
Abstract
Chronic alcohol use disorder, a major risk factor for the development of neuropsychiatric disorders including addiction to other substances, is associated with several neuropathology including perturbed neuronal and glial activities in the brain. It affects carbon metabolism in specific brain regions, and perturbs neuro-metabolite homeostasis in neuronal and glial cells. Alcohol induced changes in the brain neurochemical profile accompany the negative emotional state associated with dysregulated reward and sensitized stress response to withdrawal. However, the underlying alterations in neuro-astroglial activities and neurochemical dysregulations in brain regions after chronic alcohol use are poorly understood. This study evaluates the impact of chronic ethanol use on the regional neuro-astroglial metabolic activity using 1H-[13C]-NMR spectroscopy in conjunction with infusion of [1,6-13C2]glucose and sodium [2-13C]acetate, respectively, after 48 h of abstinence. Besides establishing detailed 13C labeling of neuro-metabolites in a brain region-specific manner, our results show chronic ethanol induced-cognitive deficits along with a reduction in total glucose oxidation rates in the hippocampus and striatum. Furthermore, using [2-13C]acetate infusion, we showed an alcohol-induced increase in astroglial metabolic activity in the hippocampus and prefrontal cortex. Interestingly, increased astroglia activity in the hippocampus and prefrontal cortex was associated with a differential expression of monocarboxylic acid transporters that are regulating acetate uptake and metabolism in the brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol use is a major concern with worldwide prevalence, contributes substantially to the global burden of diseases [1], and accounts for 3.3 million deaths every year worldwide (WHO report 2018). The transition from occasional alcohol use to dependence is accompanied by chronic perturbations in reward processing, and involves several reiterative amplifying three-stage cycles of drug abuse that include intoxication, withdrawal, and preoccupation [2,3,4]. Initial recreational alcohol use is usually rewarding, facilitates incentive salience, and a positive hedonic emotional state. Chronic alcohol-abuse, like other psychoactive substances, involves maladaptive plasticity at the cellular/neuronal circuit level that drives pathological risky behaviors of compulsive drug seeking, and produces broad cognitive, psychological and neural deficits. At later stages, alcohol-induced neuroadaptations within a neuronal circuit and between circuitries to balance reward homeostasis reset the threshold for the reward at a higher level. Besides perturbing reward circuitry, long-term alcohol use facilitates cognitive deficits, impairs memory, and perturbs neurochemical profile [5,6,7,8]. Subsequently, abstinence from alcohol drives a negative emotional state with activation of stress response that in turn drives withdrawal syndrome and relapse [9, 10]. Several preclinical and clinical studies have been performed to understand the multifactorial adverse effects of ethanol on cerebral function. Using an intravenous administration of [2-13C]ethanol in rats it was shown that ethanol is metabolized directly in neurons and astroglia [11]. Moreover, the ethanol oxidation in the astroglia was increased further in rats subjected to chronic ethanol vapor. However, the underlying molecular and neural mechanisms remain elusive. Alcohol is known to interact with the receptors of major neurotransmitter classes viz., amino acids (glutamic acid, GABA, aspartic acid and glycine), peptides (vasopressin, somatostatin and neurotensin), and monoamines (norepinephrine, dopamine, serotonin and acetylcholine), thereby perturbs homeostasis in the brain [12,13,14]. Several studies using ex vivo as well as in vivo Magnetic Resonance Spectroscopy (MRS) have been carried out to understand the impact of alcohol abuse on neuro-metabolites homeostasis in brain [15,16,17] after alcohol use.
