
Abstract
For monoclinic \(\hbox {Ag}_2\)S nanocrystals (quantum dots, \(\hbox {Ag}_2\)S/TGA QDs), the correlation between their luminescence properties and aspects of their passivation by thioglycolic acid (TGA) molecules is considered. The features of quantum confinement effect in the QDs absorption and photoluminescence spectra are analyzed for \(\hbox {Ag}_2\)S QDs with an average size of 1.7–3.1 nm. We show that, in various conditions of passivation of QDs interfaces, the luminescence mechanism of \(\hbox {Ag}_2\)S/TGA QDs is switched from recombination luminescence in the 870–1000 nm region to exciton luminescence with a band maximum at 620 nm. By means of FTIR spectra, two major types of interactions between TGA molecules and \(\hbox {Ag}_2\)S QDs, arising due to changing the [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] ratio from 1:0.9 to 1:1.43, are determined. Exciton luminescence (620 nm) occurs in case of using TGA as the sulfur source in \(\hbox {Ag}_2\)S crystallization and the interface passivation agent, with [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] = 1:1. The analysis of the FTIR spectra indicates bonding of TGA with the \(\hbox {Ag}_2\)S surface by both thiol and carboxylic groups in this case. With increasing the sulfur concentration in the synthesis of \(\hbox {Ag}_2\)S/TGA QDs, the exciton luminescence is suppressed. The employment of \(\hbox {Na}_2\)S as the sulfur source in \(\hbox {Ag}_2\)S crystallization with TGA acting as the surface passivation agent promotes the formation of recombination centers for IR luminescence. In this case, analysis of the FTIR spectra indicates passivation of \(\hbox {Ag}_2\)S QDs by TGA molecules due to adsorption of thiol groups. It is found that the photodegradation or IR luminescence of \(\hbox {Ag}_2\)S/TGA QDs upon exposure to exciting radiation is due to the photolysis of \(\hbox {Ag}_2\)S nanocrystals with formation of luminescence quenching centers as well as due to photodestruction of TGA molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
At present, semiconductor quantum dots (QDs) attract enduring scientific and practical interest. Their optical properties that depend on the QDs size make them a promising material for various implementations of luminescent sensing (Freckera et al. 2016; Yang et al. 2015; Petryayeva et al. 2013; Bera et al. 2010; Semonin et al. 2012; Galloway et al. 2012; Hu and Zhu 2015). The methods of controlling the luminescence parameters of QDs are very important for these applications. The major approach to control of QDs optical properties is based on the variation of the synthesis conditions, such as reagent ratios, mixing speed, temperature, pH, and others (Hu and Zhu 2015; Gui et al. 2015; Ratnesh and Mehataa 2015; Zhang et al. 2015; Almendral-Parra et al. 2012; Chaudhry et al. 2018; Banizi and Seifi 2017; Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012; Moradian et al. 2013; Zhang and Clapp 2011; Silva et al. 2012; Wageh et al. 2014; Tyrakowski and Snee 2014).
One of the promising techniques for the synthesis of colloidal QDs is their formation by stabilization with various substances containing functional groups, such as thiol groups, which are capable of interacting with the surface interfaces of QDs. In a thiol group, hydrogen is usually substituted by a metal ion from the QD surface (Chaudhry et al. 2018; Banizi and Seifi 2017; Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012; Moradian et al. 2013; Zhang and Clapp 2011; Silva et al. 2012; Wageh et al. 2014; Tyrakowski and Snee 2014). As a result, hybrid structures constituting an inorganic nanocrystal surrounded by multiple organic passivating molecules are formed. The considered approach provides a colloidal solution with hydrophilic properties and conditions the size-dependent absorption and luminescence properties of the QDs. In this case, enhancement of recombination luminescence and suppression of the exciton peak often occur (Chaudhry et al. 2018; Banizi and Seifi 2017; Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012; Moradian et al. 2013; Zhang and Clapp 2011; Silva et al. 2012; Wageh et al. 2014; Tyrakowski and Snee 2014).
The choice of the molecules for interface passivation is of major significance for preparing colloidal QDs solutions (Chaudhry et al. 2018; Banizi and Seifi 2017; Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012; Moradian et al. 2013; Zhang and Clapp 2011; Silva et al. 2012; Wageh et al. 2014; Tyrakowski and Snee 2014; Xiang et al. 2008; Sadovnikov et al. 2015). This choice depends on the orientation of the molecules on the QD interface (Chaudhry et al. 2018; Banizi and Seifi 2017; Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012; Moradian et al. 2013; Zhang and Clapp 2011; Silva et al. 2012; Wageh et al. 2014; Tyrakowski and Snee 2014). The free fragment of the molecule is to have high affinity to the solvent. This provides high “solubility” of QDs. Possible candidates for passivating QDs should have both free groups and active functional groups for interaction with the QD surface; they should also be capable of preventing QDs aggregation and removing extra charge at the QDs interfaces, as well as should not prevent the diffusion growth of QDs.
The studies aimed at elucidating the laws of formation of semiconductor QDs in mercapto acids have certain features (Chaudhry et al. 2018; Banizi and Seifi 2017; Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012; Moradian et al. 2013; Zhang and Clapp 2011; Silva et al. 2012; Wageh et al. 2014; Tyrakowski and Snee 2014). In most cases, authors observe quantum size effect in the spectral properties of nanocrystals of one size in the specified optimal conditions (Piven et al. 2008; Sousa et al. 2018; Sun et al. 2012). In some situations, they try to change the QDs size by annealing and examine the samples before and after it (Banizi and Seifi 2017; Moradian et al. 2013; Silva et al. 2012). The characteristic features of \(\hbox {Ag}_2\)S QDs are the low solubility of silver sulfide itself, as well as matching of excitation and luminescence regions with therapeutic transparency windows of biological tissues (650–1300 nm) (Sadovnikov and Gusev 2017; Hocaoglu et al. 2012; Duman et al. 2015; Wu et al. 2017; Zhang et al. 2017; Tang et al. 2015; Gui et al. 2014; Tan et al. 2013; Yang et al. 2013; Wang et al. 2012; Du et al. 2010).
There is no clear understanding of the size effect with regard to the absorption and luminescence spectra of \(\hbox {Ag}_2\)S QDs. For \(\hbox {Ag}_2\)S QDs passivated by mercapto acids, in particular, by thioglycolic acid (next \(\hbox {Ag}_2\)S/TGA), the size effect is analyzed mostly for exciton luminescence (Gui et al. 2015; Tang et al. 2015; Jiang et al. 2012). Recombination luminescence in the region of 800–1000 nm is still not completely studied. For example, there are literature data for \(\hbox {Ag}_2\)S QDs in gelatin (Ovchinnikov et al. 2015a, b; Perepelitsa et al. 2018). IR luminescence with a maximum in the 1000–1200 nm region was observed for these QDs. A weak size effect for this band explained as recombination of free holes with the electrons localized at the luminescence centers was found. In this case, it was proposed that the luminescence centers are of surface nature and include both gelatin fragments and dangling bonds (Ovchinnikov et al. 2015a, b; Perepelitsa et al. 2018). However, it is difficult to determine the origin of the centers due to the complexity of the gelatin structure.
In addition to the above, there are a number of works aimed at establishing the regularities of the core/shell structures formation based on \(\hbox {Ag}_2\)S QDs during synthesis in solutions of mercapto acids (TGA, 3-MPA) (Mirhosseini et al. 2019; Karimipour and Molaei 2017; Karimipour et al. 2018, 2019). In these papers, the possibility of controlling the intensity and luminescence mechanism of \(\hbox {Ag}_2\)S QDs by growing a semiconductor shell (ZnS, AgInS) on the surface of \(\hbox {Ag}_2\)S QDs was demonstrated, and the possibility of photoinduced growth of \(\hbox {Ag}_2\)S and core/shell \(\hbox {Ag}_2\)S/ZnS and \(\hbox {Ag}_2\)S/\(\hbox {AgInS}_2\) QDs under prolonged irradiation of colloidal QD solution by UV radiation for 5–200 min was shown Mirhosseini et al. (2019) and Karimipour et al. (2019). In particular, Mirhosseini et al. (2019) demonstrated the influence of Zn-doped \(\hbox {AgInS}_2\) and \(\hbox {AgInS}_2\) shells on the structure of the QD luminescence band and concluded that the donor–acceptor mechanism of radiative recombination is due to the formation of new trap states when building shell \(\hbox {AgInS}_2\) or \(\hbox {AgInS}_2\) mixed with Zn. However, in these studies, the dimensional dependences in the optical absorption and photoluminescence spectra of \(\hbox {Ag}_2\)S/TGA QDs, the mechanisms of passivator molecules interactions with QD interfaces, and also there is no analysis of the photodegradation effect of trap-state IR luminescence of \(\hbox {Ag}_2\)S/TGA QDs.
