positive value of Cauchy's pressure (C″ = C12–C44). The Poisson's ratio (v) gauges a crystal's stability versus deformation. The lower the value of Poisson's ratio, the more robust these materials are against shear stress. Table 2 summarizes the computed anisotropic factor values for A2AgMoBr6 (A = K, Rb, Cs). The departure of A from unity gauges the material's anisotropy. Table 2 depicts that the anisotropic factor is greater than one, revealing that their properties change in different crystallographic directions. We also assessed the melting point and Debye temperature, two thermodynamic variables associated with elastic response. The Debye temperature was determined by utilizing the following relationship
where h is the Planck constant, kB is the Boltzmann constant, NA is the Avogadro number, ρ is the density of the material, M is the molecular weight, and n is the number of atoms in a single cell. Table 3 provides the evaluated Debye temperature. The mean sound speed of the compounds studied was estimated utilizing (Nabi et al. 2019):
where the longitudinal and transverse components of sound velocity are computed employing shear modulus and bulk modulus, respectively, as
The melting temperature (Tm) is the temperature range over which a substance can be employed. To determine it from elastic constants, apply the relation (Nabi et al. 2019).
Table 3 depicts the observed values of vt, vl, vm, θD, and Tm.
To gauge the mechanical robustness of the double perovskites, we also projected the micro-hardness (H) (Rahman et al. 2023). The bulk or shear modulus of a material has a direct correlation with its hardness. Among them, the Cs-based double perovskites exhibit high mechanical strength, owing to the assessed H value in Table 4.
2.2.1 Electronic structure
The electronic band structure is a crucial parameter to elaborate the material’s nature and for optical and thermoelectric characteristics. The material is either a conductor or semiconductor is decided by the nature of the gap between the valence band and conduction band. If the electronic states of the valence band and conduction band overlap at the fermi energy level the material is a conductor otherwise semiconductor. The electronic band structures were addressed here by comparing the GGA and mBJ techniques, and the computed band gap values are reported in Table 5. The GGA approach is well recognized for underestimating the band gap, while the mBJ method may provide superior precise findings. As a response, we are using the mBJ technique as our conclusion to find the band gap values more exactly. Wide band gap semiconductors comprise semiconductors with band gap energy greater than 2 eV (Ji et al. 2020). The compounds A2AgMoBr6(A = K, Rb, Cs) are wide-bandgap semiconductors, implying that they have tremendous promise in various optoelectronic devices. The computed band gap (in Table 5) discovered that switching A-site elements (K/Rb/Cs) slightly changes the band gap. Figure 2 depicts the calculated band structures acquired using the GGA and GGA + mBJ functional. As can be observed in Fig. 2 all perovskite materials have a similar electronic structure in that the valence band maximum (VBM) and conduction band minimum (CBM) do not intersect at the identical high symmetry point, implying that they are all indirect band gap semiconductors.

Calculated band structure of compound A2AgMoBr6 (A = K, Rb, Cs) using GGA and GGA + mBJ approximation
We assessed the total density of states (TDOS) and projected density of states (PDOS) of these compounds using the mBJ function to identify the atomic orbital contributions to the valence and conduction bands, and the graphs are shown in Figs. 3 and 4. The TDOS shows that electronic states in the valence band and conduction band do not cross the fermi level yielding the semiconductor nature of these compounds. The electron transitions appear in the band at the Fermi level, the energy region close to the Fermi level is described as a p-type semiconductor. It is clear from the partial density of states that electronic states in the up channel are close to the fermi level exists a small band gap near the fermi level, it is because of partially filled electrons in the up channel and in the down channel the electronic states are far away from the valence band, this is because the electronic distribution is empty in the down channel results overall semiconductor nature. The p orbital of the Br atoms significantly contributes to the VBM of these perovskite compounds, whereas the CBM is constituted of hybrid states of the Ag/Mo orbitals. A-site (K/Rb/Cs) atoms pertain hardly anything near the Fermi level, corresponding to the small band gap shift caused by A-site atom substitution.