13C NMR spectroscopy has demonstrated one to one relationship between the rate of neuronal glucose oxidation and neurotransmitter cycling in the brain [18, 19]. These studies not only established that glucose oxidation in neuronal mitochondrion supports neurotransmitter energetics but are also in agreement with MRS studies in human subjects [20, 21]. Although these studies have been instrumental in advancing the understanding of biochemical aspects of alcoholism, the complete understanding of finer details for impacts of chronic alcohol use on neurochemical and neuro-energetics in the brain are still elusive, thereby limiting the success of therapeutic interventions for relapse. In previous study, we have shown that acute ethanol exposure perturbs the level of neurometabolites, and decreases the neurometabolic activity differentially across brain [17]. The study of neurometabolism under chronic ethanol exposure conditions would provide a better understanding of alcohol induced changes in the brain. In this study, we have investigated the impacts of chronic alcohol (30 days) exposure on neurometabolic and transcriptional changes in several brain regions using 1H-[13C]-NMR spectroscopy along with an infusion of [1,6-13C2]glucose and sodium [2-13C]acetate. Our findings suggest alcohol induced regional dysregulations of neuronal and astroglial metabolic activities in the brain. Additionally, there is a differential effect of chronic alcohol on the expression of monocarboxylic acid transporters (MCTs) in the hippocampus and prefrontal cortex at transcriptional level.
Materials and Methods
Animals
All experiments were performed in accordance with standard protocols and procedures approved by the Institutional Animal Ethics Committee (IAEC). C57BL/6NCrl male mice aged 8–10 weeks, weighing 22–25 g were used for the in vivo study. All mice were maintained at 12/12 h light/dark cycle, temperature: 23–25 °C, and relative humidity: 55–65%, and received chow diet and water ad libitum. Mice were divided into two groups: Alcohol (n = 25) and Control (n = 25), and further segregated for different measurements.
Alcohol Administration in Animals
An intraperitoneal alcohol administration mode was used in the study. Mice were administered a 20% ethanol (2.5 g/kg, intraperitoneal) solution prepared in normal saline for 30 days (Fig. 1A). The site of the injection was changed alternately to right and left of the abdominal area. Alcohol injections were given alternately in morning and evening sessions in order to minimize the time preference. The mice in the control group received the respective volume of normal saline for the entire duration.

Morris water maze analysis of memory after one month of alcohol administration: A Timeline of the experiment; B Time spent in the target zone; C Number of entries to the Quadrant containing platform; D Latency to escape platform; E Distance covered during probe trial. Control (n = 14) and Alcohol (n = 15) administered mice were allowed to swim and explore a hidden platform for 90 s. Symbol represents measurement from the individual animal. Values are presented as mean ± SD
Morris Water Maze (MWM) Test
All the animals were subjected for cognitive evaluation using Morris Water Maze test [22]. Briefly, a circular pool was filled with water (60–75 cm depth), and a submerged platform was used to evaluate spatial memory in animals [23]. Distinct cues on the sides of the tank and walls of the room were provided to locate the hidden platform. Animals were trained in multiple sessions each day for four days to locate the invisible submerged platform. This was followed by a memory test after a gap of 2 days. On the day of the test, animals were allowed to find the hidden platform (spatial acquisition memory), and escape latency to reach the hidden platform was recorded using Ethovision tracking software. The next day, in the probe trial test, the hidden platform was removed, and the total time spent by the animal in the platform zone was recorded. It should be noted that training/learning and memory were evaluated using the MWM test 24 h post alcohol administration to avoid any direct pharmacological impact of alcohol on learning.
Gene Expression Studies
One set of mice were used for gene expression studies. In brief, mice were euthanized after 48 h of alcohol withdrawal, and brain tissues were micro-dissected, and harvested for the extraction of RNA [24, 25]. The cDNA was synthesized using Superscript III cDNA reverse transcriptase, and the relative abundance of mRNA for the targeted gene was quantified by SYBR® Premix Ex Taq (Takara, New Delhi, India) based ΔΔCt method of real time PCR (RT-PCR) using gene-specific primers (Table S3) [22]. The microarray study (unpublished data) revealed that the expression of hypoxanthine guanine phosphoribosyl transferase (HGPT) mRNA was unchanged in alcohol-treated mice. Hence HGPT was used as an internal reference for the normalization of data.