When TGA is used as the passivating agent, the situation can be somewhat simplified since the nanocrystals are coated with molecules of a single type. FTIR spectra of the TGA molecules provide information on their interaction with the QDs. However, in this case, one should also know the dependences relating the interactions arising in the \(\hbox {Ag}_2\)S/TGA system with the synthesis conditions, as well as the data on their influence on the photoluminescence properties. An important property of luminescence, alongside with the wavelength of the luminescence maximum, Stokes shift with respect to the absorption band, full width at half maximum (FWHM), quantum yield, and luminescence decay rates, is the photostability of these parameters. The effect of the excitation light on the luminescence properties of \(\hbox {Ag}_2\)S QDs was addressed in several studies Sun et al. (2014), Rempel et al. (2017), Ovchinnikov et al. (2016), Smirnov et al. (2018) and Ovchinnikov et al. (2019). The most detailed consideration was given to the problem of photodegradation of luminescence for \(\hbox {Ag}_2\)S QDs dispersed in gelatin (Ovchinnikov et al. 2019). In this paper, the source data demonstrating the photodegradation of luminescence for \(\hbox {Ag}_2\)S/TGA QDs of one average size (2.3 nm) were provided. The authors suggested that photolysis of \(\hbox {Ag}_2\)S QDs occurs with the formation of nonradiative recombination centers. It should be noted that the photolysis of \(\hbox {Ag}_2\)S QDs was established in Sun et al. (2014). However, no correlations between this process and the luminescence properties were observed. The effect of passivating agents on the photostability of QDs luminescence properties has also not been considered yet.
In this study, for colloidal \(\hbox {Ag}_2\)S QDs of small size (1.7–3.1 nm), we establish the relationships between the conditions of their synthesis, as well as their physical and chemical properties, and quantum size effect in the luminescence together with photodegradation (fatigue) of luminescence under exposure to excitation light. To solve the stated task of investigating \(\hbox {Ag}_2\)S/TGA QDs, the most informative methods were FTIR absorption and nanosecond luminescence decay measurements.
2 Synthesis of colloidal \(\hbox {Ag}_2\)S/TGA QDs
Colloidal \(\hbox {Ag}_2\)S/TGA QDs were prepared by the aqueous synthesis technique (Smirnov et al. 2018; Ovchinnikov et al. 2019). We specially chose the synthesis conditions providing QDs formation with different luminescence and absorption bands. All reagents (\(\hbox {Na}_2\)S, \(\hbox {AgNO}_3\), TGA, NaOH) purchased from Sigma-Aldrich were of high purity.
In the first approach, \(\hbox {Na}_2\)S aqueous solution was used as the sulfur source. TGA molecules, in this case, were mainly used for the interface passivation. In the synthesis, we used an \(\hbox {Ag}^+\)/TGA precursor solution (200 ml) obtained by mixing \(\hbox {AgNO}_3\) (2.6 mmol) and TGA (2.6 mmol) with subsequent raising of pH to 10 by addition of a 1 M NaOH solution. After that, with constant stirring, 50 ml of a \(\hbox {Na}_2\)S aqueous solution were added using a peristaltic pump. The solution in the reactor changed color from light yellow to dark brown. Thus, \(\hbox {Ag}_2\)S QDs with a concentration of \(2\cdot 10^{-5}\) mol QDs/l were formed in water. The synthesis was carried out at a temperature of 30 °C.
In the second approach to the synthesis of \(\hbox {Ag}_2\)S QDs, a TGA aqueous solution was used as the only sulfur source. TGA molecules also acted as the passivating agent. In this synthesis, \(\hbox {Ag}_2\)S QDs samples were obtained in the same way as described above, but without using \(\hbox {Na}_2\)S. In this case, [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] molar ratio was 1:0.9 or 1:1.
In the considered synthesis approach, the QDs size was controlled by changing the [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] molar ratio from 1:0.9 to 1:1.43 at constant reagent injection rate. To remove the reaction by-products, the aqueous colloidal solution of QDs was diluted with acetone to 50% and centrifuged. The purification procedure was repeated several times.
3 Experimental techniques
Structural studies were performed by the methods of transmission electron microscopy (TEM), including high-resolution TEM (HR-TEM), and surface-enhanced Raman scattering (SERS). The size of the synthesized QDs ensembles was determined using a Libra 120 transmission electron microscope (CarlZeiss, Germany) with an accelerating voltage of 120 kV. The size distributions for the QDs ensemble were obtained by digital analysis of TEM images. HR-TEM images of \(\hbox {Ag}_2\)S QDs were obtained using a JEM-2100 transmission electron microscope (Jeol, Japan) with an accelerating voltage of 200 kV. Additionally, energy dispersive X-ray analysis (EDX) was performed using the built-in spectrometer of the JEM-2100 TEM (Jeol, Japan).
The SERS spectra were studied for \(\hbox {Ag}_2\)S/TGA QDs deposited from an aqueous solution to plasmonic silver foil formed on a glass substrate. Freshly deposited films were dried in horizontal orientation at room temperature. Silver nanoparticles were obtained by recovery of silver nitrate with sodium citrate. The resulting films were studied by scanning electron microscopy (SEM) using an S-806 TEM/SEM microscope (Hitachi, Japan). The films were a dense layer of spherical nanoparticles. The SERS spectra were recorded by the inverse scattering method with a Nanoflex (Solar LS, Belarus) spectrometer equipped with a 488-nm-emitting argon laser (0.2 mW) and a diffraction grating having 1200 lines per mm.
The interactions between TGA molecules and colloidal \(\hbox {Ag}_2\)S QDs in various synthesis conditions, as well as under prolonged exposure to excitation light, were studied by the FTIR spectroscopy method. We sought for changes in the characteristic frequencies of most functionally active groups of TGA molecules participating in the formation of \(\hbox {Ag}_2\)S/TGA QDs in the solution. IR absorption spectra were recorded with a Tensor 37 Fourier-transform infrared (FTIR) spectrometer (Bruker Optik GmbH., Germany). For the samples studied, cuvettes with KCl windows were used. The samples were prepared by depositing the sample solutions on KCl windows. The deposited solutions had equal volumes.
Optical absorption spectra were obtained by a USB2000+ spectrometer (OceanOptics, USA) with a USB-DT radiation source (Ocean optics).
Photoluminescence (PL) spectra and luminescence decays were obtained by the USB2000+ (OceanOptics, USA) and a TimeHarp 260 time-correlated single-photon counting board (PicoQuant, Germany) with a PMC-100-20 PMT module (Becker&Hickl, Germany) with the time resolution of the system being 0.2 ns. Deconvolution with an experimentally measured instrument response function was employed for fitting the experimental luminescence decay curves. The samples were excited by the UV radiation from an Alphalas PLDD-250 pulsed semiconductor laser (Alphalas, Germany) with a wavelength of 375 nm, pulse duration of 60 ps, and pulse repetition rate of 100 kHz. The results were obtained at room temperature.
For luminescence excitation, we used an NDV7375 laser diode (Nichia, Japan) with 405 nm emission and 200 mW optical power, an LD PLTB450 laser diode (Osram, Germany) with 445 nm emission and 440 mW optical power, a PM-G80 (CST, China) laser module with 532 nm emission and 100 mW optical power, a KLM-B635-5-5 laser module with 635 nm emission and 5 mW optical power, and an LPC-836 laser diode (Mitsubishi Electric, Japan) with 660 nm emission and 250 mW optical power.
4 Results and discussion
4.1 TEM image analysis for \(\hbox {Ag}_2\)S/TGA QDs
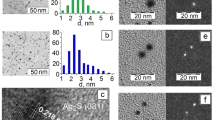
Figure 1a–e presents the TEM images of \(\hbox {Ag}_2\)S QDs most different in the average size of QD ensembles. As a result of the statistical analysis, size distribution histograms were obtained. It can be seen that the synthesized QDs are single nanocrystals with average sizes of 1.7–3.1 nm and the size distribution within 25%. The smallest QDs were obtained with the [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] ratio of 1:0.9 (TGA used as passivator and as sulfur sourse). With a slight change in the [\(\hbox {AgNO}_3\)]:[TGA]:[\(\hbox {Na}_2\)S] ratio from 1:1:0.13 to 1:1:0.16, with addition of \(\hbox {Na}_2\)S as sulfur source, the average size was changed from 2.3 to 2.5 nm with the size distribution maintained. In the approach involving TGA molecules acting simultaneously as the passivator and the sulfur source, in the crystallization reaction at a concentration ratio [\(\hbox {AgNO}_3\)]:[TGA] of 1:1, the formation of \(\hbox {Ag}_2\)S QDs with an average size of about 2.0 nm and with low size dispersion of 7–10% was observed (Fig. 1e).

TEM images and size distribution histograms for investigated samples of \(\hbox {Ag}_2\)S/TGA QDs (a–e), HR-TEM image of an \(\hbox {Ag}_2\)S/TGA nanocrystal with an interplanar distance of 0.267±0.005 nm corresponds to crystallographic plane (121) of monoclinic \(\hbox {Ag}_2\)S (f)
Thus, with the ratio of ion concentrations [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] ranging from 1:0.9 to 1:1.43, \(\hbox {Ag}_2\)S/TGA particles are formed with sizes close to the Bohr exciton radius in \(\hbox {Ag}_2\)S (1.15 nm) (Lin et al. 2015; Zhang et al. 2014).
