
Abstract
In current study, secondary ketamine derivatives (1–6) were synthesized through refluxing of 2-acetyl naphthalene, substituted phenyl and benzyl thiosemicarbazides solutions in ethanol. Different spectroscopic procedures like nuclear magnetic resonance (NMR), fourier transform infrared (FT-IR), and UV–Visible (UV–Vis) spectroscopy were used for the characterization of the synthesized compounds. Accompanying with experimental, DFT study was also made at M06/6-311 G (d,p) functional. A relative investigation of experimental and DFT spectroscopic outcomes expressed good harmony in results. Various kinds of analyses such as natural bond orbitals (NBOs), geometric optimization, molecular electrostatic potential (MEP), frontier molecular orbitals (FMOs), UV–Vis, binding energy (Eb) and transition density matrix (TDM) were conducted to explore the non-linear optical (NLO) properties of secondary ketamine compounds. Interestingly, all the synthesized compounds expressed linear polarizability [⟨α⟩ = 416.71–429.91 a.u.], first hyperpolarizability (βtot = 795.768–1201.321 a.u.), and second hyperpolarizability (γtot = 4.967–6.176 a.u.) values than that of standard urea molecule which illustrated them as efficient optoelectronic materials for hi-tech NLO applications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The potential for developing new NLO materials has recently received significant attention in the scientific community (Liu et al. 2006; Pritula et al. 2008). This incredible work has been encouraged by the significant advancement in NLO materials as well as the increasing interest in using optics and laser technologies in the processing of information and telecommunication (Paufler and Cotter 1990), sensing, imaging, microfabrication and cancer therapy (Parodi et al. 2020). Massive scientific efforts have been made in recent years to explore unique NLO compounds, containing organic and inorganic semiconductor diodes, synthetic resins, and molecular dyes etc. (Datta 2009). Organic compounds are preferred over inorganic in terms of nonlinearity owing to small dielectric constant value (Xu et al. 2020), influence of π-bonded electronic delocalization, high thermal and chemical stability, easy preparation (Berkowski et al. 2003), high values of photoelectrical coefficients, small dielectric constant value and low cost (Ambujam et al. 2006). Researchers have focused on synthesizing organic compounds with electronic characteristics and definite geometries to attain significant NLO properties (Akram et al. 2018; Makowska-Janusik 2007). It is assumed that NLO properties are emerged, due to powerful intramolecular charge transfer (ICT) in organic compounds (Khalid et al. 2019). The ICT process is inferred via donor–π–accepter (D–π–A) frame work (Abdulla 1972; Khalid et al. 2022b), where the donor acts as the hole transport and the acceptor works as an electron carrier (Khalid et al. 2019) establishing a “push–pull” mechanism (Coluccini et al. 2013). Generally, the structural building elements of NLO constituents can be divided into two categories: (1) π-conjugated elements and (2) non-π-conjugated components. π-conjugated system displays large value of hyperpolarizabilities (βtot and γ) with small value of HOMO–LUMO energy band gap (Khalid et al. 2022a). Therefore, the ELUMO–EHOMO value of π-conjugated system is generally lesser relative to non-π-conjugated system (Xiong et al. 2021). The π-conjugated system offers an effective D to A charge transmission (Elhorri 2020). There has been a significant inquisitiveness in the co-ordination chemistry of aryl hydrazones i.e., semicarbazones and thiosemicarbazones owing to their significance for organocatalysis, drug design and for the formulation of heterocyclic rings (Beraldo and Gambino 2004; Bondock et al. 2007). Due to strong ion-sensing capability, geometric variations, bonding mechanisms and biological consequences, thiosemicarbazones (TSCs) are diverse category of Schiff-based ligands comprising -S and -N acting as D-atoms (García-Tojal et al. 2011) are widely use in optical sensing.
Thiosemicarbazone analogues are interesting compounds for strong second harmonic generation (SHG) (Krishnakumar and Nagalakshmi 2005) and could be used in nonlinear optics (NLO) (Yousef et al. 2012). It is claimed that enormous electron dispersion in thiosemicarbazone entity assists the free thiosemicarbazone ligand as well as their complexes to increase the SHG efficacy (Dinçer et al. 2006, 2005; Tian et al. 1999). Thiosemicarbazide is displayed as an effective ligand for a variety of metals, comprising cadmium, zinc and nickel (Beraldo and Gambino 2004). Earlier studies propose that such ligands present an outstanding perspective to analyze the effect of ligand on the development of nanomaterials and also the significance of compounds comprising thiosemicarbazide moiety in medicinal usages (Santhakumari et al. 2010). Single crystal of acetonapthone thiosemicarbazone is amongst the impending organic NLO constituents, that are associated to the ketone group of compounds. Derivatives of thiosemicarbazone are widely explored by both theoretical and experimental societies owing to less dielectric constant, broad and ultrafast electronic responses deliberated in auspicious optoelectronic technologies (Riscob et al. 2011; Türkkan et al. 2015). In view of the thiosemicarbazone importance in NLO and biological applications with having relative study background for synthesis and computational (Adeel et al. 2017; Shahid et al. 2018), herein, thiosemicarbazone (2-acetonapthone secondary ketamine) based NLO compounds (1–6) are synthesized, and the experimentation turned out to exhibit excellent yield. Perhaps, no principal study has done previously on 2-acetonapthone secondary ketamine based NLO compounds and so the synthesis, characterization and DFT computation on 1–6 compounds is employed. Nowadays, DFT analysis coupled with experimental study has provided a comprehensive description of material characteristics. Whereas, the synthesized molecules are theoretically evaluated by utilizing IR, UV–Vis, NBOs, MEP, NPA and FMOs investigations at M06/6-311G(d,p) functional. Novel compounds are synthesized and are computationally optimized in this research and the primary objective of this report is to make an inclusive architectural and optical explanation of synthesized compounds using a variety of experimental and theoretical strategies.
2 Experimental studies materials
Chemicals and solvents were directly acquired from Merck and were utilized without further purification. The melting point instrument slip melting point (SMP) 10 was used for melting point measurement. IR (450–4000 cm−1) was performed with Bruker FTIR tensor-27. The T-80 was used for the monitoring of electronic spectrums employing UV–Vis spectrophotometer. A Bruker AM 600 spectrometer was used with dimethyl sulfoxide (DMSO) as a solvent and tetramethylsilene (TMS) as an internal standard to capture a 13C or 1H-NMR spectrum (Figures S9-S20). The spectra of proton NMR was observed at 600 MHz, 13C NMR at 75 MHz or 62.9 MHz.
3 Preparation of thiosemicarbazones
Thiosemicarbazones derivatives were prepared by using equimolar quantities of 2-acetyl naphthalene (0.01M) and substituted phenyl and benzyl thiosemicarbazides (0.01) solutions in ethanol. Both solutions were mixed and 2–3 drops of glacial acetic acid were added as a catalyst. Mixture was refluxed under condensation at 80 °C for 6 h with continuous stirring, till the precipitation occurred. Completion of reaction was monitored continuously after some fixed intervals of time. Then the product was filtered and washed several times with absolute ethyl alcohol, dried and weighed.

3.1 (E)-(N)-(2,3-dimethylphenyl)-2-(1-(naphthalene-2yl)ethylidene)hydrazinecarbothioamide (1)
Yield, (76%); White crystalline: m.p, 190 °C; IR (KBr) ѵ cm−1: 3286 (N–H), 1207 (C=S), 1590 (C=N). UV–Vis (Dimethylformamide): λmax 325 nm. 1H-NMR, δ (ppm): 7.130–8.386 (m, three phenyl rings), 10.559 (s, 1H, N–NH), 9.986 (s, 1H, Ar–NH), 2.285 (s, 3H, C–CH3), 2.121 (s, 3H, Ph-CH3), 2.491 (s, 3H, Ph-CH3). 13CNMR δ (ppm): 178.00 (C=S), 147.98 (C=N), 20.10 (C–CH3), 14.22 (Ph-CH3), 14.04 (Ph-CH3) 124.38, 125.14, 126.34, 126.57, 126.69, 126.83, 127.46, 127.48, 128.09, 128.57, 132.79, 133.32, 134.14, 135.07, 136.76, 138.26 (aromatic C and CH).