Neurometabolic Measurement
Another set of mice were subjected to neuronal or astroglial metabolic analysis using a tracer approach of 13C labeled glucose or acetate administration [26,27,28,29]. Briefly, mice were fasted for 6 h, and were administered either [1,6-13C2]glucose or sodium [2-13C]acetate for 2 min using a bolus-variable rate infusion schedule [30]. For glucose, [1,6-13C2]glucose (Cambridge Isotope Laboratories, Andover, MA, USA) dissolved in water (0.225 mol/L) was administered with an initial bolus rate of 1575 mol/kg/min for the first 20 s. The infusion rate was stepped down exponentially every 20 s to attain the final rate of 247 mol/kg/min at 100 s, and was continued till 120 s. In the case of acetate, a bolus of sodium [2-13C]acetate was administered with an initial rate of 10 mmol/kg/min during the first 15 s, and the rate was stepped down exponentially in four steps to 0.50 mmol/kg/min by 75 s, which continued till 120 s. The blood was collected for the separation of plasma within a minute before arresting the brain metabolism using a focused Beam Microwave Irradiation at 7 and 10 min for glucose and acetate experiment, respectively (4.5 kV for 1 s). The different brain regions were dissected, and stored at − 80 °C until further processing.
Plasma samples (~ 100 μL) were mixed with phosphate buffer (450 μL) prepared in deuterium oxide containing 1 mM sodium formate, and filtered through a centrifugal filter (10 kDa cutoff) to remove macromolecules. The filtrate was analyzed by 1H NMR spectroscopy for estimation of the percent 13C labeling of glucose-C1 and acetate-C3 in blood. Metabolites from brain tissue samples were extracted as described earlier [31]. Briefly, frozen brain tissues were pulverized with a handheld motorized homogenizer in 0.1N HCl/methanol (2:1 vol/wt). The [2-13C]glycine (100 ml; 2 mmol/L) was added as an internal concentration reference. The tissue was thoroughly homogenized with ice-cold ethanol (1:6 w/v; 60% ethanol). The homogenate was centrifuged at 14,000 g for 30 min. The supernatant was lyophilized, and dissolved in phosphate buffer (25 mM, pH 7.0) containing 0.25 mM sodium 3‐trimethylsilyl [2,2,3,3‐D4]‐propionate for NMR analysis.
The 1H-[13C]-NMR spectra of brain tissue extracts were acquired at a 600 MHz NMR spectrometer (AVANCE II, Bruker Biospin, Karlsruhe, Germany) using the following parameters: repetition time = 5.5 s; echo time = 8 ms; number of points in FID = 16,384; spectral width = 7212 Hz; number of averages ranging from 64 (for cerebral cortex) to 1024 (for striatum) as described previously [32, 33]. Any loss in NMR signal intensity of a particular resonance due to rapid pulsing (TR = 5.5 s) was obtained by acquiring 1H-[13C]-NMR spectra of a sample with a repetition time of 5.5 s and 20 s. The correction factor thus obtained for a particular resonance was multiplied with the measured NMR resonance intensity to correct for a loss in signal intensity. The concentration of metabolites was calculated relative to [2-13C]glycine that was added during the extraction of metabolites, and 13C enrichment of amino acids at different carbon positions was calculated from the ratio of the areas in the difference spectrum to the non-edited spectrum.
Estimation of Neuronal and Astroglial Metabolic Flux
The metabolism of [1,6-13C2]glucose via glycolysis followed by TCA cycle labels GluC4 and GABAC2 in glutamatergic and GABAergic neurons, respectively [26, 29, 34]. The labeling of GlnC4 occurs by the exchange of neurotransmitters, GABA and glutamate, into astroglia via GABA-glutamine and glutamate-glutamine neurotransmitter cycling pathways. Further metabolism of GluC4 and GABAC2 in the TCA cycles label AspC2 and AspC3 equally. AspC2/C3 on further condensation with acetyl-CoA, and metabolism in glutamatergic neurons labels GluC2 and GluC3. Similarly, GABAC3 and GABAC4 are equally labeled from AspC2/C3 in GABAergic neurons. The rate of total (neurons plus astroglia) glucose oxidation was approximated from the initial rate of 13C label accumulation into amino acids according to [28, 34]:
where fGlc is the fractional enrichment of [1,6-13C2]glucose, and AspCi, GlnC4, and GluCi are the levels of 13C labeled amino acids at 'ith' carbon in 7 min. Factor 2 in the expression is because of equal labeling of Asp at carbon 2 and 3, Glu at carbon 2 and 3, and GABA at carbon 3 and 4. The level of blood glucose was assumed to be similar during the 7 min of the study.