The analysis of the high-resolution TEM images (HR-TEM) of \(\hbox {Ag}_2\)S QDs (Fig. 1f) showed diffraction from the (121) crystallographic plane of the \(\hbox {Ag}_2\)S monoclinic crystal lattice (space group P21/c) (Sadovnikov et al. 2015) with an interplanar distance of 0.267±0.005 nm (Fig. 1). The analysis of the HR-TEM images also revealed a deviation of \(\hbox {Ag}_2\)S QDs from the spherical shape caused by faceting of the nanocrystal interfaces, as well as by imperfections arising due to crystallization.
Energy dispersive X-ray analysis (EDX) showed the presence of Ag and S atoms (Fig. 2). The presence of peaks corresponding to C atoms is due to the amorphous carbon substrate, as well as due to carbon atoms of the TGA molecule. Na atoms are detected since they are involved in the sulfur precursor (\(\hbox {Na}_2\)S) and in the buffer for stabilizing the pH level.

Results of energy dispersive X-ray analysis of \(\hbox {Ag}_2\)S/TGA QDs. The inset shows the SERS spectra of colloidal \(\hbox {Ag}_2\)S/TGA QDs with average sizes of 2.3 nm (1) and 3.1 nm (2)
4.2 SERS spectra of \(\hbox {Ag}_2\)S/TGA QDs
The obtained SERS spectra of \(\hbox {Ag}_2\)S/TGA QDs (Fig. 2, insert) are characterized by a wide, intense band with a maximum in the region of 225 \(\hbox {cm}^{-1}\). This band is due to optical phonons localized in the \(\hbox {Ag}_2\)S nanoparticles. It is overlapped with the phonon peak from silver nanoparticles at 154 \(\hbox {cm}^{-1}\). Therefore, the exact position of the maximum can hardly be determined. At the same time, a similar peak was observed near 212 \(\hbox {cm}^{-1}\) for samples prepared on gold substrates with the peak from gold nanoparticles located at 137 \(\hbox {cm}^{-1}\). A peak in the region of 212–225 \(\hbox {cm}^{-1}\) indicates the formation of \(\hbox {Ag}_2\)S particles with a monoclinic lattice (Milekhin et al. 2011). The considerable FWHM of the observed fundamental mode in the SERS spectrum is due to the non-stoichiometry of silver sulfide, as well as due to the presence of nanoparticles of different sizes in the ensemble. This conclusion is consistent with the data of other authors, who noted that the structure of \(\hbox {Ag}_2\)S can involve both amorphous and polycrystalline phases (Minceva-Sukarova et al. 1997).
4.3 FTIR spectra of TGA molecules passivating \(\hbox {Ag}_2\)S QDs
Figure 3 shows the FTIR spectra of pure TGA at pH = 10, as well as the spectra of the molecules adsorbed on \(\hbox {Ag}_2\)S nanocrystals. For \(\hbox {Ag}_2\)S/TGA QDs synthesized at pH=10 under different conditions and at different [\(\hbox {Ag}^-\)]:[\(\hbox {S}^{2+}\)] ratios of ion concentrations ranging from 1:0.9 to 1:1.43, the FTIR spectra differ from each other and from the spectrum of the reference sample [pure TGA at pH=10 (Fig. 3, curve 1)].

FTIR spectra of TGA molecules (1) and colloidal \(\hbox {Ag}_2\)S/TGA QDs with different sizes before exposure (2, 4, 6) and after exposure (3, 5, 7)
The values of the wave numbers corresponding to the peaks of the characteristic frequencies in the FTIR spectra of TGA molecules are given in Fig. 3 (curve 1) and Table 1.
For all FTIR spectra of the \(\hbox {Ag}_2\)S/TGA QD samples, we observe that the peak corresponding to the stretching vibrations of the S–H-group of TGA (2559 \(\hbox {cm}^{-1}\)) vanishes, which indicates the appearance of interactions between the thiol terminal group of TGA and dangling bonds at the \(\hbox {Ag}_2\)S QD interfaces (Banizi and Seifi 2017; Silva et al. 2012; Wageh et al. 2014; Hocaoglu et al. 2012; Zhang et al. 2017; Gui et al. 2014; Chung and Lee 2004; Miao et al. 2015).
Passivation of \(\hbox {Ag}_2\)S QDs by TGA molecules gives rise to peaks of asymmetric and symmetric stretching vibrations of the carboxylic group (\(\hbox {COO}^-\)) (1584 and 1387 \(\hbox {cm}^{-1}\), respectively), which indicates adsorption of TGA molecules on the \(\hbox {Ag}_2\)S interfaces in the ionic form with a free carboxylic terminal group \(\hbox {COO}^-\) (Banizi and Seifi 2017; Silva et al. 2012; Wageh et al. 2014; Hocaoglu et al. 2012; Zhang et al. 2017; Gui et al. 2014; Chung and Lee 2004; Miao et al. 2015). With increasing the concentration of [\(\hbox {S}^{2-}\)] ions from \(\hbox {Na}_2\)S during formation of \(\hbox {Ag}_2\)S/TGA QDs (Fig. 3, curve 2 and 3), a slight high-frequency shift of 7–10 \(\hbox {cm}^{-1}\) was observed for the peak of asymmetric stretching vibrations \(\nu ^{as}\)(\(\hbox {COO}^-\)) = 1567 \(\hbox {cm}^{-1}\) together with a low-frequency shift by 3–5 \(\hbox {cm}^{-1}\) for symmetric stretching vibrations \(\nu ^{s}\)(\(\hbox {COO}^-\))=1388 \(\hbox {cm}^{-1}\), probably caused by the interaction of thiol with the \(\hbox {Ag}_2\)S QD surface. The intensity ratio for the peaks of asymmetric and symmetric stretching vibrations of the carboxylic group \(\hbox {COO}^-\) in the two cases under consideration (Fig. 3, curves 2, 3 and 4) corresponds to the situation when the carboxylic groups are not attached to the surface of the \(\hbox {Ag}_2\)S QD interface (Banizi and Seifi 2017; Silva et al. 2012; Wageh et al. 2014; Hocaoglu et al. 2012; Zhang et al. 2017; Gui et al. 2014; Chung and Lee 2004; Miao et al. 2015). For the third sample (Fig. 3, curve 4, 5) synthesized at the precursor ratio [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] starting from 1:0.9 (without \(\hbox {Na}_2\)S), a significant low-frequency shift of the peak of symmetric stretching vibrations \(\nu ^{s}\)(\(\hbox {COO}^-\)) from 1388 \(\hbox {cm}^{-1}\) to 1359 \(\hbox {cm}^{-1}\) was observed together with a high-frequency shift of the peak of asymmetric stretching vibrations \(\nu ^{as}\)(\(\hbox {COO}^-\)) from 1567 \(\hbox {cm}^{-1}\) to 1579 \(\hbox {cm}^{-1}\). Such spectral shifts indicate that the \(\hbox {COO}^-\) functional groups participate in the intermolecular interactions with, e.g., \(\hbox {Ag}_2\)S QD interfaces. In addition, a change in the intensity ratio for asymmetric and symmetric stretching vibrations \(\hbox {COO}^-\) was noted (Fig. 3, curve 6, 7). This feature is due to the change in the symmetry of vibrations of TGA molecules upon their adsorption by carboxylic groups on a solid substrate and is characteristic of the formation of carboxylate complexes with dangling bonds of \(\hbox {Ag}_2\)S QDs (Chung and Lee 2004).
Adsorption of TGA molecules on \(\hbox {Ag}_2\)S QD interfaces is accompanied by the appearance of peaks at 1788 \(\hbox {cm}^{-1}\) and 1724 \(\hbox {cm}^{-1}\) (Fig. 3, curves 3 and 4) with their intensity being lower than that for \(\nu ^{as(s)}\)(\(\hbox {COO}^-\)). For the samples having the maximum concentration of [\(\hbox {S}^{2-}\)] at crystallization, these peaks disappear, and only the peak at 1743 \(\hbox {cm}^{-1}\) remains. These peaks are associated with vibrations \(\nu ^{as}\)(C=O) of the COOH–group (Silva et al. 2012; Castro et al. 2004). The occurrence of such vibrations is also a sign of the interaction of TGA molecules with \(\hbox {Ag}_2\)S QDs, in which deprotonation of the thiol group is accompanied by protonation of the carboxylic group and appearance of H-bond with the \(\hbox {Ag}_2\)S QD interface. Proton transfer of this kind is characteristic of TGA molecules, mainly in the acidic environment (Castro et al. 2004). In our case, the decisive role is likely to belong to the charge of the QD interface. In addition to the mentioned peaks, weak peaks at 2766 \(\hbox {cm}^{-1}\), 2620 \(\hbox {cm}^{-1}\) were observed, which are due to the compound vibration arising from the interaction of the stretching vibrations C–O (1294 \(\hbox {cm}^{-1}\)) and in-plane \(\delta \)(C–O–H) (1400 \(\hbox {cm}^{-1}\)). The occurrence of these peaks is uncharacteristic of TGA at pH = 10 and seems to be caused by the specifity of adsorption on \(\hbox {Ag}_2\)S QDs. The complex nature of this peak was confirmed in Ihs and Liedberg (1991) and Chen and Kimura (1999).