3.2 (E)-(N)-(2,4-dimethylphenyl)-2-(1-(naphthalene-2yl)ethylidene)hydrazinecarbothioamide (2)
Yield (82%); White crystalline: m.p, 187 °C; IR (KBr) v cm−1: 3286 (N–H), 1218 (C=S) 1600 (C=N). UV–Vis (Dimethylformamide): λmax 325 nm. 1H-NMR, δ (ppm): 7.003–8.096 (m, three phenyl ring s), 10.479 (s, 1H, N–NH), 9.845 (s, 1H, Ar–NH), 2.170 (s, 3H, C–CH3), 2.286 (s, 3H, Ph-CH3), 3.316 (s, 3H, Ph-CH3). 13CNMR δ (ppm): 177.94 (C=S), 163.73 (C=N), 14.23 (C–CH3), 20.65 (Ph-CH3), 17.79 (Ph-CH3) 114.96, 115.10, 126.44, 128.45, 129.13, 129.19, 130.59, 134.08, 134.10, 135.06, 135.73, 135.76, 147.18, 162.09 (aromatic C and CH).
3.3 (E)-(N)-(2,5-dimethylphenyl)-2-(1-(naphthalene-2yl)ethylidene)hydrazinecarbothioamide(3)
Yield (72%); White crystalline: m.p, 198 °C; IR (KBr) ѵ cm−1: 3338 (N–H), 1160 (C=S) 1517 (C=N). UV–Vis (Dimethylformamide): λmax 325 nm. 1H-NMR, δ (ppm): 7.519–8.484 (m, three phenyl rings), 10.594 (s, 1H, N–NH), 9.883 (s, 1H, Ph-NH), 3.321 (s, 3H, C–CH3), 2.490 (s,3H, Ph-CH3), 2.209 (s, 3H, Ph-CH3). 13CNMR δ (ppm): 177.80 (C=S), 147.73 (C=N), 13.94 (C–CH3), 19.18 (Ph-CH3), 18.15 (Ph-CH3), 124.47, 126.31, 126.65, 126.79, 126.90, 127.44, 127.45, 127.60, 128.57, 132.79, 133.32, 135.12, 136.47, 137.52 (aromatic C and CH).
3.4 (E)-(N)-(2,6-dimethylphenyl)-2-(1-(naphthalene-2yl)ethylidene)hydrazinecarbothioamide(4)
Yield (76%); White crystalline: m.p, 189 °C; IR (KBr) ѵ cm−1: 3300 (N–H), 1208 (C=S) 1600 (C=N). UV–Vis (Dimethylformamide): λmax 325 nm. 1H-NMR, δ (ppm): 7.017–8.421 (m, three phenyl rings), 10.574 (s, 1H, N–NH), 9.948 (s, 1H, Ar–NH), 2.201 (s, 3H, C–CH3), 2.489 (s, 3H, Ph-CH3), 2.290 (s, 3H, Ph-CH3). 13CNMR δ (ppm): 177.70 (C=S), 148.01 (C=N), 14.06 (C–CH3), 20.52 (Ph-CH3), 17.49 (Ph-CH3), 124.34, 126.35, 126.70, 126.84, 127.30, 127.46, 127.49, 128.57, 128.95, 129.91, 132.18, 132.78, 133.33, 134.96, 135.06, 138.07 (aromatic C and CH).
3.5 (E)-N-(4-methoxybenzyl)-2-(1-naphthalen-2-yl)ethylidene)hydrazinecarbothioamide (5)
White crystalline: Yield, 86.2%; mp, 182 °C; IR (KBr), ѵ (cm−1): 3317 (N–H), 1203 (C=S), 1598 (C=N). UV–Vis (ethanol): 361 nm. 1H-NMR, δ (ppm): 4.9384 (d, J = 5.12 Hz, 2H, CH2–NH), 8.0183 (s, 1H, CS-NH), 8.8971 (s, 1H, NHN), 2.3364 (s, 3H, CH3), 2.7111(s, 3H, OCH3), 7.1571–7.8581 (m, three phenyl rings). 13CNMR, δ (ppm): 137.5302 (C = N), 177.8169 (C=S), 29.7178 (CH2–N), 13.5656 (C–CH3), 48.3456 (OCH3), 123.1847, 126.7175, 127.7346, 129.5077, 132.9803 (aromatic C and CH).
3.6 (E)-N-(2,5-dichlorobenzyl)-2-(1-(naphthalen-2-yl)ethylidene)hydrazinecarbothioamide (6)
Off white crystalline: Yield, 54%; mp, 169 °C; IR (KBr), ѵ (cm−1): 3310 (N–H), 1203 (C=S), 1590 (C=N). UV–Vis (ethanol): 361 nm. 1H-NMR, δ (ppm): 5.028–5.0207 (d, J = 5.84 Hz, 2H, CH2–NH2), 8.8467 (s, 1H, NHN), 2.3763 (s, 3H, CH3), 7.2104–8.0495 (m, three phenyl rings). 13CNMR, δ (ppm): 134.2115 (C=N), 178.3254 (C=S), 45.4818 (CH2–N), 13.4519 (C–CH3), 123.0843, 123.9207, 126.7778, 127.3666, 129.4207, 131.1671, 132.9937 (aromatic C and CH).
4 Computational procedure
The vibrational, NBOs, MEP, NPA and NLO properties of novel thiosemicarbazone derivatives (1–6) were measured by employing quantum chemical algorithms at the M06/6-311G(d,p) level (Poirier et al. 1985) with Gaussian 09 (Frisch 2016). In addition, Gauss View 5.0 (Frisch et al. 2000) was utilized for input files generation, whereas the software Chemcraft (Zhurko 2014), Gaussum 5.0 (O’boyle et al. 2008), and Avogadro (Hanwell et al. 2012) were utilized to examine data from output files. Chemcraft provided elucidation of NBOs, Avogadro generated FMOs surface diagrams showing electronic charge density along with energy gap (Egap) determination, and Gaussum interpreted UV–Vis spectral data. Furthermore, for UV–Vis and FMOs investigations, time-dependent density functional theory (TD-DFT) (Burke et al. 2005; Hirata and Head-Gordon 1999) computations at the aforementioned level were employed. The global reactivity parameters were determined with the aid of Koopmans’s theorem (Fukui 1982) utilizing the HOMO/LUMO bandgaps. NBO 3.1 program package (Glendening et al. 1998) was potentially employed with second order perturbation theory access, to investigate the hyper conjugative interactions within the entitled compounds.
5 Results and discussion
Thiosemicarbazones derivatives were prepared by using equimolar quantities of 2-acetyl naphthalene (0.01M) and substituted phenyl and benzyl thiosemicarbazides (0.01M) through condensation method. Purity of compounds was tested by thin layer chromatography (TLC). Ligands were characterized by UV–Vis, FTIR and NMR spectroscopy, in addition nonlinear optical (NLO) properties were also investigated. The comparable results were found in the vibrational frequencies (Tables S8–S13) and NMR data (Tables S24–S35) by changing the various group and their position in the synthesized compounds.
5.1 1H and 13C NMR analyses
1H and 13C NMR spectra also assisted to determine the structure of titled products. The 1H–NMR spectra of compound 1–6 showed absorption signal of aromatic carbons in the region of 7.003–8.484 ppm. The downfield singlet peak exhibited by N–NH proton in the region of 8.847–10.594 ppm. The slightly up field singlet peak for Ph-NH of compounds (1–4) occurred at 9.845–9.986 ppm and C–NH in compounds 5 and 6 appeared as singlet at 8.018 and 8.208 ppm. The up field peak for C-CH3 is displayed in the region of 2.201–2.490 ppm in compounds (1–6). The protons of Ph-CH3 in compounds 1–4 showed singlet signal in the region 2.121–2.489 ppm, while the methoxy protons appeared as singlet signal at 2.711 ppm in compound 5. Moreover, the doublet peak in compounds 5 and 6 for CH2–N protons displayed at 4.938 and 5.021 ppm, correspondingly. The 13C NMR of compounds (1–6) disclosed peaks in the region 177.700–178.325 ppm for C=S and C=N peak seemed in the region of 134.21–163.73 ppm. The peak emerged in the region of 14.04–20.65 ppm for Ph-CH3 in compounds (1–4), while the methoxy carbon in compound 5 exhibited the peak at 29.718 ppm. The peaks appeared at region 13.452–20.100 ppm for C–CH3, while The CH2–N carbon in compound 5 and 6 showed peak at 48.346 and 45.482 ppm, respectively. A comparative analysis between DFT and experimental NMR data is illustrated in Tables S24–S35.