The rate of glucose oxidation in glutamatergic neurons (CMRGlc(Glu)) was estimated as follows:
where AspCi and GluCi are the levels of 13C labeled amino acids at 'ith' carbon in 7 min. Factor 0.82 represents the fractional pool of glutamate in the glutamatergic neurons in the cerebral cortex [29]. The neuronal fraction of aspartate (0.84) was distributed equally (0.42) into glutamatergic and GABAergic neurons.
The rate of glucose oxidation in GABAergic neurons (CMRGlc(GABA)) was determined using the following expression:
where GABACi represents the concentration of labeled GABA at 'ith' carbon from [1,6-13C2]glucose. Factor 0.02 is the fractional pool of glutamate in the GABAergic neurons.
Unlike glucose, [2-13C]acetate is transported and preferentially metabolized in astroglia [35,36,37]. Metabolism of [2-13C]acetate in astroglial TCA cycle transfers 13C label to the small pool of glutamate-C4 (GluC4) followed by glutamine-C4 (GlnC4) by astroglial specific enzyme glutamine synthetase [29, 36, 38]. The labeling of GABAC2 and GluC4 in neurons occurs by glutamine-GABA and glutamine-glutamate pathways. The cerebral metabolic rate of acetate oxidation (CMRAce(Ox)) was calculated based on the 13C label trapped into different amino acids from [2-13C]acetate using the following expression [28]:
where fAce is the fractional enrichment of [2-13C]acetate, and AspCi, GABACi, GlnCi and GluCi are the concentrations of 13C labeled amino acids at 'ith' carbon from [2-13C]acetate in 10 min.
Statistics
GraphPad Prism and Microsoft Excel were used for statistical analysis. Two-tailed Student’s t test with confidence intervals of 95% was performed for assessing the significance of the difference between the ethanol and control groups. Outliers in data were determined using Grubbs’ test to remove the extreme values. Benjamin Hochberg method was used to adjust p value for multiple comparisons, and an adjusted p value of < 0.05 was considered for statistical significance between groups. For gene expression analysis, two-way ANOVA was used to estimate the statistical significance between the groups. Unless specified, results are expressed as Mean ± SD.
Results
Chronic Alcohol Results in Impaired Memory in Mice
Animals were administered ethanol (2.5 g/kg, intraperitoneal) daily for 30 days. The training and memory test were conducted using MWM during the last week of the ethanol treatment (Fig. 1A). Alcohol-exposed animals (22.1 ± 2.8 s) spent significantly (t(27) = 2.65, p = 0.013) less amount of time in the virtual target as compared to the saline-treated controls (27.2 ± 6.9 s) (Fig. 1B). The memory retention specific to the exact platform (Fig. S1A) location containing zone was also significantly (t(27) = 3.80, p = 0.0007) reduced in the alcohol-treated mice (1.2 ± 0.5 s) as compared to the normal saline-treated controls (2.6 ± 1.3 s). Furthermore, the number of entries to the platform containing quadrant (12.7 ± 3.3 vs 17.5 ± 5.2, t(27) = 2.98, p = 0.006) and zone (2.6 ± 1.12 vs 4.5 ± 2.0, t(27) = 3.20, p = 0.003) were significantly reduced in ethanol treated mice (Fig. 1C, Fig. 1SB). In addition, latency to platform was increased (t(27) = 2.04, p = 0.052) in alcohol treated mice (29.5 ± 23.7 s) when compared with controls (15.8 ± 8.1 s) (Fig. 1D). It is noteworthy that the deficits in performance in MWM were not due to reduced locomotor activity as there was no significant difference (t(27) = 0.14, p = 0.89) in total distance covered during the test between alcohol (2331 ± 367 cm) and control mice (2316 ± 187 cm) (Fig. 1E). These data suggest chronic alcohol exposure induces cognitive deficits in the spatial retention memory (Fig. 1, Fig. S1). Overall, Morris water maze behavioral results suggest memory deficits in alcohol-administered mice.