For symmetric and asymmetric \(\hbox {CH}_2\) stretching vibrations in the region of 2980–2850 \(\hbox {cm}^{-1}\)), as well as for all types of \(\hbox {CH}_2\) bending vibrations [1220–1230 \(\hbox {cm}^{-1}\), 1125–1133 \(\hbox {cm}^{-1}\), 910–750 \(\hbox {cm}^{-1}\) (Banizi and Seifi 2017; Silva et al. 2012; Wageh et al. 2014; Hocaoglu et al. 2012; Zhang et al. 2017; Gui et al. 2014; Chung and Lee 2004; Miao et al. 2015; Castro et al. 2004)], variations within 5–15 \(\hbox {cm}^{-1}\) were detected. This feature is also a sign of the interaction between TGA and the \(\hbox {Ag}_2\)S QD surface. However, CH-groups are not directly involved in the adsorption.
In each of the analyzed FTIR spectra of \(\hbox {Ag}_2\)S/TGA QD samples, changes were noted for the stretching and bending vibrations of OH-bonded groups. It was found that with an increase in [\(\hbox {S}^{2-}\)] ions concentration when using \(\hbox {Na}_2\)S, a high-frequency feature arises in the range of 3350–3500 \(\hbox {cm}^{-1}\) (Fig. 3, curve 2). This feature is related to OH-groups of water molecules located near TGA molecules and \(\hbox {Ag}_2\)S/TGA QDs (Banizi and Seifi 2017; Silva et al. 2012; Wageh et al. 2014; Miao et al. 2015; Ihs and Liedberg 1991; Chen and Kimura 1999; Kaur and Tripathi 2014; Koneswaran and Narayanaswamy 2009). These OH-groups provide the H-bond bridges formation, in the case of dimerization and especially \(\hbox {Ag}_2\)S QD interfaces passivation with TGA molecules. These molecules are less bonding with TGA molecules (Koneswaran and Narayanaswamy 2009). This hypothesis is also confirmed by a high-frequency shift for 30–45 \(\hbox {cm}^{-1}\) of the OH bending vibrations peak (1646 \(\hbox {cm}^{-1}\)) under \(\hbox {Ag}_2\)S QD interfaces passivation with TGA molecules. Similar shift indicates the occurrence of H-bonds between \(\hbox {Ag}_2\)S QD interface, COOH functional groups, and \(\hbox {H}_2\)O molecules.
Thus, the data obtained from FTIR spectra indicate two predominantly realized forms of TGA molecules adsorption on the \(\hbox {Ag}_2\)S QD interfaces (Fig. 3). In the case of \(\hbox {Ag}_2\)S/TGA QDs synthesis in the absent of \(\hbox {Na}_2\)S, predominantly adsorption by two functional groups (thiol and carboxylic) was noted. When \(\hbox {Na}_2\)S is used as an additional sulfur source during \(\hbox {Ag}_2\)S QDs crystallization, TGA adsorption is predominantly carried out by the thiol group. It is accompanied by molecule deprotonation.
4.4 Size effect in optical absorption spectra of colloidal \(\hbox {Ag}_2\)S/TGA QDs
The optical absorption spectra of investigated samples are present by wide bands characteristic for semiconductor QDs with a feature located in the region from 1.55 to 3.10 (±0.01) eV (Fig. 4) and caused by ground state exciton absorption. For all seven studied samples the position of ground state exciton absorption exceeded by 0.55–2.00 eV the band gap of bulk \(\hbox {Ag}_2\)S crystals with a monoclinic crystal structure (1.09 eV) (Lin et al. 2015). The observed difference is caused by quantum confinement in the optical absorbtion spectra.

Optical absorption and photoluminescence spectra of colloidal \(\hbox {Ag}_2\)S/TGA QDs with various sizes
The optical absorption spectrum of \(\hbox {Ag}_2\)S/TGA QD samples prepared without \(\hbox {Na}_2\)S addition was the most structured. An exciton peak at 2.1 eV was observed in the optical absorption spectrum (Fig. 4). This type of QD ensembles is characterized by the smallest size distribution (7–10%). In other cases, when the QD size distribution was 25%, the ground state exciton absorption band did not have a pronounced maximum. This confirms the conclusion that the increase in nanoparticles size distribution is one of the main reasons for the loss of the exciton absorption band structure of \(\hbox {Ag}_2\)S/TGA QDs.
The position of ground state exciton absorption in each case was found from the minimum of the second derivative for the optical density spectra with respect to the incident radiation energy \(d^2D/d(\hbar \omega )^2\). These values are shown in Fig. 4 near the arrow marking their position. Thus, the selected conditions for the samples synthesis provide a change in the size effect in the optical absorbtion spectra of \(\hbox {Ag}_2\)S/TGA QDs synthesized in the presence of \(\hbox {Na}_2\)S.
The ground state exciton absorption energies in optical absorption are 1.55 eV, 1.65 eV, 1.81 eV, 1.90 eV, 2.29 eV, and 3.10 eV, respectively (Fig. 4). They were used to estimate average sizes. Estimates of QDs average size were realized using the blue shift value for the ground state exciton absorption band maximum in the framework of strong confinement approximation (Efros et al. 1982) of the effective mass approximation. This approximation does not take into account the Coulomb interaction similar to it was done in Kayanuma (1988). However, this approach is justified for the small nanoparticles sizes, which are presented in TEM images (nanocrystal sizes are 1.7–3.1 nm). Its using let to estimate separately the size effect for the states of electrons and holes (Fig. 5). It was used further in Sect. 4.5 to establish a trap state luminescence mechanism.
Ground state energies of electrons and holes discrete levels with the main quantum number n = 1 and orbital quantum number l = 0 (S-like lower state) were calculated using the equation
The energy changes values of electrons \(\varDelta E^{e}_{0,1}\) and holes \(\varDelta E^{h}_{0,1}\) due to quantum confinement are given in Table 2. For estimation the average QD size using the effective band gap, it was used the equation
where \(E^{bulk}_g\) = 1.00 eV (Lin et al. 2015) is a band gap of bulk \(\hbox {Ag}_2\)S crystal; \(\mu =\frac{m_em_h}{m_e+m_h}\) is a reduced mass, R is QD radius, \(m_e=0.42m_0\) and \(m_h=0.81m_0\) are effective masses of electron and hole, respectively (Lin et al. 2015).
The results of theoretical estimates of \(\hbox {Ag}_2\)S/TGA QDs average sizes are presented in Table 2. It turned out that the appearance of characteristic features for all studied samples is caused by \(\hbox {Ag}_2\)S nanoparticles with an average size of 3.14 nm (1.55 eV), 2.90 nm (1.65 eV), 2.58 nm (1.81 eV), 2.46 nm (1.90 eV), 2.22 nm (2.10 eV ), 2.04 nm (2.29 eV), 1.60 nm (3.10 eV). Slight differences of the obtained results from the data on TEM analysis are due to the calculation error in the effective mass approximation. We observed similar deviations in the analysis of \(\hbox {Ag}_2\)S QD size dependence, synthesized by various methods, which presented in Perepelitsa et al. (2018).

Size dependences of recombination luminescence peak (experimental) and for energy states of conduction electrons (estimations) and fully filled states of valence band (estimations by Eq. 1)
Thus, the results of average sizes estimates indicate that the used method of \(\hbox {Ag}_2\)S/TGA QDs synthesis ensures the formation of nanoparticles with a size close to/or smaller than the Bohr radius of the Wannier–Mott exciton (1.15 nm) (Zhang et al. 2014; Lin et al. 2015). In this case, insignificant changes in the nanoparticles average sizes lead to a noticeable size effect in the optical absorbtion spectra.
4.5 Size effect in photoluminescence spectra of colloidal \(\hbox {Ag}_2\)S/TGA QDs
The synthesized \(\hbox {Ag}_2\)S/TGA QD samples show photoluminescence excited by light from the ground state exciton absorption region (Fig. 4). The position and FWHM of luminescence bands, as well as the value of the Stokes shift for the studied \(\hbox {Ag}_2\)S QD samples, substantially depend on the their synthesis conditions. Luminescence bands were observed with peaks at 620 nm (2.22 nm), 660 nm (1.60 nm), 880 nm (2.04 nm), 900 nm (2.46 nm), 934 nm (2.58 nm), 960 nm (2.90 nm) and 1000 nm (3.14 nm) (nanoparticle sizes are shown in parentheses). The blue shift of the luminescence spectrum peak with a decrease in the QD size is a size effect manifestation in luminescence.