Total density of states of compound A2AgMoBr6 (A = K, Rb, Cs) using GGA and GGA + mBJ approach


Partial density of states of compound A2AgMoBr6 (A = K, Rb, Cs) using mBJ approach
2.3 Optical properties
The optical properties of semiconductor materials are highly correlated to their electronic band structures; it is usually assessed from the dielectric function utilizing the formula (Parvin et al. 2016; Fahad et al. 2015) ε(ω) = ε1(ω) + iε2(ω), where ω is the angular frequency and ε1(ω) and ε2(ω) are the real and imaginary aspects of the complex dielectric function, respectively.
Figure 5 portrays the dielectric function of A2AgMoBr6(A = K, Rb, Cs), with the real part in Fig. 5a and the imaginary part in Fig. 5b. The polarization behavior of the materials is described by the real part ε1(ω). K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 have static dielectric constants ε1(0) of 3.75, 3.80, and 3.92, respectively. The ε1(0) slowly rises as the Alkaline atom (K/Rb/Cs) changes. Hefty static dielectric constants could even endorse low-level charge defects and slow the rate of radiation electron–hole replication, both of which are desirable for light absorption. A significant proportion of impact on various interband transitions between the valence and conduction bands can be identified by the imaginary dielectric function ε2(ω). The imaginary portion of the dielectric function ε2(ω) offers evidence of the materials' light absorption. The specific absorption peaks can be seen in the K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 imaginary parts at energies of 2.48, 2.54, and 2.49 eV.

Variation of optical properties with a real (ε1(ω)) and imaginary (ε2(ω)) dielectric coefficient of double perovskites A2AgMoBr6 (A = K, Rb, Cs) compounds, respectively
The refractive index n(ω), extinction coefficient k(ω), absorption coefficient α(ω), reflectivity coefficient R(ω), optical conductivity k(ω), and energy loss function L(ω) are computed and displayed in Figs. 6, 7 and 8 respectively.

Variation of absorption coefficient (α(ω)) and optical conductivity (σ(ω)) with a photon energy of compounds A2AgMoBr6 (A = K, Rb, Cs) respectively

Variation of Refractive index n(ω) and Extinction coefficient K(ω) with a photon energy of compounds A2AgMoBr6 (A = K, Rb, Cs) respectively

Variation of Energy Loss and Reflectivity with a photon energy of compounds A2AgMoBr6 (A = K, Rb, Cs) respectively
The absorption coefficient for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 materials gives information on how these materials would react to radiation. In response to the absorption coefficient, the frequency significantly affects how incoming photons communicate with electrons, leading the electrons to move from the VB to the CB. The absorption coefficient discloses a material's capacity for absorbing incident photons of a particular energy. Each semiconductor material has a light absorption threshold below which it does not operate. The photon can interfere with valence electrons and be competent in light absorption above this threshold limit. The relationship α = 4πk/λ symbolizes the highest rate of light decay (Hassan et al. 2018; Mahmood et al. 2019; Rashid et al. 2021). Figure 6(a) reveals the computed values of the absorption coefficient α(ω)for these compounds. For A2AgMoBr6(A = K, Rb, Cs), the absorption rate begins at 2 eV. The highest elevation values for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 are around 13.00 eV. Higher energy regions exhibit more fluctuations when electrons absorb the incident photon’s energy with a discrete resonant rate. The electrons are expelled from the valence band into the conduction band due to the interaction between photons and electrons. Due to their high absorption and low energy loss, A2AgMoBr6(A = K, Rb, Cs) are ideal for optoelectronic applications largely owing to extensive research on optical behavior.