5.2 Geometric parameters
The optimized geometries of 1–6 compounds are expressed in Figure S21. The simulated bond angles for naphthalene in compounds 1–6 fall within the range of 118.6–122.6°, with bond lengths ranging from 1.361 to 1.423 Å which showed good harmony with experimentally reported bond angles: 133–142, bond lengths: 1.40–1.44 Å (Coulson et al. 1951) Likewise, for benzene, the simulated bond lengths are noted to be in the range of 1.382–1.397 Å, along with bond angles ranging from 119.4° to 121.2°. These values also showed closeness with experimental data (bond lengths: 1.382–1.393 Å, bond angle r: 119–123°) (Demaison et al. 2013; Heo et al. 2022). In all compounds (1–6), the bond lengths between functional groups such as C=S and N−C were found to be in the range of 1.646 Å and 1.284–1.383 Å, respectively, along with bond angles of 124.1–125.2° and 116–124.3°, respectively. All these DFT values showed good harmony with experimental values (Ashfaq et al. 2022; Khalid et al. 2020; Liu and Hoffmann 1995).
5.3 UV–vis analysis
The absorbance spectra of all compounds is acquired to examine maximum absorption wavelengths (λmax), molecular orbital contributions, oscillator strength (fos), and excitation energies (E). All these parameters were computed utilizing TD-DFT approach at aforementioned functional and the obtained outcomes are formulated in Table 1, while Tables S14–S19 represented six singlet excitations. Additionally, a comparative experimental and DFT study of λmax was also conducted for current synthesized compounds (Figure S7).
The computed results ascribed that the λmax (DFT) of the entitled compounds lies in the close proximity of each other (324.3–330.7 nm). Compound 1–6 displayed λmax (DFT) at 329.3, 329.0, 330.7, 328.4, 327.1 and 324.3 nm, correspondingly, observed to be in agreement with experimentally obtained λmax; 325, 325, 325, 325, 361 and 361 nm, correspondingly (Table 1). The highest experimental λmax is detected in compounds 5 and 6 i.e., 361 nm, while the maximum simulated λmax is observed in compound 3 at 330.7 nm. Compound 6 possessed the maximum excitation energy of 3.823 eV amongst all synthesized compounds with 0.989 fos and 93% HOMO to LUMO orbital contribution at simulated absorption maxima of 324.3 nm. Of all the synthesized compounds, 3 is found to have the maximum fos of 1.092 with E at 3.749 eV and HOMO to LUMO orbitals contribution of 90% at λmax of 330.7 nm. A comparative analysis is also made between these synthesized compounds and our previously reported work (Jawaria et al. 2022). Interestingly, our compounds (1–6) exhibited bathochromic shift (324.3–330.7 nm) with greater value of oscillation strength (0.986–1.092) and lower excitation energy (3.749–3.823 eV) than that of TMOH-DCMH (λmax= 314.9–316 nm, fos= 0.643–0.732 and E = 3.922 -3.937 eV). It has been seen that the molecule that displays lower excitation energy and greater absorbance shows better charge transfer and higher efficiency (Mahmood et al. 2019, 2015, 2014). Overall, UV–Vis analysis showed that all the synthesized molecules (1–6) expressed redshift spectrum which showed greater charge transference therefore, can be used as NLO materials.
5.4 Vibrational analysis
To study vibrational modes nature of compounds (1–6), theoretically and experimentally vibrational studies were carried out. Tables S8−S13 presents values of absorption frequencies and Figures S1–S6 presents experimental spectra. Acetonaphtone was taken as reference and all the vibrational peaks were appeared at expected region. A momentous absorption band in the functional group band region was seen in the infrared spectra of new compounds (1–6). In compounds (1–6), the –NH group was correspondingly responsible for intense bands at frequency of 3286–3338 cm−1. The distinctive band associated to –CO at 1680 cm−1 disappeared, and new band associated to C=N appeared in the frequency range of 1600–1517 cm−1, confirming the new functional group (C=N) introduce in these compounds. The notable absorption band peak of C=S and the free aliphatic CH group existed in the 1203–1517 and 2863–2870 cm−1 ranges, respectively.
5.4.1 C–H vibrations
Experimental and DFT studies are used to detect vibrational modes nature for compounds (1–6). In the region of 3300–2800 cm−1, C–H stretching vibrations are found. In the present study, absorption bands are originated at 3005, 3005, 3011, 3002, 2979 and 3015 cm−1 (DFT) and on account of symmetric stretching vibrations for compounds 1, 2, 3, 4, 5 and 6, correspondingly. Experimentally obtained C–H vibrational bands are found at 2963, 2869, 2915, 2870, 2863 and 2863 cm−1 for compounds 1–6, correspondingly.
5.4.2 C=N vibrations
Vibrational spectra of C=N is perceived in the range of 1700–1500 cm−1, correspondingly which are detected at 1534, 1676, 1538, 1658, 1547 and 1535 cm−1 in (DFT) calculations for compounds 1–6, respectively. Furthermore, these DFT spectrum corresponds to the experimental outcomes at 1590, 1600, 1517, 1600, 1598 and 1590 cm−1, respectively.
5.4.3 N–H vibrations
Vibrational spectra of (N–H) is normally seen between 3600 and 3250 cm−1 at 3557 to 3489, 3455 to 3490, 3555 to 3489, 3546 to 3488, 3566 to 3488 and 3572 to 3482 cm−1 in (DFT) calculations for compounds 1–6, correspondingly. Furthermore, these DFT spectrum corresponds to the experimental outcomes at 3286, 3286, 3338, 3300, 3317 and 3310 cm−1 for compounds 1–6, correspondingly.
5.4.4 C=S vibrations
The C=S bonds are identified in the range of 1500–1200 cm−1 and vibrational peaks in current investigation are observed at 1287, 1498, 1509, 1228, 1238 and 1237 cm−1, while the experimental values are detected at 1207, 1437, 1517, 1208, 1203 and 1203 cm−1, correspondingly for compounds 1–6, correspondingly.
5.5 Frontier molecular orbitals (FMOs) study
The FMOs inquiry is significant to determine the chemical stability, electronic and optical properties of the compounds (Li et al. 2018; Sung and Hsu 1998). The capability electron acceptance is ascertained by the lowest unoccupied molecular orbital (LUMO) energy values while electron donation is estimated by the highest occupied molecular orbital (HOMO) energy values. Both of these orbitals play a dynamic role to conclude UV–Vis spectra and reaction mechanism. Moreover, the HOMO–LUMO gap also govern the parameters for the calculation of the chemical reactivity and the kinetic stability of the molecules (Nan et al. 2020). The compounds with high band gaps are considered as chemically stable and hard. Whereas, the compounds with greater degree of softness are considered to be less stable and have minor energy gap. Furthermore, the molecules with low HOMO/LUMO band gap typically display high polarizability and good NLO response (Dini and Hanack 2004). The FMOs of 1–6 are determined at the abovementioned functional and the ELUMO, EHOMO and energy gaps (Egap) are tabulated in Table 2 and for higher orbitals the values are tabulated in Table S1.