In the luminescence spectrum of \(\hbox {Ag}_2\)S/TGA QDs synthesized without using \(\hbox {Na}_2\)S a narrow peak was observed at 620 nm with a FWHM of 50 nm under excitation of 532 nm. The slight Stokes shift (0.1 eV) and small FWHM indicate an exciton luminescence for this sample. In this case, there are no recombination luminescence bands.
A wider luminescence bands with peaks position depends on the QD size were observed for colloidal \(\hbox {Ag}_2\)S/TGA QD solutions obtained using \(\hbox {Na}_2\)S under excitation. Photoluminescence bands for these samples are also shown on Fig. 4. They are distinguished by the significant Stokes shift of luminescence spectra peak relative to the position of ground state exciton absorption, which increases with decreasing the nanocrystal size. The Stokes shift values lie at 0.29–1.22 eV. This feature indicates the recombination nature of observed luminescence. Also it is a size effect manifestation in luminescence.
The size dependence of the recombination luminescence peaks are closest in slope to the curve showing the size effect for the holes energy states. The size effect for the electrons energy states was sharper than for the luminescence peak (Fig. 5). The existing regularities indicate that the recombination of a localized electron with a free hole is most probable. A similar conclusion was made by us earlier for \(\hbox {Ag}_2\)S QDs dispersed in gelatin (Ovchinnikov et al. 2015b; Perepelitsa et al. 2018). Later, the authors of Mir et al. (2015) came to the same conclusion, but for \(\hbox {Ag}_2\)S QDs caped by 3-MPA. The size dependence of the luminescence band peaks for \(\hbox {Ag}_2\)S QDs dispersed in gelatin is also shown in Fig. 5 (red curve).
Thus, the recombination luminescence centers for \(\hbox {Ag}_2\)S QDs stabilized with various organic molecules (TGA, 3-MPA, gelatin) have a similar structure, providing the same type of recombination. However, their position in the spectrum at similar sizes may differ. Probably, a significant polarizing effect of the molecules surrounding QDs, or their inclusion in the centers structure of the trap state luminescence, having an interface nature. In addition, the available data indicate the influence of synthesis conditions on the nature of \(\hbox {Ag}_2\)S QDs luminescence.
4.6 Luminescence decay
For all investigated samples of colloidal \(\hbox {Ag}_2\)S/TGA QDs the different luminescence decay was observed (Fig. 6). The observed luminescence decay curves for all samples in the time range of 0.1–200 ns have a complex multiexponential dependence and different average luminescence lifetimes (Table 3). Approximation of experimental curves with a sum of four exponents let to determine the average luminescence decay time
where \(f(t)\Delta {dt}\) is a probability of a light quantum emission from \(\hbox {Ag}_2\)S/TGA QDs at time t, \(a_i\) and \(\tau _i\) are the amplitude and lifetime of i-th component luminescence.

Luminescence decay of \(\hbox {Ag}_2\)S/TGA QDs
Characteristic average luminescence lifetimes for the bands were 620 nm (1.0 ns), 660 nm (32.8 ns), 880 nm (2.0 ns), 900 nm (2.6 ns), 934 nm (3.2 ns), 960 nm (2.5 ns), and 1000 nm (3.3 ns) (Table 3). A qualitative analysis of the luminescence decay curves shows the presence of the initial (fast) component with characteristic times of a few nanoseconds and slow component on a time scale of hundreds nanoseconds.
For the \(\hbox {Ag}_2\)S/TGA QD sample with an exciton luminescence band with a peak near 620 nm, the fast component of luminescence decay has a maximum contribution. The initial time constant is 0.8 ns. For \(\hbox {Ag}_2\)S/TGA QDs with a recombination luminescence band, the which peak is located near 880 nm, the initial time of decay curve has a time constant \(\sim \) 2 ns, and luminescence decays no more than 50 times. For \(\hbox {Ag}_2\)S/TGA QDs with a recombination luminescence band with peak near 660 nm, the contribution of the fast component is minimal. The luminescence decays only 5 times. The average luminescence lifetime is maximum. Its value is 32.8 ns.
The complex luminescence decay law for QDs is usually due to several reasons. This is a quenching by solvent molecules or impurities. The decay law takes a multiexponential form due to the distribution on the recombination quenching rate constants (Tachiya 1982; Sadhu et al. 2009). The second reason is QDs size distribution. This also leads to the multiexponential decay law (Ovchinnikov et al. 2019; Smirnov et al. 2018; Ovchinnikov et al. 2016). However, the luminescence decay law turns out to be multiexponential even in the case of a high quantum yield when quenching by solvent and ligands is ineffective, and the size distribution of QDs is small. The reason, apparently, is the statistical distribution of luminescence centers in QDs (Ye et al. 2010; Shea-Rohwer and Martin 2007; Kima et al. 2005; Martin and Shea-Rohwer 2006).
Thus, the differences found in the law of luminescence decay are largely determined by the QDs size distribution and various luminescence quenching channels that formed under different crystallization conditions. In the future, the data on the average times of luminescence decay will be used to study the nature of \(\hbox {Ag}_2\)S QD luminescence properties degradation under the influence of exciting radiation.
4.7 Photostability of IR luminescence of \(\hbox {Ag}_2\)S/TGA QDs
An important found property of \(\hbox {Ag}_2\)S/TGA QDs luminescence is a decrease in its intensity with increasing the exposure time by exiting radiation. Long exposure of the samples leads to a decrease in the luminescence intensity over the entire spectrum (Fig. 7). The characteristic decay times of \(\hbox {Ag}_2\)S QDs IR luminescence intensity under the exposure with a wavelength, corresponded to the exciton absorption region at an incident power of about 100 mW, are 200–300 s. \(\hbox {Ag}_2\)S/TGA QDs with exciton luminescence with peak near 620 nm turned out to be the least susceptible to luminescence photodegradation. A similar dependence was found for \(\hbox {Ag}_2\)S/TGA QDs with recombination luminescence with peak at 660 nm. For other QDs with recombination IR luminescence in the range of 880–1000 nm, the decrease in luminescence intensity exceeded 50% under the same exposure conditions.

Photodegradation of \(\hbox {Ag}_2\)S/TGA QD luminescence properties
The photostimulated decrease in the luminescence intensity of \(\hbox {Ag}_2\)S QDs turned out to be reversible mainly for samples with IR luminescence in the range of 880–1000 nm (Fig. 7, dotted line). The luminescence intensity is restored almost to its initial value in the dark within 22–24 h. The activation energy of the luminescence intensity restoring process was about 0.9–1.0 eV.
Simultaneously with the decrease in \(\hbox {Ag}_2\)S/TGA QDs luminescence intensity, a decrease in average QDs luminescence lifetime was found. In some cases it reaches 50–60% (Table 2). The observed regularities indicates an additional channels of a luminescence quenching. Non-radiative resonance energy transfer is possible directly from the IR luminescence centers in colloidal \(\hbox {Ag}_2\)S/TGA QDs to the quenching centers arising under samples photoexposure.
The formation of new centers of non-radiative recombination is also supported by a decrease in the degree of the luminescence intensity degradation and slowdown in its rate at low (77 K) temperatures. Apparently, \(\hbox {Ag}_2\)S nanocrystals photolysis, established in Rempel et al. (2017), plays a significant role. The initial stage of this photochemical reactions kind on the interface of photosensitive crystals was considered in Ievlev et al. (2006) and Ovchinnikov et al. (2007). In these works, the participation of photolytic ions and silver atoms in the photostimulated formation of low-atomic clusters due to photodiffusion is substantiated.
Also the higher concentration of sulfur ions used in the QD crystallization is feature of \(\hbox {Ag}_2\)S/TGA QD samples with luminescence in the range of 880–1000 nm. In this case, the formation of low-atomic silver clusters is more possible. Their formation leads to the luminescence quenching. In the case of \(\hbox {Ag}_2\)S/TGA QDs with luminescence in the range of 880–1000 nm, a decrease in the intensity is accompanied by an acceleration of the recombination luminescence lifetime during samples photoexposure (Table 2).
In the case of QDs, TGA molecules play role of a sulfur precursor. The interaction of TGA and low-atomic silver clusters leads to the formation of silver sulfide and healing of silver defects. This achieves the reversibility of the luminescence intensity degradation.
However, there are possible situations in which the luminescence lifetime varies little. So for \(\hbox {Ag}_2\)S/TGA QDs sample with exciton luminescence, the photodegradation of luminescence was not accompanied by a noticeable decrease in the lifetime luminescence. At the same time the luminescence quenching was irreversible. Its intensity did not recover when the samples were kept in the dark at room temperature. A similar picture was also noted for \(\hbox {Ag}_2\)S/TGA QDs, which have recombination luminescence near 660 nm (Fig. 7). This fact indicates the existence of another process leading to degradation of \(\hbox {Ag}_2\)S/TGA QD luminescence intensity. It dominates in these two cases. This process is associated with a structural change in the QD interface under the action of exciting radiation since QDs luminescence properties are very sensitive to the interface structure, which is determined by synthesis methods.