The term “optical conductivity” σ(ω) refers to the number of free carriers that emerge because of bond breakdown caused by photon electron interaction and is shown in Fig. 6b. The highest peaks for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 of values 5.62 × 1015 s−1, 5.57 × 1015 s−1 and 5.25 × 1015 s−1 can be seen at 12.96 eV, 12.98 eV, and 12.75 eV respectively. With increasing light absorption, optical conductivity appears to rise as photons transmit energy to dissipate bonds.
The refractive index is vital for portraying a material's optical characteristics and plays a key role in optical devices like solar cells and detectors. (Zhao et al. 2015) The plots for the refractive indices of K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 are shown in Fig. 7a, and they all suit the real part of the dielectric functions. The static refractive index, in this case, is 2.70, 2.75, and 2.80 for all three compounds under investigation A2AgMoBr6 (A = K, Rb, Cs) and are shown in Figure 7a. The extinction coefficient k (ω) is employed to illustrate how an electromagnetic wave attenuates in a material and is directly linked to the imaginary dielectric constant ε2(ω) and absorption coefficient α(ω) (Hassan et al. 2018). As a result, Figure 7b indicates how the plot of the extinction coefficient implements the same pattern as ε2(ω) and α(ω).
The energy loss function is integral to portraying the energy loss perceived by electrons as they move through a dielectric. The apex of the loss function is linked to plasma oscillation, and the function is intrinsically about the real and imaginary parts of dielectric functions (Fatima et al. 2023; Abbas et al. 2022a). The energy loss function as a function of photon energy is exhibited in Figure 8(a). There is a significant energy loss in the two regions, with the maximum energy loss points for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 being at 3.48, 12.40, and 12.48 eV, respectively. When the energy exceeds 12.95 eV, the electronic energy loss for these three compounds is almost zero.
The reflectivity R(ω) of K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 compounds, as seen in Fig. 8b indicate light reflection from any surface. The reflectivity's zero frequency, R(0) of K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 are 4.9, 5.2, and 5.8 respectively. Our findings show that K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 compounds have high reflectivity at lower energies due to the interband transition.
2.4 Thermoelectric properties
The choice of promising material for their commercial use in thermoelectric devices has been heavily inspired by the enormously important transport properties. Numerous factors, such as the Seebeck coefficient (S), electrical conductivity (σ/τ), electron thermal conductivity (k/τ), and figure of merit (zT = S2σT /k), where T is the absolute temperature and τ is the relaxation time, may be utilized to assess a material's thermoelectric properties (Ruleova et al. 2010; Mir and Gupta 2021a; Abdullah and Gupta 2023; Khandy and Gupta 2021; Manzoor et al. 2022). Specifically, it has been confirmed that double perovskite halides displayed superior thermoelectric properties (Roy et al. 2016; Sugahara et al. 2008).
Among the most decisive thermoelectric parameters is electrical conductivity (σ/τ), which is influenced by the number of charge carriers. The temperature-dependent electrical conductivity (σ/τ) for the A2AgMoBr6 (A = K, Rb, Cs) compounds is shown in Fig. 9 It should be acknowledged that as the temperature rises, electrical conductivity grows consistently. Because there is a direct correlation between electrical conductivity and temperature, the carrier's movement and concentration enhancement in the conduction band is the cause (Haleoot and Hamad 2019). Because the electrical conductivity is proportional to the number of charge carriers as σ = neμ. At 300 K (1000 K) the electrical conductivity is 0.58 (2.99 × 1019), 0.42 (3.19 × 1019) and 0.74 (3.17 × 1019) for K, Rb, and Cs respectively.