The HOMO/LUMO energy values of compounds (1–6) are observed to be − 6.323/− 1.763, − 6.317/− 1.757, − 6.329/− 1.770, − 6.337/− 1.751, − 6.290/− 1.699 and − 6.330/− 1.695 eV, respectively while their Egap values are 4.560, 4.560, 4.559, 4.586, 4.591 and 4.635 eV, correspondingly (Table 2). The topmost value of Egap (4.635 eV) is perceived in compound 6 possessing chloro substituent around the benzene ring. Compound 5 depicted a slightly lesser Egap value as compared to compound 6 which might be due to the + resonance effect of methoxy substituent. Further decline in Egap is detected in compound 4 due to the methyl group at 2 and 6 positons of benzene ring. Likewise, compounds (3, 2 and 1) demonstrated the least values of Egap attributed to methyl group presence at ortho, para and meta positions. The lowest values of Egap in these compounds might be due to the hyper conjugative effect of methyl substituent. The escalating Egap of the entitled compounds is: 3 < 1 = 2 < 4 < 5 < 6. The HOMO/LUMO orbital charge densities are displayed in Fig. 1. It is clearly obvious from surface diagrams that the electronic densities were significantly placed over the (E)-2-(1-(naphthalen-2-yl)ethylidene)hydrazinecarbothioamide moiety in every entitled compound.

The FMOs of the synthesized compounds (1–6)
5.6 Global reactivity parameters (GRPs)
Various global reactivity descriptors like electrophilicity index (ω), global hardness (η), ionization potential (IP), electronegativity (X), global softness (σ), chemical potential (μ) and electron affinity (EA) can be attributed to the energy band gaps (Egap = ELUMO–EHOMO)(Khan et al. 2021; Saleem et al. 2022). The electron affinity and ionization energy values represented the electron accepting and donating capability of the molecules. Usually, the molecular stability is signified by the negative values of the chemical potential and the electron attracting capability is denoted by electronegativity. GRPs are essentially calculated via Koopman’s theorem (Fukui 1982), the utilized Eqs. (1–6) (Chattaraj and Roy 2007; Koopmans 1934, 1933; Parr et al. 1978; Parr and Pearson 1983; Pearson 1986) are mentioned below, while the computed values of GRPs are shown in Table 3.
An IP of 6.323, 6.327, 6.339, 6.347, 6.290 and 6.330 eV is observed in 1–6, respectively. The increasing trend for the IP is; 4 > 3 > 6 > 2 > 1 > 5. While the increasing EA order for the entitled compounds is: 6 (1.695) < 5(1.699) < 4(1.751) < 2(1.757) < 1 (1.763) < 3(1.770) in eV. Furthermore, the increasing trend for the electronegativity (X) values in eV is noticed as: 5(3.995) < 6(4.013) < 2(4.037) < 1(4.043) < 4(4.044) < 3(4.050). The global electrophilicity index (ω) can be recorded in terms of reduced energy upon maximized electron transfer from donor towards acceptor moiety (Ramesh and Reddy 2022). Eventually, the highest electrophilicity index has been estimated for compound 3 with the maximum electronegativity i.e., (X = 4.050, ω = 3.597 eV).
The calculated hardness values of the synthesized compounds are in the following ascending order as: 1 = 2 = 3 < 5 < 4 < 6. The softness trend is reverse of the abovementioned hardness trend. The increasing order of the softness is as follows: 6 < 5 = 4 < 3 = 2 = 1. Furthermore, the highest hardness (4.635 eV) and the lowest softness (0.216 eV) values are observed for compound 6 which manifested it as chemically hard, less reactive and kinetically more stable among all the synthesized compounds (1–6).
Surprisingly, the greatest softness (0.219 eV) values have been evaluated for the compounds 1–3 which demonstrated that these compounds are kinetically less stable, soft and chemically more reactive than rest of molecules all. Moreover, the negative chemical potential values and higher magnitudes of hardness as compared to softness, indicated the synthesized compounds as kinetically stable, thus enhancing the efficiency of their synthesis. Surprisingly, the calculated GRPs are in coherence with the band gap trends along with the computed absorption maxima. It is concluded that compound 3 is found to have the smallest band gap (4.559 eV), the largest simulated λmax (330.7 nm), the smallest hardness (4.569 eV) and the largest softness (0.219 eV), making it a remarkable NLO entity that would be potentially employed in optoelectronics.
5.7 Natural bonding orbitals (NBOs) analysis
The NBOs investigation has been proved to be a valuable method for examining the interactions among different bonds and for charge transference across unfilled and filled orbitals (Bal et al. 2014; Khalid et al. 2020). It has also been used to offer a good visual overview of intramolecular delocalization in chemical structures. The second-order perturbation stabilization energy of any molecule in NBOs analysis could be determined by Eq. 7 (Khalid et al. 2021).
where E(2) stands for the stabilization energy, qi refers to the donor−orbital occupancy, Fi,j indicates the off-diagonal NBO Fock matrix elements, and Ei and Ej point out the diagonal elements. NBOs examination of compounds 1 to 6 has been carried out and results are showed in Tables S2–S7.
For compound 1, π → π* electronic transition having the highest and the lowest values of stabilization energy as 24.6 kcal/mol and 0.51 kcal/mol examined in π(C31–C33) → π*(C30–C32) and π(C26–S27) → π*(C18–N23) transitions. Furthermore, σ → σ* transition σ(C5–C18) → σ*(N23–N24) is noticed with the highest E(2) of 5.67 kcal/mol, whereas σ(C5–C18) → σ*(C9–H21) exhibited 0.51 kcal/mol stability energy which is the lowest computed energy.
In compound 2, π → π* electronic transition with the maximum E(2) of 26.11 kcal/mol is observed in π(C31–C41) → π*(C33–C35) transition whereas, π(C26–S27) → π*(C26–S27) has the lowest E(2) of 2.00 kcal/mol. In σ → σ* electronic transitions, σ(C5–C18) → σ*(N23–N24) and σ(C19–H21) → σ*(C19–H20) are examined with the greatest and the smallest stabilization energies of 5.63 kcal/mol and 0.51 kcal/mol, respectively. π → π* transition in compound 3 possess the maximum E(2) of 23.13 kcal/mol which is examined in π(C31–C41) → π*(C30–C32), while π(C26–S27) → π*(C26–S27) electronic transition with minimum energy of 1.98 kcal/mol. Furthermore, σ → σ* transitions of σ(C5–C18) → σ*(N23–N24) are perceived with maximum stability energy of 5.66 kcal/mol, however σ(C19–H20) → σ*(C18–N23) transition displayed 0.51 kcal/mol which is the lowest computed energy.
For compound 4, π → π* transition displays the maximum E(2) of 24.70 kcal/mol which is detected in π(C31–C41) → π*(C33–C34) electronic transition, although π(C26–S27) → π*(C18–N23) transition exhibited the least E(2) of 0.53 kcal/mol. Moreover, σ → σ* transitions of σ (C30–C32) → σ*(C30–C31) is obtained with the largest value of stabilization energy 6.10 kcal/mol, but σ(C5–C18) → σ*(C19–H21) exhibited 0.50 kcal/mol which is the lowest calculated energy.
For compound 5, π → π* transition demonstrated the greatest E(2) of 24.41 kcal/mol which is detected in π(C28–C33) → π*(C29–C30) transition, while π(C28–C33) → π*(C28–C33) electronic transition possess least E(2) of 0.63 kcal/mol. Furthermore, σ → σ* transition of σ(C14–C18) → σ*(N19–N20) is achieved with the energy value of 5.90 kcal/mol, and σ(C3–H7) → σ*(C2–C3) electronic transition unveiled the stability energy of 0.50 kcal/mol which is the minimum calculated energy.
π → π* transition for compound 6 showed highest E(2) of 22.37 kcal/mol in π(C31–C32) → π*(C29–C30) electronic transition however, π(C18–N19) → π*(C18–N19) transition possess lowest stability energy of 0.78 kcal/mol. Furthermore, σ → σ* transition of σ(C14–C18) → σ*(N19–N20) is achieved with the utmost energy value of 6.41 kcal/mol and σ(C29–C30) → σ*(C25–H27) transition shown 0.50 kcal/mol E(2) which is the lowermost computed energy. It is clear from the above discussion that hyper conjugative and strong intera molecular interactions play major role to stabilize these systems.