Photoprocesses involving interfaces cannot be identical for different QDs. This conclusion is confirmed in our case by FTIR spectra analysis (Fig. 3). The restructuring/destruction reactions of surface ligands usually modify the physicochemical states of QDs surface atoms and dramatically change various QDs properties, including the photostability of QD surface atoms, resistance to the oxidative dissolution process, and tendency to coagulation/deposition. Many researchers Miao et al. (2015) and Carrillo-Carrión et al. (2009), Cordero et al. (2000), Guo et al. (2005), Wang et al. (2004) and Ma et al. (2007) report that thiol-coated QDs begin to precipitate at continuous UV or visible radiation exposure. A photooxidation of QDs surfaceis possible Miao et al. (2015), Carrillo-Carrión et al. (2009), Cordero et al. (2000), Guo et al. (2005), Wang et al. (2004) and Ma et al. (2007). At the same time, an increase/decrease in the luminescence intensity was noted. In our experiments, QDs precipitation was observed neither visually nor spectrally.
However, the effect of exciting radiation on thiol-coated QDs can lead to its photodestruction. In particular, the photodissociation of the C–OH-bond for TGA with the formation of \(\alpha \)-thiol-substituted acyl radical (S–\(\hbox {CH}_2\)–CO\(\cdot \)) is known (Attar et al. 2011). A similar photochemical reaction is supported by our FTIR spectra of \(\hbox {Ag}_2\)S/TGA QD samples subjected to photo-exposure (Fig. 3, dotted curves). A change in the high-frequency form of the OH-bonding mode is observed in the region of 3600–3500 \(\hbox {cm}^{-1}\). After exposure, a peak arises from the edge from this band near 3600 \(\hbox {cm}^{-1}\) due to vibrations of free or weakly bound OH-groups. At the same time, a change in the intensities ratio of asymmetric (1574–1579 \(\hbox {cm}^{-1}\)) and symmetric (1383–1388 \(\hbox {cm}^{-1}\)) stretching vibrations of \(\hbox {COO}^-\)-groups was observed. Probably it is caused by a change in the nature of the interaction with \(\hbox {Ag}_2\)S QDs surface. Along with adsorption by thiol groups, it is possible that carboxylic groups attached to the \(\hbox {Ag}_2\)S QDs surfaces, including with the participation of OH-groups (Banizi and Seifi 2017; Silva et al. 2012; Wageh et al. 2014; Hocaoglu et al. 2012; Zhang et al. 2017; Gui et al. 2014; Chung and Lee 2004; Miao et al. 2015).
5 Conclusion
Thus, the paper presents the results of studies of synthesis conditions influence on optical properties of colloidal \(\hbox {Ag}_2\)S QDs, coated with thioglycolic acid molecules. It is shown that it is possible to change the QD sizes in the range of 1.7–3.1 nm by changing the precursors [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] molar ratio from 1:0.9 to 1:1.43. The formation of \(\hbox {Ag}_2\)S QDs with a monoclinic crystal structure (space group P21/c) is proven using HR TEM and SERS methods. Peaks in the Raman spectra were found in the region of 212–225 \(\hbox {cm}^{-1}\) for gold and silver plasmon substrates corresponded to the LO phonons absorption.
Synthesized \(\hbox {Ag}_2\)S QDs at different ratios of precursors have fundamentally different luminescence properties. So for samples synthesized without using \(\hbox {Na}_2\)S, luminescence is observed in the visible region. At [\(\hbox {AgNO}_3\)]:[TGA] molar ratio equal to 1:1, QDs samples have exciton luminescence with the luminescence peak near 620 nm and FWHM of less than 50 nm. When the [\(\hbox {Ag}^+\)]:[\(\hbox {S}^{2-}\)] molar ratio varies from 1:0.9 to 1:1.43, a trap state luminescence arises with the band maximum located in the region of 660–1000 nm and FWHM more than 100 nm. The addition of \(\hbox {Na}_2\)S at the synthesis stage in the reaction mixture lets shift the luminescence bands in NIR spectral region from 880 nm to 1000 nm.
FTIR spectra show that TGA molecules are adsorbed on the \(\hbox {Ag}_2\)S QDs interfaces at maximum of \(\hbox {Na}_2\)S concentrations mainly by the thiol group. With a decrease in \(\hbox {Na}_2\)S concentration during synthesis, the attachment to the \(\hbox {Ag}_2\)S QDs interfaces is carried out by two active groups. They are thiol and carboxylic groups.
For all samples of colloidal \(\hbox {Ag}_2\)S QDs, it was found that exciting radiation leads to the degradation of luminescence properties. Moreover, for \(\hbox {Ag}_2\)S QDs synthesized without using \(\hbox {Na}_2\)S, this degradation is irreversible. The keeping of colloidal \(\hbox {Ag}_2\)S QDs synthesized in the presence of \(\hbox {Na}_2\)S in the dark at room temperature leads to a partial recovery of IR photoluminescence intensity.
According to the data obtained with use the time correlated single photon counting method, it was shown that a decrease in IR luminescence intensity during photodegradation is accompanied by a decrease in the average luminescence lifetime. This fact is interpreted as the formation of non-radiative recombination channels arising during the initial stage of silver sulfide photolysis. Data on FTIR spectra show that IR luminescence photodegradation is also accompanied by changes in the thioglycolic acid molecules covering \(\hbox {Ag}_2\)S QDs.
The presented data demonstrate the key role of the chosen approach to the \(\hbox {Ag}_2\)S/TGA QDs synthesis in the formation of size-dependent absorption and luminescence properties, as well as their photostability to exciting radiation.
References
Almendral-Parra, M.J., Alonso-Mateos, Á., Sánchez-Paradinas, S., Boyero-Benito, J.F., Rodríguez-Fernández, E., Criado-Talavera, J.J.: Procedures for controlling the size, structure and optical properties of CdS quantum dots during synthesis in aqueous solution. J. Fluoresc. 22(1), 59–69 (2012)
Attar, A.R., Blumling, D.E., Knappenberger, K.L.: Photodissociation of thioglycolic acid studied by femtosecond time-resolved transient absorption spectroscopy. J. Chem. Phys. 134, 024514 (2011)
Banizi, Z.T., Seifi, M.: Optical properties of hydrothermally synthesized TGA-capped CdS nanoparticles: controlling crystalline size and phase. Mater. Res. Express 4(10), 105007 (2017)
Bera, D., Qian, L., Tseng, T.-K., Holloway, P.H.: Quantum dots and their multimodal applications: a review. Materials 3(4), 2260–2345 (2010)
Carrillo-Carrión, C., Cárdenas, S., Simonet, B.M.: Valcárcel, M.: Quantum dots luminescence enhancement due to illumination with UV/Vis light. Chem. Commun. 35, 5214–5226 (2009)
Castro, J.L., López-Ramírez, M.R., Arenasand, J.F., Otero, J.C.: Surface-enhanced Raman scatteringof 3-mercaptopropionic acid adsorbedon a colloidal silver surface. J. Raman Spectrosc. 35, 997–1000 (2004)
Chaudhry, M., Lim, D.-K., Qamar, R., Bhatti, A.S.: The adverse role of excess negative ions in reducing the photoluminescence from water soluble MAA-CdSe/ZnS quantum dots in various phosphate buffers. Phys. Chem. Chem. Phys. 20, 29446–29451 (2018)
Chen, S., Kimura, K.: Synthesis and characterization of carboxylate-modified gold nanoparticle powders dispersible in water. Langmuir 15(4), 1075–1082 (1999)
Chung, Ch., Lee, M.: Self-assembled monolayers of mercaptoacetic acid on Ag powder: characterization by FT-IR diffuse reflection spectroscopy. Bull. Korean Chem. Soc. 25(10), 1461–1462 (2004)
Cordero, S.R., Carson, P.J., Estabrook, R.A., Strouse, G.F., Buratto, S.K.: Photo-activated luminescence of CdSe quantum dot monolayers. J. Phys. Chem. B 104(51), 12137–12142 (2000)