Variation of electrical conductivity and see beck coefficient with temperature For A2AgMoBr6 (A = K, Rb, Cs)
The Seebeck coefficient (S) is usually portrayed as a response to temperature displays in Fig. 9. Within the temperature range of (100–1000 K), the Seebeck coefficient (S) displayed a decreasing trend with an increase in temperature because the Seebeck coefficient and charge carriers are inversely proportional to each other. The electrical conductivity depicts an increasing pattern with an increase in charge carrier so the Seebeck coefficient must decrease with a temperature rise which is evident from Fig. 9 revealing its semiconductor nature. At 50 K(1000 K) the Seebeck coefficient values for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 are 638.31(202.90) µVK−1, 914.91(205.87) µVK−1 and 479.71(198.33) µVK−1. The values of the Seebeck coefficient indicate a reduction with rising temperature. The substantial rise in carrier density caused by rising temperature leads to a declining Seebeck coefficient (S) value.
A material's potential to transfer heat as a result of lattice vibrations is referred to as thermal conductivity. Quantitatively, thermal conductivity can be expressed as κel = κe + κl, where κel is the resultant of the electronic component (κe) and lattice conductivity (κl). The electron and holes that convey heat are symbolized by the expected electronic component. Figure 10 reveals the thermal conductivity (κe) as a response to temperature. As the temperature increases, κe begins to rise, as shown in Fig. 10. At 1000 K the electronic thermal conductivity values for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 are 1.54 × 1015 WK−1 m−1 s−1, 1.65 × 1015 WK−1 m−1 s−1, and 1.59 × 1015 WK−1 m−1 s−1 respectively.

Variation of electronic thermal conductivity and figure of merit zT with temperature For A2AgMoBr6 (A = K, Rb, Cs)
The most vital component for assessing the effectiveness of thermoelectric materials is the dimensionless figure of merit (zT) (Mir and Gupta 2021b). We deduced the outcome of the figure of merit zT versus temperature to examine the thermoelectric efficiency of A2AgMoBr6 (X = K, Rb, Cs) compounds, as shown in Fig. 10. Numerous parameters attributed to the flow of heat and electricity through materials are mapped by the figure of merit zT. At 50 K (1000 K) the figure of merit zT for K2AgMoBr6, Rb2AgMoBr6, and Cs2AgMoBr6 are 0.96 (0.79), 0.98 (0.81) and 0.93 (0.78) respectively.Our materials have a zT value that is nearest to 1. The essence of augmented achievement for thermoelectric power generators is this quality. Thus, it describes that these materials have the potential to be employed within the thermoelectric industry. As the value of zT gets closer to its maximum, the material becomes considerably more effective at transforming heat energy into usable electrical energy (Saxena and Maiti 2017; Abbas et al. 2022b, 2023).
3 Conclusion
In conclusion, the optoelectronic and thermoelectric properties of the A2AgMoBr6 (X = K, Rb, Cs) halide perovskite have been thoroughly examined using the mBJ potential. Numerous parameters, such as the tolerance factor, formation energy, binding energy, and Born Huang criterion, have been used to confirm thermodynamic reliability and mechanical stability. Additionally, the outcomes of the elastic constants demonstrate the ductility of all the materials and their suitability for use in flexible and portable devices. The ductile nature of the materials is predicted by the calculated Paugh's value, which is greater than 1.75 of A2AgMoBr6 (X = K, Rb, Cs). All three materials have indirect bandgaps corresponding to the bandgap structure results. The examined materials pose as potential candidates for optical devices based on the significant value of the absorption coefficient α(ω) in the UV and visible region. In addition, A2AgMoBr6 (X = K, Rb, Cs) has a ZT value of 0.89, 0.92, and 0.85 respectively at room temperature, making it a good choice for thermoelectric purposes.
Data availability
The datasets generated during and/or analyzed during the current study would be available from the corresponding author upon reasonable request.