5.8 Transition density matrix (TDM)
TDM calculations are utilized for the interpretation of transitions process in the aforementioned compounds. The approximation of the behavior of transitions, especially form ground to excited state (S0–S1) as well as the electron–hole localization, was executed at M06/6-311G(d,p) functional. This investigation offers a pictographic demonstration of interaction amid different parts of in S1 state with three-dimensional heat maps having adequate color discrepancy i.e., indicated by blue area. Owing to the trivial impact of hydrogen atom in transitions, the effect of hydrogen atoms is neglected. The outcomes obtained for TDM investigation for all the synthesized compounds (1–6) are represented in Fig. 2. In the examined compounds (1–6), the maximum electronic cloud is observed at (E)-2-ethylidene-N,1-dimethylhydrazinecarbothioamide region at N13, N14, C15, S16, N17, C18 in compound 1–4 while at N12, N13, C14, S15, N16, C17 at compound 5 and 6 owing to electronegativity of these atoms. Hence, the charge transfer is majorly noticed in the aforementioned region which is also validated by the FMOs analysis.

TDM graphs of entitled compounds (1–6)
In order to compute charge transfer in entitled compounds, electron–hole coupling is also studied. It is very valuable and widely utilized technique to comprehend the movement of charge density in a molecule (Hassan et al. 2022). In compound 1 and 3, hole transition is observed at benzene ring having highest charges (0.144) as demonstrated in Figure S22. In compound 5 and 6, three transitions are noticed at C2, C9 and N13. In investigated compounds i.e., 2 and 4, the utmost electron transition is noticed at C4, C7, N13, C15 and S16 (Figure S22). Form the above discussion, it can be summarized that hole intensity is majorly observed at benzene and somewhat on naphthalene whereas electron intensity is noticed at thiosemicarbazide in all the compounds.
5.9 Exciton binding energy (E b)
The exciton binding energy (Eb) is an essential factor in ascertaining the rate of charge transference and is directly associated to the coulombic force between electron and hole (Stefan et al. 2017). The lesser Eb value designates the weaker electron–hole interactions, hence proliferating the charge transmission process (Mahmood and Irfan 2020). Equation 8 signifies the relationship among energy gap (EH–L), optimization energy (\(E_{{{\text{opt}}}}\)) and binding energy (\(E_{b}\)) (Köse 2012).
The outcomes acquired from Eq. 3 are arranged in Table 4. The Eb values of all the compounds are found comparable to each other. The descending trend of Eb of all the compounds are as: 6 > 3 = 4 > 5 > 1 = 2. Compounds 1 and 2 exhibited reduced value (0.79 eV) of Eb amid all the entitled compounds explicating greater optoelectronic characteristics with elevated exciton dissociation rate. Lesser Eb demonstrate weaker interactions between atoms that contribute to the enhanced optical properties. The compounds having 1.9 eV value of Eb are considered as appropriate candidates for optical activity (Kraner et al. 2015). Fascinatingly, all the studied compounds have displayed Eb values lesser than 1.9 eV and are observed as best candidates for range of NLO applications.
5.10 Natural population analysis (NPA)
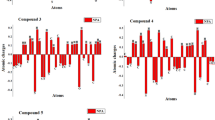
Natural population studies of atomic charges of any chemical molecule are used to investigate the hydrogen bonding pathways. More electronegative atoms including N in the entitled compounds contributed to the asymmetrical distribution of electronic density as per Mulliken statistics (Gómez-Jeria 2013). The aforementioned functional is utilized to gain the fundamental NPA values of 1–6. Natural charges on nitrogen atoms like N23, N24, S27 and N28 are observed to be negative. The negative atomic charges on N atoms compiled the presence of strong nucleophilic region. Moreover, the fundamental values of carbon and hydrogen atoms are monitored to be positive. These positive magnitudes of atomic charge demonstrate the allocation of high electrophilic area over there. Conclusively, higher positively and negatively charged atoms are taken as core electrophilic and nucleophilic regions, in addition to the intense hydrogen bonded areas. As an outcome, natural charges maintained the system of bonds present in compound 1–4 (N24–H25, N23–C18 and S27–C26 and N28–H29) and compound 5–6 (N20–H21, N19–C18 and S23–C22 and N24–H36). NPA graphs of synthesized compounds are represented in Figure S8.
5.11 Nonlinear optical properties
NLO materials are significantly employed due to their wide applications in various disciplines like optical signal modulation, telecommunications, electronics and medical. Organic compounds appeared as highly capable entities imparting strong NLO characteristics (Muhammad et al. 2013, 2009). The heterocyclic organic compounds exhibiting greater hyper polarizability have gained much attention towards them owing to their applicability as NLO material (Li et al. 2016). DFT studies have performed a significant role in the analysis of experimental data, particularly those linked to NLO response (Yeung et al. 1998). Mainly due to the manifestation of electron withdrawing groups on the heterocyclic rings, enlarged hyperpolarizabilities were discovered. Undoubtedly, superior charge transfer resulted in the greater linear polarizability ⟨α⟩, first hyper polarizability (βtot) and second hyper polarizability (γtot). All of the calculations point to a reliable NLO response, leading to successful NLO findings. The calculated dipole moments μtot, polarizability ⟨α⟩, first hyper polarizability (βtot), and second order hyper polarizability γtot of synthesized compounds (1–6) were presented in Table 5, while their major contributed tensors were exhibited in Tables S20–S23. While, carrying out NLO investigation, Eq. 9 was used to determine the dipole moment values collectively with its contributing tensors (Li et al. 2017).
The average polarizability ⟨α⟩ was calculated with Eq. 10 (Alparone 2013).
Investigation of the Gaussian output file yields nine hyper polarizability tensors oriented along x, y, and z directions: βxxx, βxyy, βxzz, βyyy, βxxy, βyzz, βzzz, βxxz and βyyz. With the assistance of Eq. 11, the total first hyper polarizability was computed (Plaquet et al. 2008).
The second order hyper polarizability was calculated by utilizing the Eq. 12 (Liebman 2009).
Dipole moment values of all the synthesized compounds are in the range of 3.1348–3.6312 D. The highest value of dipole moment (3.6312 D) is shown by compound 5 and the lowermost value (3.135 D) is observed in compound 1. According to the literature, urea is considered as standard molecule for the analysis of dipole moment as well as first hyperpolarizability. The μtot of all the entitled compounds are found greater than urea (Barone and Cossi 1998). The major dipole polarizability is located along y-axis for 1–6, indicated by the dominant values of y-tensor i.e., 3.064, 3.154, 3.253, 3.354, 3.212 and 3.236 D, respectively. The values of linear polarizability ⟨α⟩ lied in the range of 416.71–429.91 a.u. Maximum value of [⟨α⟩ = 429.91 a.u.] is unveiled by compound 6, while the lowest value (416.71 a.u) is calculated in case of compound 4. Overall, the increasing order of ⟨α⟩ values is in the following order: 4 < 1 < 2 < 3 < 5 < 6. In the linear polarizability, the αxx is found with the dominant values i.e., 655.59, 666.92, 662.31, 647.60, 649.79, and 652.04 a.u., respectively, for compounds 1–6 depicting that αxx contributed the most to the average linear polarizability values. The values of βtot existed in the range of 437.698–1201.321 a.u. The highest value of βtot is calculated for compound 5 to be 1201.321 a.u, while a reduced value of 884.450 a.u. is shown by compound 3 which further declined to 839.238 a.u. in case of compound 1 due to presence of 2,3-dimethyl group as substituent. Subsequently, the lowest calculated first hyperpolarizability value is for compound 6 (437.698 a.u). Conclusively, the ascending order for the βtot values is noted as: 6 < 4 < 2 < 1 < 3 < 5. While, taking into account, the first hyperpolarizability tensors, it is deduced that for compounds 1–4, βxxy dominated having magnitude of 1022.150, 942.281, 1019.390, and 903.439 a.u., respectively, thus influenced βtot to a maximum extent. However, for compound 5, βxxx was the authoritative tensor and for compound 6, βxyy assisted the most. By summarizing the overall outcomes of this analysis, it is concluded that the hyper polarizability of the synthesized molecules has been highly facilitated by the presence of π-electrons which assisted in the delocalization of electronic charge. The βtot values of compounds 1–6 are 20, 19, 21, 19, 28 and 10 times greater than standard molecule (urea). Second hyperpolarizability (γtot) is also calculated for all the synthesized compounds. The γtot values lie in the range of 4.967–6.175 × 105 a.u. The highest value (6.175 × 105 a.u) is observed in compound 3, then the value declines to 6.081 × 105 a.u. in 2 which is further decreased to 5.944 × 105 a.u. in 1 and the lowest value of 4.967 × 105 a.u. was exhibited by compound 6. Overall, the escalating order of the γtot values is as: 6 < 5 < 4 < 1 < 2 < 3. The determined second hyperpolarizability tensors nominated γx as the most dominant tensor, owing to the enlarged magnitudes of γx (5.534 × 105, 5.687 × 105, 5.755 × 105, 5.068 × 105, 5.024 × 105 and 4.954 × 105 a.u., respectively) in all the fore-said compounds. It is manifested that second hyperpolarizability values are inversely related to the band gaps, the highest γtot is depicted by compound with smallest band gap i.e., 3 (γtot = 6.176 × 105 a.u., Egap = 4.56 eV) and vice versa. Predicted by the detailed theoretical analysis and well-supported by various NLO parameters, it’s been concluded that compound 3 can be best utilized as efficient NLO material in modern technological domains.