Chronic Alcohol Decreased Glucose Metabolism in Brain
The level of β-hydroxybutyrate (BHB) in the plasma was measured in 1H NMR-spectrum. The CH3 resonance of BHB could be seen at 1.21 ppm (Fig. S2). There was no significant difference (p = 0.89) in the BHB level in alcohol (0.08 ± 0.05 mmol/L, n = 7) treated mice when compared with controls (0.08 ± 0.04 mmol/L, n = 7) (Table S1), suggesting that ketogenesis is unpertubed following chronic ethanol treatment.
For the assessment of glucose metabolism, alcohol treated mice were administered [1,6-13C2]glucose, and the concentration of 13C labeled amino acids in brain tissue extracts were measured using 1H-[13C]-NMR spectroscopy (Fig. 2A). Although, there was a trend for a reduction in the levels of NAA (Ethanol: 6.2 ± 0.5 μmol/g; Control: 6.6 ± 0.3 μmol/g) in the striatum, aspartate (2.6 ± 0.2 vs 2.4 ± 0.1 μmol/g) in the hippocampus, and myo-inositol (6.5 ± 0.4 vs 6.2 ± 0.2 μmol/g) in the cerebral cortex, none of these changes survive for the statistical correction for the multiple comparisons (Table S2). These data are suggestive of no specific effects of chronic alcohol use on the neuro-metabolites homeostasis in brain.

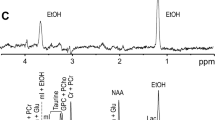
Concentration of 13C labeled amino acids from [1,6-13C2]glucose in Control and ethanol administered mice: A 1H-[13C]-NMR spectrum from a hippocampal extract of alcohol-treated mouse showing 13C labeled neurometabolites from [1,6-13C2]glucose. The top spectrum depicts the total level of metabolites, and spectrum in the lower panel shows 13C labeled metabolites. The concentration of 13C labeled amino acids from [1,6-13C2]glucose in B Hippocampus, and C Prefrontal cortex. Mice were infused with [1,6-13C2]glucose for 2 min, euthanized at 7 min, and 1H-[13C]-NMR spectra were recorded from the hippocampal tissue extracts. The 13C concentrations of amino acids were measured in 1H-[13C]-NMR spectra of brain tissue using [2-13C]glycine as the concentration standard. Values are presented as mean ± SD, and the symbol represents measurement from the individual mouse
There was no significant change (p = 0.964) in the percent 13C labeling of blood [1-13C]glucose in ethanol-treated mice (22.8 ± 2.7%, n = 6) when compared with saline-treated controls (22.0 ± 1.8% n = 6) suggesting that plasma glucose homeostasis is not perturbed in ethanol-treated mice. The concentration of 13C labeled metabolites was measured in edited 1H-[13C]-NMR spectrum (lower panel, Fig. 2A). The well-resolved signals of AlaC3, AspC3, GluC4/C3, GlnC4, GABAC2 and LacC3, which are labeled from [1,6-13C2]glucose are seen in the hippocampus of alcohol treated mice. The percent 13C labeling of LacC3 was found to be similar in ethanol (25.2 ± 3.1%, n = 6) and normal-saline treated mice (25.1 ± 3.7%, n = 6). There was a small but significant (padj < 0.05) reduction in the 13C labeling of hippocampal GluC4 (Ethanol: 1.90 ± 0.14 μmol/g, Control: 2.09 ± 0.11 μmol/g, t(10) = 2.74, padj = 0.031), GluC3 (0.38 ± 0.06 vs 0.48 ± 0.02 μmol/g, t(10) = 3.55, padj = 0.016), GABAC2 (0.32 ± 0.02 vs 0.36 ± 0.02 μmol/g, t(10) = 4.52, padj = 0.006) and GlnC4 (0.31 ± 0.05 vs 0.37 ± 0.02 μmol/g, t(10) = 2.95, padj = 0.029) amino acids from [1,6-13C2]glucose in alcohol-treated mice when compared to controls (Fig. 2B). Moreover, there was a trend for decrease (padj = 0.07) in striatal GluC4 (1.95 ± 0.07 vs 2.17 ± 0.17 μmol/g, t(10) = 2.96, padj = 0.07), GlnC4 (0.29 ± 0.03 vs 0.33 ± 0.03 μmol/g, t(10) = 2.62, padj = 0.07) and GluC3 (0.36 ± 0.06 vs 0.44 ± 0.05 μmol/g, t(10) = 2.41, padj = 0.07) in the alcohol-administered mice as compared to controls (Fig. S3A). Similar to the striatum, there was a decreasing trend in 13C labeling of GluC4 (2.43 ± 0.25 vs 2.74 ± 0.20 μmol/g, t(9) = 2.3, padj = 0.15) and GluC3 (0.45 ± 0.07 vs 0.57 ± 0.07 μmol/g, t(9) = 2.90, padj = 0.10) in the prefrontal cortex of alcohol-treated mice (Fig. 2C). However, these trends could not survive the statistical corrections for multiple comparisons. There were no significant differences in the 13C labelling of metabolites in the cerebral cortex (Fig. S3B). The reduced 13C labeling of amino acids is suggestive of a decreased rate of glucose oxidation through the neuronal TCA cycle in the hippocampus. Additionally, the reduced 13C labeling of GlnC4 is suggestive of impaired neurotransmitter cycling in the hippocampus of alcohol-treated mice.