References
Abbas, Z., Fatima, K., Abubakr, M., Gorczyca, I., Alshahrani, T., Muhammad, S., Al-Sehemi, A.G.: A DFT+ U study of the effect of transition metal replacements on optoelectronic and elastic properties of TmCu3S4 (Tm= V, Ta, Nb). Optik 250, 168289 (2022a)
Abbas, Z., Fatima, K., Gorczyca, I., Irfan, M., Alotaibi, N., Alshahrani, T., Muhammad, S.: Proposition of new stable rare-earth ternary semiconductor sulfides of type LaTlS2 (La= Er, Eu, Tb): Ab-initio study and prospects for optoelectronic, spintronic and thermoelectric applications. Mater. Sci. Semicond. Process. 146, 106662 (2022b)
Abbas, Z., Fatima, K., Gorczyca, I., Jaffery, S.H.A., Ali, A., Irfan, M., Jung, J.: First-principles calculations to investigate electronic, optical, and thermoelectric properties of Na2GeX3 (X= S, Se, Te) for energy applications. Mater. Sci. Semicond. Process. 154, 107206 (2023)
Abdullah, D., Gupta, D.C.: Structural, mechanical and dynamical stabilities of K2NaMCl6 (M: Cr, Fe) halide perovskites along with electronic and thermal properties. J. Magn. Magn. Mater. 569, 170474 (2023)
Andrianov, A.V., Aleshin, A.N., Matyushkin, L.B.: Terahertz Vibrational Modes in CH3 NH3 PbI3 and CsPbI3 Perovskite films. JETP Lett. 109, 28–32 (2019)
Bhamu, K.C., Soni, A., Sahariya, J.: Revealing optoelectronic and transport properties of potential perovskites Cs2PdX6 (X= Cl, Br): a probe from density functional theory (DFT). Sol. Energy 162, 336–343 (2018)
Birch, F.: Finite elastic strain of cubic crystals Phys. Rev. 71, 809–824 (1947)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: Techn. An Augmented Plane Wave+ Local Orbitals Program for Calculating Crystal Properties, (2001).
Chettri, S., Rai, D.P., Shankar, A., Khenata, R., Ghimire, M.P., Thapa, R.K., Bin Omran, S.: GGA+ U and mBJ+ U study of the optoelectronic, magnetic and thermoelectric properties of the SmAlO3 compound with spin–orbit coupling Int. J. Mod. Phys. B 30(12), 1650078 (2016)
Dahbi, S., Tahiri, N., El Bounagui, O., Ez-Zahraouy, H.: Electronic, optical, and thermoelectric properties of perovskite BaTiO3 compound under the effect of compressive strain. Chem. Phys. 544, 111105 (2021)
Dehkordi, A.M., Zebarjadi, M., He, J., Tritt, T.M.: Thermoelectric power factor: enhancement mechanisms and strategies for higher performance thermoelectric materials. Mater. Sci. Eng. R 97, 1–22 (2015)
Elsheikh, M.H., Shnawah, D.A., Sabri, M.F.M., Said, S.B.M., Hassan, M.H., Bashir, M.B.A., Mohamad, M.: A review on thermoelectric renewable energy: principle parameters that affect their performance. Renew. Sustain. Energy Rev. 30, 337–355 (2014)
Fahad, S., Murtaza, G., Ouahrani, T., Khenata, R., Yousaf, M., Omran, S.B., Mohammad, S.: Structural, elastic, electronic, bonding, and optical properties of BeAZ2 (A= Si, Ge, Sn; Z= P, As) chalcopyrites. J. Alloy. Compd. 646, 211 (2015)
Fatima, K., Abbas, Z., Butt, F., Butt, K., Hussain, S., Ali, A., Al-Sehemi, A.G.: First-principles quantum analysis of promising double perovskites Z2SiF6 (Z= K, Li, Na, Rb) as prospective light harvesting materials: optoelectronic, structural and thermodynamic properties. Int. J. Quant. Chem. 123, e27179 (2023)