5.12 Molecular electrostatic potential (MEP)
MEP maps are used to envisage global electron density in three-dimensional structure of molecules. It is performed to express the chemical reactivity by predicting multiple electrophilic and nucleophilic sites in compounds (Akram et al. 2018). MEP drawings displays different colored density cloud in their map. All these colors have a distinct identity to illustrate the specific characteristics of molecules. For instance, positively charged areas indicates the blue color, green color shows the neutral region (highest possible potential area) negative region occupies the red color representation. The intensity of electrostatic potential amplified in the following order: red < orange < yellow < green < blue (Luque et al. 1981). MEP analysis was executed at M06-6-311 G(d,p) functional to investigate the reactivity of compounds and their graphical demonstration is shown in Fig. 3.

MEP diagrams of studied compounds (1–6)
In compounds (1–6), red color indicates S atoms and it is the most perfect area for electrophilic interaction. Nevertheless, blue color is majorly located on C and H atoms which shows that nucleophilic attack most probably occurs on these positions. The green area that is localized around hydrogen atoms indicates the average of the two high values of electrostatic potential. Yellow color symbolizes the intermediate potential between opposites (red and blue).
6 Conclusion
A series of secondary ketamine of 2-acetonapthone (1–6) is synthesized and their structure elucidation is done with various spectroscopic analyses. A comparative analysis between DFT and experimental results of spectroscopic techniques exhibits good harmony between these results. The band gap calculated through FMOs is found in the range of 4.560–4.635 eV along with 324.3–330.7 nm value of λmax. Effective charge transference from HOMO to LUMO is also seen. GRP revealed that the molecules (1–6) possessed high IP (6.323–6.347 eV) and EA (1.695–1.770 eV) which suggests their stronger electron-accepting capability. High hardness (2.280–2.318 eV) and low softness (0.216–0.219 eV) values attributed the kinetically more stable and less reactive nature of 1–6 compounds. A sufficient ICT is observed through NBO findings in all the synthesized compounds, endorsed by the electrophilic/nucleophilic interactions and closely related band gap values. NLO calculations manifested that all the synthesized compounds have greater values of linear and non-linear polarizability. Among all of the newly synthesized compounds, the uppermost value (429.91 a.u) of ⟨α⟩ is exhibited by compound 6 as 2,5-dichloro substituent is utilized as axial acceptor. Similarly, 2,5-dimethyl group is present at axial position of compound 3 which shows 6.17 × 105 a.u value for \(\gamma\) tot, while the maximum βtot is found in compound 5 (1201.321 a.u.) due to utilization of 4-methoxy group as axial donor moiety. Based on a comparability assessment with a standard compound (urea), molecules 1–6 appears to possess promising NLO characteristics and it is suggested for further exploration owing to their potential use in NLO devices.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Abdulla, R.F.: Evidence of d pi-acceptor resonance in halogen substituents. J. Chem. Educ. 49, 64 (1972)
Adeel, M., Braga, A.A., Tahir, M.N., Haq, F., Khalid, M., Halim, M.A.: Synthesis, X-ray crystallographic, spectroscopic and computational studies of aminothiazole derivatives. J. Mol. Struct. 1131, 136–148 (2017)
Akram, M., Adeel, M., Khalid, M., Tahir, M.N., Khan, M.U., Asghar, M.A., Ullah, M.A., Iqbal, M.: A combined experimental and computational study of 3-bromo-5-(2, 5-difluorophenyl) pyridine and 3, 5-bis (naphthalen-1-yl) pyridine: Insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties. J. Mol. Struct. 1160, 129–141 (2018)
Alparone, A.: Linear and nonlinear optical properties of nucleic acid bases. Chem. Phys. 410, 90–98 (2013)
Ambujam, K., Selvakumar, S., Prem Anand, D., Mohamed, G., Sagayaraj, P.: Crystal growth, optical, mechanical and electrical properties of organic NLO material γ-glycine. Cryst. Res. Technol. J. Exp. Ind. Crystallogr. 41, 671–677 (2006)
Ashfaq, M., Khalid, M., Tahir, M.N., Ali, A., Arshad, M.N., Asiri, A.M.: Synthesis of crystalline fluoro-functionalized imines, single crystal investigation, Hirshfeld surface analysis, and theoretical exploration. ACS Omega 7, 9867–9878 (2022)
Bal, M., Ceyhan, G., Avar, B., Köse, M., Kayraldiz, A., Kurtoğlu, M.: Synthesis and X-ray powder diffraction, electrochemical, and genotoxic properties of a new azo-Schiff base and its metal complexes. Turk. J. Chem. 38, 222–241 (2014)
Barone, V., Cossi, M.: Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 (1998)
Beraldo, H., Gambino, D.: The wide pharmacological versatility of semicarbazones, thiosemicarba-zones and their metal complexes. Mini. Rev. Med. Chem. 4, 31–39 (2004)
Berkowski, M., Fink-Finowicki, J., Diduszko, R., Byszewski, P., Aleksiyko, R., Kikalejshvili-Domukhovska, R.: Growth and structure of SrAl0.5Ta0.5O3:LaAlO3 solid solutions single crystals. J. Cryst. Growth 257, 146–152 (2003)
Bondock, S., Khalifa, W., Fadda, A.A.: Synthesis and antimicrobial evaluation of some new thiazole, thiazolidinone and thiazoline derivatives starting from 1-chloro-3, 4-dihydronaphthalene-2-carboxaldehyde. Eur. J. Med. Chem. 42, 948–954 (2007)
Burke, K., Werschnik, J., Gross, E.K.U.: Time-dependent density functional theory: past, present, and future. J. Chem. Phys. 123, 062206 (2005)
Chattaraj, P.K., Roy, D.R.: Update 1 of: electrophilicity index. Chem. Rev. 107, PR46–PR74 (2007)
Coluccini, C., Sharma, A.K., Caricato, M., Sironi, A., Cariati, E., Righetto, S., Tordin, E., Botta, C., Forni, A., Pasini, D.: Switching of emissive and NLO properties in push–pull chromophores with crescent PPV-like structures. Phys. Chem. Chem. Phys. 15, 1666–1674 (2013)
Coulson, C.A., Daudel, R., Robertson, J.M.: Bond lengths in naphthalene and anthracene. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 207, 306–320 (1951)
Datta, A.: Role of metal ions (M = Li+, Na+, and K+) and pore sizes (Crown-4, Crown-5, and Crown-6) on linear and nonlinear optical properties: new materials for optical birefringence. J. Phys. Chem. C 113, 3339–3344 (2009)
Demaison, J., Rudolph, H.D., Császár, A.G.: Deformation of the benzene ring upon fluorination: equilibrium structures of all fluorobenzenes. Mol. Phys. 111, 1539–1562 (2013). https://doi.org/10.1080/00268976.2013.793843
Dinçer, M., Özdemir, N., Cukurovalı, A., Yılmaz, İ: Benzoin thiosemicarbazone. Acta Crystallogr. Sect. E Struct. Rep. Online 61, o880–o883 (2005)
Dinçer, M., Özdemir, N., Çukurovalı, A., Yılmaz, İ, Büyüükgüngör, O.: Benzoin 4-ethylthiosemicarbazone. Acta Crystallogr. C 62, o13–o15 (2006)