The rates of glucose oxidation in Glutamatergic (Ethanol: 0.34 ± 0.04 μmol/g/min, Control: 0.39 ± 0.01 μmol/g/min, t(10) = 3.18, padj = 0.01) and GABAergic neurons (0.11 ± 0.01 vs 0.13 ± 0.008 μmol/g/min, t(10) = 2.77, padj = 0.02) were reduced significantly with an overall reduction (0.57 ± 0.06 vs 0.66 ± 0.02 μmol/g/min, t(10) = 3.23, padj = 0.01) in the rates of glucose oxidation in the hippocampus of alcohol-administered mice when compared with saline-treated controls (Fig. 3A). Similarly, there was a decrease (0.34 ± 0.02 vs 0.39 ± 0.03 μmol/g/min, t(10) = 2.78, padj = 0.04) in the rate of glucose oxidation Glutamatergic neurons along with a decrease (0.56 ± 0.04 vs 0.63 ± 0.06 μmol/g/min, t(10) = 2.57, padj = 0.04) in the total glucose oxidation in the striatum (Fig. 3B). The glucose oxidation in the GABAergic neurons was unperturbed (0.11 ± 0.01 vs 0.12 ± 0.01 μmol/g/min, t(10) = 1.51, padj = 0.16) in ethanol-treated mice (Fig. 3B). Likewise, there was a trend of reduction in the rate of glucose oxidation in the prefrontal cortex, however, it did not reach the statistical significance (0.70 ± 0.08 vs 0.80 ± 0.06 μmol/g/min, t(9) = 2.21, padj = 0.08) (Fig. 3C). There was no change in rate of glucose oxidation in the cerebral cortex (0.64 ± 0.07 vs 0.67 ± 0.05 μmol/g/min, t(9) = 0.75, padj = 0.91) (Fig. S4). These results show hypo-glucose metabolism in the hippocampus and striatum after chronic alcohol use. This attenuated neurometabolic activity might be responsible for memory deficits in ethanol administered mice.