Gets, D.S., Tiguntseva, E.Y., Berestennikov, A.S., Lyashenko, T.G., Pushkarev, A.P., Makarov, S.V., Zakhidov, A.A.: Photoinduced migration of ions in optically resonant perovskite nanoparticles. JETP Lett. 107, 742–748 (2018)
Goldsmid, H.J.: The Physics of Thermoelectric Energy Conversion. Morgan and Claypool, San Rafael (2017)
Haleoot, R., Hamad, B.: Thermodynamic and thermoelectric properties of CoFeYGe (Y= Ti, Cr) quaternary Heusler alloys: first principle calculations. J. Phys. Condens. Matter 32(7), 075402 (2019)
Haq, A.U., Mustafa, G.M., Amin, M., Ramay, S.M., Mahmood, A.: Ab‐initio study of opto‐electronic and thermoelectric properties of direct bandgap double perovskites Rb2XGaBr6 (X, Na, K). Int. J. Energy Res. 45(6), 9241–9251 (2021)
Haque, E., Hossain, M.A.: Origin of ultra-low lattice thermal conductivity in Cs2BiAgX6 (X= Cl, Br) and its impact on thermoelectric performance. J. Alloys Compd. 748, 63 (2018)
Hassan, M., Shahid, A., Mahmood, Q.: Structural, electronic, optical and thermoelectric investigations of antiperovskites A3SnO (A= Ca, Sr, Ba) using density functional theory. Solid State Commun. 270, 92–98 (2018)
Houari, M., Bouadjemi, B., Abbad, A., Lantri, T., Haid, S., Benstaali, W., Matougui, M., Bentata, S.: Lead-free semiconductors with high absorption: insight into the optical properties of K2GeSnBr 6 and K2GeSnI6 halide double perovskites. JETP Lett. 112, 364–369 (2020)
Ji, F., Klarbring, J., Wang, F., Ning, W., Wang, L., Yin, C., Gao, F.: Lead‐free halide double perovskite Cs2AgBiBr6 with decreased band gap. Angew. Chem. 132(35), 15303–15306 (2020)
Khandy, S.A., Gupta, D.C.: New isostructural halide double perovskites Cs2GeNiX6 (X= Cl, Br) for semiconductor spintronics and thermoelectric advancements. J. Solid-State Chem. 300, 122196 (2021)
Mahmood, Q., et al.: Density functional theory-based study of the magnetic and optical properties of PbMO3 (M= Cr, Fe) using the modified BeckeJohnson mBJ functional. J. Phys. Chem. Solid 128, 275–282 (2019)
Maiti, T., Saxena, M., Roy, P.: Double perovskite (Sr2B′ B ″O6) oxides for high-temperature thermoelectric power generation: a review. J. Mater. Res. 34, 107–125 (2019)
Manzoor, M., Bahera, D., Sharma, R., Tufail, F., Iqbal, M.W., Mukherjee, S.K.: Vibrational, mechanical, electronic and thermodynamic properties of rhenium-based perovskites XReO3 (X= Li, Be) by an ab-initio computation. Int. J. Energy Res. 46(15), 1–17 (2022)
Mingo, N.: Thermoelectric figure of merit and maximum power factor in III–V semiconductor nanowires. Appl. Phys. Lett. 84(2004), 2652–2654 (2004)
Mir, S.A., Gupta, D.C.: Understanding the origin of semiconducting ferromagnetic character along with the high figure of merit in Cs2NaMCl6 (M= Cr, Fe) double perovskites. J. Magn. Magn. Mater. 519, 167431 (2021a)
Mir, S.A., Gupta, D.C.: Scrutinizing the stability and exploring the dependence of thermoelectric properties on band structure of 3 d-3 d metal-based double perovskites Ba2FeNiO6 and Ba2CoNiO6. Sci. Rep. 11(1), 10506 (2021b)
Mousa, A.A., Al-Qaisi, S., Abu-Jafar, M., Al Azar, S., Jaradat, R., Khalifeh, J.M., Khenata, R.: Ab initio studies of the structural, elastic, electronic and optical properties of the Ni3In intermetallic compound. Mater. Chem. Phys. 249, 123104 (2020)