Dini, D., Hanack, M.: Phthalocyanines as materials for advanced technologies: some examples. J. Porphyr. Phthalocyanines 8, 915–933 (2004)
Elhorri, A.M.: Theoretical study of new push–pull molecules based on transition metals for NLO applications and determination of ICT mechanisms by DFT calculations. J. Theor. Comput. Chem. 19, 2050026 (2020)
Frisch, M.J.: Gaussian 16. Gaussian Inc., Wallingford (2016)
Frisch, A., Nielson, A.B., Holder, A.J.: Gaussview User Manual, p. 556. Gaussian Inc., Pittsburgh (2000)
Fukui, K.: Role of frontier orbitals in chemical reactions. Science 218, 747–754 (1982)
García-Tojal, J., Gil-García, R., Gómez-Saiz, P., Ugalde, M.: Pyridine-2-carbaldehyde thiosemicarbazonecopper system: extending some findings to other thiosemicarbazone and coordination compounds. Curr. Inorg. Chem. 1, 189–210 (2011)
Glendening, E.D., Reed, A.E., Carpenter, J.E., Weinhold, F.: Nbo Version 3.1, Tci, p. 65. University of Wisconsin, Madison (1998)
Gómez-Jeria, J.S.: A new set of local reactivity indices within the Hartree–Fock–Roothaan and density functional theory frameworks. Can. Chem. Trans. 1, 25–55 (2013)
Hanwell, M.D., Curtis, D.E., Lonie, D.C., Vandermeersch, T., Zurek, E., Hutchison, G.R.: Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 1–17 (2012)
Hassan, T., Hussain, R., Khan, M.U., Habiba, U., Irshad, Z., Adnan, M., Lim, J.: Development of non-fused acceptor materials with 3D-Interpenetrated structure for stable and efficient organic solar cells. Mater. Sci. Semicond. Process. 151, 107010 (2022)
Heo, I., Lee, J.C., Özer, B.R., Schultz, T.: Structure of benzene from mass-correlated rotational Raman spectroscopy. RSC Adv. 12, 21406–21416 (2022)
Hirata, S., Head-Gordon, M.: Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 314, 291–299 (1999)
Jawaria, R., Khalid, M., Khan, J., Khan, M.U., Braga, A.A.C., Zahoor, S., Alam, M.M., Imran, M.: A facile synthesis and structural elucidation for furfural based chromophores: prediction of linear and nonlinear optical properties. J. Mol. Struct. 1249, 131543 (2022)
Khalid, M., Hussain, R., Hussain, A., Ali, B., Jaleel, F., Imran, M., Assiri, M.A., Usman Khan, M., Ahmed, S., Abid, S.: Electron donor and acceptor influence on the nonlinear optical response of diacetylene-functionalized organic materials (DFOMs): density functional theory calculations. Molecules 24, 2096 (2019)
Khalid, M., Arshad, M.N., Tahir, M.N., Asiri, A.M., Naseer, M.M., Ishaq, M., Khan, M.U., Shafiq, Z.: An efficient synthesis, structural (SC-XRD) and spectroscopic (FTIR, 1HNMR, MS spectroscopic) characterization of novel benzofuran-based hydrazones: an experimental and theoretical studies. J. Mol. Struct. 1216, 128318 (2020)
Khalid, M., Lodhi, H.M., Khan, M.U., Imran, M.: Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: a DFT approach. RSC Adv. 11, 14237–14250 (2021)
Khalid, M., Arshad, M.N., Murtaza, S., Shafiq, I., Haroon, M., Asiri, A.M., de AlcântaraMorais, S.F., Braga, A.A.: Enriching NLO efficacy via designing non-fullerene molecules with the modification of acceptor moieties into ICIF2F: an emerging theoretical approach. RSC Adv. 12, 13412–13427 (2022)
Khalid, M., Naz, S., Mahmood, K., Hussain, S., Braga, A.A.C., Hussain, R., Ragab, A.H., Al-Mhyawi, S.R.: First theoretical probe for efficient enhancement of optical nonlinearity via structural modifications into phenylene based D–π–A configured molecules. RSC Adv. 12, 31192–31204 (2022)
Khan, M.U., Khalid, M., Shafiq, I., Khera, R.A., Shafiq, Z., Jawaria, R., Shafiq, M., Alam, M.M., Braga, A.A.C., Imran, M.: Theoretical investigation of nonlinear optical behavior for rod and T-shaped phenothiazine based D–π–A organic compounds and their derivatives. J. Saudi Chem. Soc. 25, 101339 (2021)
Koopmans, T.: Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1, 104–113 (1933)
Koopmans, T.: Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1, 104–113 (1934)
Köse, M.E.: Evaluation of acceptor strength in thiophene coupled donor–acceptor chromophores for optimal design of organic photovoltaic materials. J. Phys. Chem. A 116, 12503–12509 (2012)
Kraner, S., Scholz, R., Plasser, F., Koerner, C., Leo, K.: Exciton size and binding energy limitations in one-dimensional organic materials. J. Chem. Phys. 143, 244905 (2015)
Krishnakumar, V., Nagalakshmi, R.: Crystal growth and vibrational spectroscopic studies of the semiorganic non-linear optical crystal—bisthiourea zinc chloride. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 61, 499–507 (2005)
Li, Z., Kim, H., Chi, S.-H., Hales, J.M., Jang, S.-H., Perry, J.W., Jen, A.K.-Y.: Effects of counterions with multiple charges on the linear and nonlinear optical properties of polymethine salts. Chem. Mater. 28, 3115–3121 (2016)
Li, B., Xu, C., Xu, X., Zhu, C., Gu, F.L.: Remarkable nonlinear optical response of excess electron compounds: theoretically designed alkali-doped aziridine M–(C2NH5)n. Phys. Chem. Chem. Phys. 19, 23951–23959 (2017)
Li, T., Dai, S., Ke, Z., Yang, L., Wang, J., Yan, C., Ma, W., Zhan, X.: Fused tris (thienothiophene)-based electron acceptor with strong near-infrared absorption for high-performance as-cast solar cells. Adv. Mater. 30, 1705969 (2018)
Liebman, J.F.: Book Review: Density Functional Theory: A Practical Introduction, by David S. Sholl and Janice A. Steckel. Wiley, London (2009)
Liu, Q., Hoffmann, R.: Theoretical aspects of a novel mode of hydrogen–hydrogen bonding. J. Am. Chem. Soc. 117, 10108–10112 (1995)
Liu, J., Huang, X., Li, Y., Sulieman, K.M., He, X., Sun, F.: Self-assembled CuO monocrystalline nanoarchitectures with controlled dimensionality and morphology. Cryst. Growth Des. 6, 1690–1696 (2006)
Luque, F.J., López, J.M., Orozco, M.: Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects.” Chem Phys 55, 117 (1981)
Mahmood, A., Irfan, A.: Effect of fluorination on exciton binding energy and electronic coupling in small molecule acceptors for organic solar cells. Comput. Theor. Chem. 1179, 112797 (2020)
Mahmood, A., Abdullah, M.I., Nazar, M.F.: Quantum chemical designing of novel organic non-linear optical compounds. Bull. Korean Chem. Soc. 35, 1391–1396 (2014)
Mahmood, A., Abdullah, M.I., Khan, S.U.-D.: Enhancement of nonlinear optical (NLO) properties of indigo through modification of auxiliary donor, donor and acceptor. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 139, 425–430 (2015)
Mahmood, A., Khan, S.U.-D., Rana, U.A., Tahir, M.H.: Red shifting of absorption maxima of phenothiazine based dyes by incorporating electron-deficient thiadiazole derivatives as π-spacer. Arab. J. Chem. 12, 1447–1453 (2019)
Makowska-Janusik, M.: Influence of the polymeric matrix on the NLO molecular response in guest-host materials. Nonlinear Opt. Quantum Opt. Concepts Mod. Opt. 37, 75–85 (2007)