Du, Y., Xu, B., Fu, T., Cai, M., Li, F., Zhang, Y., Wang, Q.: Near-infrared photoluminescent \(\text{ Ag }_2\)S quantum dots from a single source precursor. J. Am. Chem. Soc. 132, 1470–1471 (2010)
Duman, F.D., Hocaoglu, I., Ozturk, D.G., Gozuacik, D., Kiraz, A., Acar, H.Y.: Highly luminescent and cytocompatible cationic \(\text{ Ag }_2\)S NIR-emitting quantum dots for optical imaging and gene transfection. Nanoscale 7(26), 11352–11362 (2015)
Efros, A.L., Efros, L.: Interband absorption of light in a semiconductor sphere. Sov. Phys. Semicond. 16(7), 772–775 (1982)
Freckera, T., Baileya, D., Arzeta-Ferrera, X., McBridea, J., Rosenthal, S.J.: Review-quantum dots and their application in lighting, displays, and biology. ECS J. Solid State Sci. Technol. 5(1), R3019–R3031 (2016)
Galloway, J.F., Park, J., Dvoracek, ChM, Dallas, M., Konstantopoulos, K., Maitra, A., Searson, P.C., Lee, K.H.: Quantitative molecular profiling of biomarkers for pancreatic cancer with functionalized quantum dots, nanomedicine: nanotechnology. Biol. Med. 8(7), 1043–1051 (2012)
Gui, R., Sun, J., Liu, D., Wang, Y., Jinc, H.: A facile cation exchange-based aqueous synthesis of highly stable and biocompatible \(\text{ Ag }_2\)S quantum dots emitting in the second near-infrared biological window. Dalton Trans. 43, 16690–16697 (2014)
Gui, R., Jin, H., Wang, Z., Tan, L.: Recent advances in synthetic methods and applications of colloidal silver chalcogenide quantum dots. Coord. Chem. Rev. 296, 91–124 (2015)
Guo, J., Yang, W., Wang, C.: Systematic study of the photoluminescence dependence of thiol-capped CdTe nanocrystals on the reaction conditions. J. Phys. Chem. B 109(37), 17467–17473 (2005)
Hocaoglu, I., Cizmeciyan, M.N., Erdem, R., Ozen, C., Kurt, A., Sennaroglud, A., Acar, H.Y.: Development of highly luminescent and cytocompatible near-IR-emitting aqueous \(\text{ Ag }_2\)S quantum dots. J. Mater. Chem. 22, 14674 (2012)
Hu, M.Z., Zhu, T.: Semiconductor nanocrystal quantum dot synthesis approaches towards large-scale industrial production for energy applications. Nanosc. Res. Lett. 10, 469 (2015)
Ievlev, V.M., Latyshev, A.N., Ovchinnikov, O.V., Smirnov, M.S.: Photostimulated formation of anti-stokes luminescence centers in ionic covalent crystals. Dokl. Phys. 51(8), 400–402 (2006)
Ihs, A., Liedberg, B.: Chemisorption of L-cysteine and 3-mercaptopropionic acid on gold and copper surfaces: an infrared reflection-absorption study. J. Colloid Interface Sci. 144(1), 282–292 (1991)
Jiang, P., Zhu, C.-N., Zhang, Z.L., Tian, Z.Q., Pang, D.-W.: Water-soluble \(\text{ Ag }_2\)S quantum dots for near-infrared fluorescence imaging in vivo. Biomaterials 33, 5130–5135 (2012)
Karimipour, M., Molaei, N.M.M.: Strong NIR luminescent \(\text{ Ag }_2\)S@ZnS core-shells synthesized by a novel one pot pulsed microwave irradiation. J. Luminesc. 182, 91–97 (2017)
Karimipour, M., Bagheri, M., Johansson, E.M.J., Molaei, M.: Excellent growth of ZnS shell on \(\text{ Ag }_2\)S QDs using a photochemical-microwave irradiation approach and fabrication of their indoor QD thin film solar cells. Mater. Technol. 33(12), 784–792 (2018)
Karimipour, M., Bagheri, M., Molaei, M.: One pot, rapid and green photo-chemical synthesis of \(\text{ Ag }_2\)S and \(\text{ Ag }_2\)S-ZnS core–shells. J. Electron. Mater. 48(4), 2555–2562 (2019)
Kaur, G., Tripathi, S.K.: Size tuning of MAA capped CdSe and CdSe/CdS quantum dots and their stability in different pH environments. Mater. Chem. Phys. 143, 514–523 (2014)
Kayanuma, Y.: Quantum-size effects of interacting electrons and holes in semiconductor microcrystals with spherical shape. Phys. Rev. B 38, 9797–9805 (1988)
Kima, D., Miyamoto, M., Mishima, T., Nakayama, M.: Strong enhancement of band-edge photoluminescence in CdS quantum dots prepared by a reverse-micelle method. J. Appl. Phys. 98, 083514 (2005)
Koneswaran, M., Narayanaswamy, R.: Mercaptoacetic acid capped CdS quantum dots as fluorescence single shot probe for mercury(II). Sens. Actuat. B 139, 91–96 (2009)
Lin, S., Feng, Y., Wen, X., Zhang, P., Woo, S., Shrestha, S., Conibeer, G., Huang, S.: Theoretical and experimental investigation of the electronic structure and quantum confinement of wet-chemistry synthesized \(\text{ Ag }_2\)S nanocrystals. J. Phys. Chem. 119, 867–872 (2015)
Ma, J., Chen, J.-Y., Zhang, Y., Wang, P.-N., Guo, J., Yang, W.-L., Wang, C.-C.: Photochemical instability of thiol-capped CdTe quantum dots in aqueous solution and living cells: process and mechanism. J. Phys. Chem. B 111(41), 12012–12016 (2007)
Martin, J.E., Shea-Rohwer, L.E.: Lifetime determination of materials that exhibit a stretched exponential luminescent decay. J. Lumin. 121(2), 573–587 (2006)
Miao, Ya., Yang, P., Zhao, J., Du, Y., He, H., Liu, Yu.: Photodegradation of mercaptopropionic acid- and thioglycollic acid-capped CdTe quantum dots in buffer solutions. J. Nanosci. Nanotechnol. 15(6), 4462–4469 (2015)
Milekhin, A., Sveshnikova, L., Duda, T., Surovtsev, N., Adichtchev, S., Zahn, D.R.T.: Optical phonons in nanoclusters formed by the Langmuir–Blodgett technique. Chin. J. Phys. 49(1), 63–70 (2011)
Minceva-Sukarova, B., Najdoski, M., Grozdanov, I., Chunnilall, C.J.: Raman spectra of thin solid films of some metal sulfides. J. Mol. Struct. 410–411, 267–270 (1997)
Mir, W.J., Swarnkar, A., Sharma, R., Katti, A., Adarsh, K.V., Nag, A.: Origin of unusual excitonic absorption and emission from colloidal \(\text{ Ag }_2\)S nanocrystals: ultrafast photophysics and solar cell. J. Phys. Chem. Lett. 6, 3915–3922 (2015)
Mirhosseini, H.N., Karimipour, M., Bagheri, M.M.M.: One pot and room temperature photochemical synthesis of high quantum yield NIR emissive \(\text{ Ag }_2\)S@Ag(In, Zn)\(\text{ S }_2\) core-shells at the presence of air in water. J. Photochem.Photobiol. A Chem. 380, 111854 (2019)
Moradian, R., Elahi, M., Hadizadeh, A., Roshani, M., Taghizadeh, A., Sahraei, R.: Structural, optical, and electrical properties of thioglycolic acid-capped CdTe quantum dots thin films. Int. Nano Lett. 3, 56 (2013)
Ovchinnikov, O.V., Smirnov, M.S., Latyshev, A.N., Stasel’ko, D.I.: Photostimulated formation of sensitized anti-stokes luminescence centers in AgCl(I) microcrystals. Opt. Spectrosc. 103(3), 482–489 (2007)
Ovchinnikov, O.V., Smirnov, M.S., Perepelitsa, A.S., Shatskikh, T.S., Shapiro, B.I.: Optical power limiting in ensembles of colloidal \(\text{ Ag }_2\)S quantum dots. Quant. Electron. 45(2), 1143–1150 (2015)
Ovchinnikov, O.V., Smirnov, M.S., Shapiro, B.I., Shatskikh, T.S., Perepelitsa, A.S., Korolev, N.V.: Optical and structural properties of ensembles of colloidal \(\text{ Ag }_2\)S quantum dots in gelatin. Semiconductors 49(3), 373–379 (2015)
Ovchinnikov, O.V., Kondratenko, T.S., Grevtseva, I.G., Smirnov, M.S., Pokutnyi, S.I.: Sensitization of photoprocesses in colloidal \(\text{ Ag }_2\)S quantum dots by dye molecules. J. Nanophoton. 10(3), 033505(1-12) (2016)
Ovchinnikov, O.V., Smirnov, M.S., Korolev, N.V., Golovinski, P.A., Vitukhnovsky, A.G.: The size dependence recombination luminescence of hydrophilic colloidal CdS quantum dots in gelatin. J. Lumin. 179, 413–419 (2016)
Ovchinnikov, O.V., Grevtseva, I.G., Smirnov, M.S., Kondratenko, T.S.: Reverse photodegradation of Infrared luminescence of colloidal \(\text{ Ag }_2\)S quantum dots. J. Luminesc. 207, 626–632 (2019)