Murtaza, G., Alshahrani, T., Khalil, R.M.A., Mahmood, Q., Flemban, T.H., Althib, H., Laref, A.: Lead free double perovsites halides X2AgTlCl6 (X= Rb, Cs) for solar cells and renewable energy applications. J. Solid State Chem. 297, 121988 (2021)
Nabi, M., Bhat, T.M., Gupta, D.C.: Effect of pressure on electronic, magnetic, thermodynamic, and thermoelectric properties of tantalum‐based double perovskites Ba2MTaO6 (M= Mn, Cr). Int. J. Energy Res. 43(9), 4229–4242 (2019)
Nemtsev, A.V., Zhandun, V.S., Zinenko, V.I.: Ab initio study of the polarization, electronic, magnetic, and optical properties of perovskite SrMO3 (M= Fe, Mn) crystals and thin films containing magnetic ions. J. Exp. Theor. Phys. 126, 497–505 (2018)
Noor, N.A., Iqbal, M.W., Zelai, T., Mahmood, A., Shaikh, H.M., Ramay, S.M., Al-Masry, W.: Analysis of direct band gap A2ScInI6 (A= Rb, Cs) double perovskite halides using DFT approach for renewable energy devices. J. Matter. Res. Technol. 13, 2491–2500 (2021)
Parvin, R., Parvin, F., Ali, M.S., Islam, A.K.M.A.: Band structure, fermi surface, elastic, thermodynamic, and optical properties of AlZr3, AlCu3, and AlCu2Zr: First-principles study. Chin. Phys. b. B 25, 083101 (2016)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996)
Petersen, M., Wagner, F., Hufnagel, L., Scheffler, M., Blaha, P., Schwarz, K.: Improving the efficiency of FP-LAPW calculations. Comput. Phys. Commun. 126, 294–309 (2000)
Pugh, S.F.: Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 45, 823–843 (1954)
Radja, K., Farah, B.L., Ibrahim, A., Lamia, D., Fatima, I., Nabil, B., Mohamed, A., Al-Douri, Y., Abd-El-Rehim, A.F.: Investigation of structural, magneto-electronic, elastic, mechanical and thermoelectric properties of novel lead-free halide double perovskite Cs2AgFeCl6: First-principles calcuations. J. Phys. Chem. Solids 167, 110795 (2022)
Rahman, M.A., Mousumi, K., Ali, M.L., Rahman, M.Z., Hasan, S.S., Hasan, W., Hasan, M.Z.: First-principles calculations to investigate elastic, electronic, optical and thermodynamic properties of Pt3X (X= Ti, Cu). Results Phys. 44, 106141 (2023)
Rashid, M., et al.: Pressure-induced modifications in the optoelectronic and thermoelectric properties of MgHfO3 for renewable energy applications. Arab. J. Sci. Eng. 47, 777–785 (2021)
Ravi, V.K., Singhal, N., Nag, A.: Initiation and future prospects of colloidal metal halide double-perovskite nanocrystals: Cs2 AgBiX6 (X= Cl, Br, I). J. Mater. Chem. A 6, 21666–21675 (2018)
Roy, P., Bose, I., Maiti, T.: Synthesis and characterization of Sr2TiMO6 (M= Fe, Co) double perovskites for high temperature thermoelectric applications. Integr. Ferroelectr. 174(1), 34–42 (2016)
Ruleova, P., Drasar, C., Lostak, P., Li, C.P., Ballikaya, S., Uher, C.: Thermoelectric properties of Bi2O2Se. Mater. Chem. Phys. 119(1–2), 299–302 (2010)
Saeed, M., Haq, I.U., Saleemi, A.S., Rehman, S.U., Haq, B.U., Chaudhry, A.R., Khan, I.: First-principles prediction of the ground-state crystal structure of double-perovskite halides Cs2AgCrX6 (X= Cl, Br, and I). J. Phys. Chem. Solids 160, 110302 (2022)