Marder, S.R.: Organic nonlinear optical materials: where we have been and where we are going. Chem. Commun. (2), 131–134 (2006)
Muhammad, S., Janjua, M.R.S.A., Su, Z.: Investigation of dibenzoboroles having π-electrons: toward a new type of two-dimensional NLO molecular switch? J. Phys. Chem. C 113, 12551–12557 (2009)
Muhammad, S., Xu, H.-L., Zhong, R.-L., Su, Z.-M., Al-Sehemi, A.G., Irfan, A.: Quantum chemical design of nonlinear optical materials by sp 2-hybridized carbon nanomaterials: issues and opportunities. J. Mater. Chem. C 1, 5439–5449 (2013)
Nan, M.I., Lakatos, E., Giurgi, G.-I., Szolga, L., Po, R., Terec, A., Jungsuttiwong, S., Grosu, I., Roncali, J.: Mono-and di-substituted pyrene-based donor-π-acceptor systems with phenyl and thienyl π-conjugating bridges. Dyes Pigm. 181, 108527 (2020)
O’boyle, N.M., Tenderholt, A.L., Langner, K.M.: Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845 (2008)
Parodi, V., Jacchetti, E., Osellame, R., Cerullo, G., Polli, D., Raimondi, M.T.: Nonlinear optical microscopy: From fundamentals to applications in live bioimaging. Front. Bioeng. Biotechnol. 8, 585363 (2020)
Parr, R.G., Pearson, R.G.: Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516 (1983)
Parr, R.G., Donnelly, R.A., Levy, M., Palke, W.E.: Electronegativity: the density functional viewpoint. J. Chem. Phys. 68, 3801–3807 (1978)
Paufler, P.N., Cotter, D.: The Elements of Nonlinear Optics. Cambridge University Press, Cambridge (1990)
Pearson, R.G.: Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. 83, 8440–8441 (1986)
Plaquet, A., Guillaume, M., Champagne, B., Castet, F., Ducasse, L., Pozzo, J.-L., Rodriguez, V.: In silico optimization of merocyanine-spiropyran compounds as second-order nonlinear optical molecular switches. Phys. Chem. Chem. Phys. 10, 6223–6232 (2008)
Poirier, R., Kari, R., Csizmadia, I.G.: Handbook of Gaussian Basis Sets. Elsevier, London (1985)
Pritula, I., Kosinova, A., Kolybayeva, M., Puzikov, V., Bondarenko, S., Tkachenko, V., Tsurikov, V., Fesenko, O.: Optical, structural and microhardness properties of KDP crystals grown from urea-doped solutions. Mater. Res. Bull. 43, 2778–2789 (2008)
Ramesh, G., Reddy, B.V.: Investigation of barrier potential, structure (monomer & dimer), chemical reactivity, NLO, MEP, and NPA analysis of pyrrole-2-carboxaldehyde using quantum chemical calculations. Polycycl. Aromat. Compd. (2022). https://doi.org/10.1080/10406638.2022.2086889
Riscob, B., Shakir, M., Sundar, J.K., Natarajan, S., Wahab, M.A., Bhagavannarayana, G.: Synthesis, growth, crystal structure and characterization of a new organic material: glycine glutaric acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 78, 543–548 (2011)
Saleem, T., Khan, S., Yaqub, M., Khalid, M., Islam, M., ur Rehman, M.Y., Rashid, M., Shafiq, I., Braga, A.A., Syed, A.: Novel quinoline-derived chemosensors: synthesis, anion recognition, spectroscopic, and computational study. New J. Chem. 46, 18233–18243 (2022)
Santhakumari, R., Ramamurthi, K., Vasuki, G., Yamin, B.M., Bhagavannarayana, G.: Synthesis and spectral characterization of acetophenone thiosemicarbazone: a nonlinear optical material. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 76, 369–375 (2010)
Shahid, M., Salim, M., Khalid, M., Tahir, M.N., Khan, M.U., Braga, A.A.C.: Synthetic, XRD, non-covalent interactions and solvent dependent nonlinear optical studies of Sulfadiazine-Ortho-Vanillin Schiff base:(E)-4-((2-hydroxy-3-methoxy-benzylidene) amino)-N-(pyrimidin-2-yl) benzene-sulfonamide. J. Mol. Struct. 1161, 66–75 (2018)
Stefan, K., Giacomo, P., Gianaurelio, C.: Exciton binding energy in molecular triads. J. Phys. Chem. C 121, 17088–17095 (2017)
Sung, P.-H., Hsu, T.-F.: Thermal stability of NLO sol–gel networks with reactive chromophores. Polymer 39, 1453–1459 (1998)
Tian, Y.-P., Yu, W.-T., Jiang, M.-H., Shanmuga Sundara Raj, S., Yang, P., Fun, H.-K.: Diiodobis (4-methoxybenzaldehyde thiosemicarbazone-S) cadmium(II). Acta Crystallogr Sect. C Cryst. Struct. Commun. 55, 1639–1641 (1999)
Türkkan, B., Ülküseven, B., Eroğlu, E.: Zinc(II) complexes of acetophenone and 5-chloro-2-hydroxy-benzophenone thiosemicarbazones. Synthesis, characterization, and nonlinear optical properties from quantum chemical calculations. Phosphorus Sulfur Silicon Relat. Elem. 190, 53–65 (2015)
Xiong, L., Wu, L.-M., Chen, L.: A general principle for DUV NLO materials: π-conjugated confinement enlarges band gap. Angew. Chem. 133, 25267–25271 (2021)
Xu, K., Cao, L., You, F., Zhong, D., Wang, T., Yu, Z., Hu, C., Tang, J., Teng, B.: Crystal growth and characterization of optical, thermal and electrical properties of organic NLO crystal OHB-T. J. Cryst. Growth 547, 125757 (2020)
Yeung, M., Ng, A.C., Drew, M.G., Vorpagel, E., Breitung, E.M., McMahon, R.J., Ng, D.K.: Facile synthesis and nonlinear optical properties of push- pull 5, 15-diphenylporphyrins. J. Org. Chem. 63, 7143–7150 (1998)
Yousef, T.A., El-Reash, G.A., El-Gammal, O.A., Bedier, R.A.: Co(II), Cu(II), Cd(II), Fe(III) and U(VI) complexes containing a NSNO donor ligand: synthesis, characterization, optical band gap, in vitro antimicrobial and DNA cleavage studies. J. Mol. Struct. 1029, 149–160 (2012)
Zhurko, G.A.: Chemcraft: http://www.chemcraftprog.com. Accessed 22 Oct 2014
Acknowledgements
Dr. Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (Project No. 20-14703/NRPU/R&D/HEC/2021). A.A.C.B. acknowledges the financial support of the São Paulo Research Foundation (FAPESP) (Grants 2014/25770-6 and 2015/01491-3), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) of Brazil for academic support (Grant 309715/2017-2), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) that partially supported this work (Finance Code 001). The authors thank the Researchers Supporting Project Number (RSP2023R29), King Saud University, Riyadh, Saudi Arabia.
Funding
Dr. Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (Project No. 20-14703/NRPU/R&D/HEC/2021). The authors also thank the Researchers Supporting Project Number (RSP2023R29), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
MK: software; project administration. RJ: Data curation; formal analysis; supervision. SAD: Conceptualization; methodology. IS: Data curation; formal analysis. AACB: Data curation; formal analysis; validation. SMA: Conceptualization; methodology; software. MAA: Resources; software; supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khalid, M., Jawaria, R., Ditta, S.A. et al. Synthesis, characterization and exploration of optical non-linearity of secondary ketamine derivatives via DFT study. Opt Quant Electron 55, 1121 (2023). https://doi.org/10.1007/s11082-023-05361-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05361-z