Perepelitsa, A.S., Smirnov, M.S., Ovchinnikov, O.V., Latyshev, A.N., Kotko, A.S.: Thermostimulated luminescence of colloidal \(\text{ Ag }_2\)S quantum dots. J. Luminesc. 198, 357–363 (2018)
Petryayeva, E., Russ Algar, W., Medintz, I.L.: Quantum dots in bioanalysis: a review of applications across various platforms for fluorescence spectroscopy and imaging. Appl. Spectros. 67, 215–252 (2013)
Piven, N.G., Khalavka, YuB, Shcherbak, L.P.: Effect of SH-containing ligands on the growth of CdS nanoparticles. Inorg. Mater. 44(10), 1047 (2008)
Ratnesh, R.K., Mehataa, M.S.: Controlled synthesis and optical properties of tunable CdSe quantum dots and effect of pH. AIP Adv. 5, 097114 (2015)
Rempel, S.V., Kuznetsova, Y.V., Rempel’, A.A., Gerasimov, E.Y.: The irradiation influence on the properties of silver sulfide (\(\text{ Ag }_2\)S) colloidal nanoparticles. Phys. Solid State 59(8), 1629–1636 (2017)
Sadhu, S., Tachiya, M., Patra, A.: A stochastic model for energy transfer from CdS quantum dots/rods (donors) to nile red dye (acceptors). J. Phys. Chem. C 113, 19488–19492 (2009)
Sadovnikov, S.I., Gusev, A.I.: Recent progress in nanostructured silver sulfide: from synthesis and nonstoichiometry to properties. J. Mater. Chem. A 5, 17676–17704 (2017)
Sadovnikov, S.I., Gusev, A.I., Gerasimov, EYu., Rempel, A.A.: Facile synthesis of \(\text{ Ag }_2\)S nanoparticles functionalized by carbon-containing citrate shell. Chem. Phys. Lett. 642, 17–21 (2015)
Sadovnikov, S.I., Gusev, A.I., Rempel, A.A.: Nonstoichiometry of nanocrystalline monoclinic silver sulfide. Phys. Chem. Chem. Phys. 17, 12466–12471 (2015)
Semonin, O.E., Luther, J.M., Beard, M.C.: Quantum dots for next-generation photovoltaics. Materialstoday 15, 508–515 (2012)
Shea-Rohwer, L.E., Martin, J.E.: Luminescence decay of broadband emission from CdS quantum dots. J. Lumin. 127(2), 499–507 (2007)
Silva, F.O., Carvalho, M.S., Mendonça, R., Macedo, W.A.A., Balzuweit, K., Reiss, P., Schiavon, M.A.: Effect of surface ligands on the optical properties of aqueous soluble CdTe quantum dots. Nanoscale Res Lett. 7(1), 536 (2012)
Smirnov, M.S., Ovchinnikov, O.V., Grevtseva, I.G., Zvyagin, A.I., Perepelitsa, A.S., Ganeev, R.A.: Photoinduced degradation of the optical properties of colloidal \(\text{ Ag }_2\)S and CdS quantum dots passivated by thioglycolic acid. Opt. Spectrosc. 124(5), 681–686 (2018)
Sousa, J.C.L., Vivas, M.G., Vale, B.R.C., Ferrari, J.L., Schiavo, M.A.: Effect of mercaptosuccinic acid stabilizer agent on the optical properties of colloidal CdTe quantum dots. J. Nanosci. Nanotechnol. 18(1), 651–658 (2018)
Sun, Q., Fu, Sh, Dong, T., Liu, Sh, Huang, Ch.: Aqueous synthesis and characterization of TGA-capped CdSe quantum dots at freezing temperature. Molecules 17, 8430–8438 (2012)
Sun, J., Yu, W., Usman, A., Isimjan, T.T., DGobbo, S., Alarousu, E., Takanabe, K., Mohammed, O.F.: Generation of multiple excitons in \(\text{ Ag }_2\)S quantum dots: single high-energy versus multiple-photon excitation. J. Phys. Chem. Lett. 5, 659–665 (2014)
Tachiya, M.: Kinetics of quenching of luminescent probes in micellar systems. II. J. Chem. Phys. 76, 340–348 (1982)
Tan, L., Wan, A., Li, H.: \(\text{ Ag }_2\)S quantum dots conjugated chitosan nanospheres toward light-triggered nitric oxide release and near-infrared fluorescence imaging. Langmuir 29(48), 15032–15042 (2013)
Tang, R., Xue, J., Xu, B., Shen, D., Sudlow, G.P., Achilefu, S.: Tunable ultrasmall visible-to-extended near-infrared emitting silver sulfide quantum dots for integrin-targeted cancer imaging. ACS Nano 9(1), 220–230 (2015)
Tyrakowski, ChM, Snee, P.T.: A primer on the synthesis, water-solubilization, and functionalization of quantum dots, their use as biological sensing agents, and present status. Phys. Chem. Chem. Phys. 16, 837–855 (2014)
Wageh, S., Higazy, A.A., Al-Ghamdi, A.A., Hassouna, A.S.: Effect of environment on the preparation of CdSe quantum dots capped with mercaptoacetic acid. J. Nanosci. Nanotechnol. 14, 6442–6451 (2014)
Wang, Y., Tang, Z., Correa-Duarte, M.A., Pastoriza-Santos, I., Giersig, M., KotovLuis, N.A., Liz-Marzán, M.: Mechanism of strong luminescence photoactivation of citrate-stabilized water-soluble nanoparticles with CdSe cores. J. Phys. Chem. B 108(40), 15461–15469 (2004)
Wang, C., Wang, Y., Xu, L., Zhang, D., Liu, M., Li, X., Sun, H., Lin, Q., Yang, B.: Facile aqueous-phase synthesis of biocompatible and fluorescent \(\text{ Ag }_2\)S nanoclusters for bioimaging: tunable photoluminescence from red to near infrared. Small 8(20), 3137–3142 (2012)
Wu, Q., Zhou, M., Shi, J., Li, Q., Yang, M., Zhang, Z.: Synthesis of water-soluble \(\text{ Ag }_2\)S quantum dots with fluorescence in the second near-infrared window for turn-on detection of Zn(II) and Cd(II). Anal. Chem. 89(12), 6616–6623 (2017)
Xiang, J., Cao, H., Wu, Q., Zhang, S., Zhang, X., Watt, A.A.R.: L-cysteine-assisted synthesis and optical properties of \(\text{ Ag }_2\)S nanospheres. J. Phys. Chem. C 112, 3580–3584 (2008)
Yang, H.Y., Zhao, Y.W., Zhang, Z.Y., Xiong, H.M., Yu, S.N.: One-pot synthesis of water-dispersible \(\text{ Ag }_2\)S quantum dots with bright fluorescent emission in the second near-infrared window. Nanotechnology 24(5), 055706(1-10) (2013)
Yang, Y., Zheng, Y., Cao, W., Titov, A., Hyvonen, J., Manders, J.R., Xue, J., Holloway, P.H., Qian, L.: High-efficiency light-emitting devices based on quantum dots with tailored nanostructures. Nat. Photon. 9, 259–266 (2015)
Ye, O., Rayevska, G., Ya.Grodzyuk, V.M. Dzhagan, Stroyuk, O.L., Kuchmiy, S. Ya., Plyusnin, V.F., Grivin, V.P, Valakh, M. Ya.: Synthesis and characterization of white-emitting CdS quantum dots stabilized with polyethylenimine. J. Phys. Chem. C 114, 22478–22486 (2010)
Zhang, Y., Clapp, A.: Overview of stabilizing ligands for biocompatible quantum dot nanocrystals. Sensors 11, 11036–11055 (2011)
Zhang, Y., Liu, Y., Li, C., Chen, X., Wang, Q.: Controlled synthesis of \(\text{ Ag }_2\)S quantum dots and experimental determination of the exciton bohr radius. J. Phys. Chem. C 118, 4918–4923 (2014)
Zhang, J., Crisp, R.W., Gao, J., Kroupa, D.M., Beard, M.C., Luther, J.M.: Synthetic conditions for high-accuracy size control of PbS quantum dots. J. Phys. Chem. Lett. 6(10), 1830–1833 (2015)
Zhang, Y., Xia, J., Li, C., Zhou, G., Yang, W., Wang, D., Zheng, H., Du, Yu., Li, X., Li, Q.: Near-infrared-emitting colloidal Ag2S quantum dots excited by an 808 nm diode laser. J. Mater. Sci. 52(16), 9424–9429 (2017)
Acknowledgements
Results of TEM investigations with the Libra 120 TEM were obtained on the equipment of the Center of collective usage of scientific equipment of Voronezh State University. High-resolution TEM images were obtained on the equipment of the Center of collective usage of Moscow Institute of Physics and Technology. This study was supported by the Russian Science Foundation under Project No. 19-12-00266.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Fundamentals of Laser Assisted Micro- & Nanotechnologies.
Guest edited by Tigran Vartanyan, Vadim Veiko, Andrey Belikov and Eugene Avrutin.
Rights and permissions
About this article

Cite this article
Ovchinnikov, O.V., Grevtseva, I.G., Smirnov, M.S. et al. Effect of thioglycolic acid molecules on luminescence properties of \(\hbox {Ag}_2\)S quantum dots. Opt Quant Electron 52, 198 (2020). https://doi.org/10.1007/s11082-020-02314-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-020-02314-8