Saxena, M., Maiti, T.: Effect of Ba-doping on high temperature thermoelectric properties of Sr2TiMoO6 double perovskites. J. Alloy. Compd. 710, 472–478 (2017)
Schwarz, K., Blaha, P., Madsen, G.K.H.: Electronic structure calculations of solids using the WIEN2k package for material sciences. Comput. Phys. Commun. 147, 71–76 (2002)
Shafiq, M., Ahmad, I., Asadabadi, S.J.: Theoretical studies of strongly correlated rare-earth intermetallics RIn3 and RSn3 (R= Sm, Eu, and Gd). J. Appl. Phys. 116, 103905 (2014)
Shafiq, M., Arif, S., Ahmad, I., Jalali Asadabadi, S., Maqbool, M., Aliabad, H.A.R.: Elastic and mechanical properties of lanthanide monoxides. J. Alloys Compd. 618, 292 (2015)
Shi, W., Cai, T., Wang, Z., Chen, O.: The effects of monovalent metal cations on the crystal and electronic structures of Cs2MBiCl6 (M= Ag, Cu, Na, K, Rb, and Cs) perovskites. J. Chem. Phys. 153, 141101 (2020)
Sugahara, T., Ohtaki, M., Souma, T.: Thermoelectric properties of double-perovskite oxide Sr2-xMxFeMoO6 (M= Ba, La). J. Ceram. Soc. Jpn. 116(1360), 1278–1282 (2008)
Tran, F., Blaha, P.: Generalized gradient approximation made simple. Phys. Rev. Lett. 102, 226401 (2009)
Tritt, T.M., Subramanian, M.A.: Thermoelectric materials, phenomena, and applications: a bird’s eye view. MRS Bull. 31, 188–198 (2006)
Varadwaj, P.R.: A2AgCrBr6 (A= K, Rb, Cs) and Cs2AgCrX6 (X= Cl, I) double perovskites: a transition-metal-based semiconducting material series with remarkable optics. Nanomaterials 10(5), 973 (2020)
Varadwaj, P.R., Marques, H.M.: The Cs2AgRhCl6 halide double perovskite: a dynamically stable lead-free transition-metal driven semiconducting material for optoelectronics. Front. Chem. 8, 796 (2020)
Yan, L., Zhao, L., Zhao, C., Lin, S.: Theoretical Understanding of thermoelectric energy conversion efficiency in Lead-Free halide double perovskites showing intrinsic defect tolerance. Appl. Therm. Eng. 215, 119024 (2022)
Zhao, S., Lan, C., Ma, J., Pandey, S.S., Hayase, S., Ma, T.: First principles study on the electronic and optical properties of B-site ordered double perovskite Sr2MMoO6 (M= Mg, Ca, and Zn). Solid State Commun. 19, 213–214 (2015)
Zheng, X.F., Liu, C.X., Yan, Y.Y., Wang, Q.: A review of thermoelectrics research–recent developments and potentials for sustainable and renewable energy applications. Renew. Sustain. Energy Rev. 32, 486–503 (2014)
Zhou, X., Jankowska, J., Dong, H., Prezhdo, O.V.: Recent theoretical progress in the development of perovskite photovoltaic materials. J. Energy Chem. 27, 637–649 (2018)
Funding
The research work is not funded by any agency.
Author information
Authors and Affiliations
Contributions
Both authors have significantly contributed to the research. DA: conceptualization, methodology, writing—original draft, revision; and DCG: supervision, software, validation, writing—review, and modification.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abdullah, D., Gupta, D.C. Scrutinizing the structural, opto-electronic, mechanical, and thermoelectric properties of semiconductor lead-free double perovskites A2AgMoBr6 (A = K, Rb, Cs). Opt Quant Electron 55, 1277 (2023). https://doi.org/10.1007/s11082-023-05493-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05493-2