Cerebral metabolic rates of glucose oxidation (CMRGlc(Ox)) in: A Hippocampus, B Striatum, and C Prefrontal cortex of ethanol-treated mice. The 13C labeling of amino acids from [1,6-13C2]glucose was measured in the brain tissue extracts using 1H-[13C]-NMR spectroscopy. The CMRGlc(Ox) was calculated from the concentration of 13C labeled amino acids from [1,6-13C2]glucose. The vertical bar represents the mean ± SD of the group, while the symbols depict measurement from individual mouse
Chronic Alcohol Increased Acetate Oxidation in the Brain
A representative 1H-[13C]-NMR spectrum from the hippocampus of mouse infused with sodium [2-13C]acetate is presented in Fig. 4A. The 13C labeling of GluC4, GlnC4 and GABAC2 is seen in the edited spectrum (lower panel, Fig. 4A). The concentration of GluC3 from [2-13C]acetate in the hippocampus of alcohol administered mice (0.17 ± 0.02 μmol/g) increased significantly (t(10) = 3.38, padj = 0.04) when compared with the saline-treated controls (0.12 ± 0.03 μmol/g) (Fig. 4B). Similarly, the concentration of GluC4 increased in the prefrontal cortex of alcohol-administered mice (Ethanol: 0.60 ± 0.02 μmol/g, Control: 0.55 ± 0.02 μmol/g, t(10) = 1.67, padj = 0.046)) (Fig. 4C). There was no significant (padj > 0.05) difference in the 13C labeling of amino acids in the striatum and cerebral cortex in alcohol administered mice (Fig. S5A&B). Consequently, the rate of acetate oxidation, estimated by accounting for the 13C label trapped into different amino acids, was found to be significantly increased in the hippocampus (0.17 ± 0.01 vs 0.15 ± 0.01 μmol/g/min, t(10) = 2.96, padj = 0.03) and prefrontal cortex (0.18 ± 0.005 vs 0.17 ± 0.004 μmol/g/min, t(10) = 3.69, padj = 0.02) of alcohol-treated mice (Fig. 5). As acetate is believed to be preferentially transported and metabolized in astroglia, the findings of increased rate of acetate oxidation in the hippocampus and PFC in alcohol treated mice suggests increased metabolic activity of astroglia.

Concentration of 13C labeled amino acids from [2-13C]acetate in Control and Ethanol administered mice. A 1H-[13C]-NMR spectrum of hippocampal extract of alcohol-treated mice infused with sodium [2-13C]acetate. The top spectrum depicts the total level of metabolites, while that in the lower panel exhibits 13C labeled metabolites from [2-13C]acetate. The concentration of 13C labeled amino acids in: B Hippocampus and C Prefrontal Cortex. Mice were infused with sodium [2-13C]acetate for 2 min, euthanized at 10 min, and the concentrations of 13C labeled metabolites were measured in 13C edited 1H-[13C]-NMR spectra of brain tissue extracts using glycine as the concentration standard. Values are presented as mean ± SD

Cerebral metabolic rates of acetate oxidation in alcohol-treated mice. Animals were infused with sodium [2-13C]acetate for 2 min, and cerebral metabolism was arrested at 10 min using focused microwave beam directed to head. The concentration of 13C label trapped into various neurometabolites were measured in brain tissue extracts, and were used for the estimation of the rates of acetate oxidation. Values are presented as mean ± SD, and symbols depict measurement from individual mouse
It is noteworthy that the energy equivalence of acetate (9 ATP) is low compared to that of glucose (32 ATP). As a result, although there was an increase in the acetate oxidation in the hippocampus of alcohol-treated mice, the total ATP synthesis rate is still lower in ethanol treated mice than in controls, and does not compensate the deficiency associated with neuronal glucose metabolism in alcohol treated mice (Table S4).
Chronic Alcohol Affected Gene Expression in a Brain Region-Specific Manner
To get further insights about the impact of chronic alcohol use on the gene expression changes associated with astroglia, the levels of mRNA in the hippocampus and prefrontal cortex were evaluated using RT-qPCR. There were small but significant alcohol-induced changes in the expression of several genes involved in the astrocytic metabolism of acetate (Fig. 6). The expression of Glial Fibrillary Acidic Protein (Gfap) increased significantly in both the hippocampus (1.59x, p = 0.007) and prefrontal cortex (1.85x, p < 0.0001) of alcohol-administered mice as compared to saline-treated controls. The Gfap expression is specific to astroglia, hence increased Gfap expression is indicative of either an increase in the astroglial population or increased neuroinflammation in alcohol-treated mice. Alcohol-induced changes in Gfap mRNA levels were more prominent in the prefrontal cortex. Furthermore, there was a significant increase in mRNA level of a Monocarboxylate Transporter 1 (MCT1 or Slc16a1) (1.63x, p = 0.002) and Monocarboxylate Transporter 4 (MCT4 or Slc16a4) (2.12x, p < 0.0001) in the hippocampus. The level of MCTs were unperturbed in the prefrontal cortex (p ≥ 0.05). Moreover, the expressions of MCT2 in both brain regions were unchanged (p ≥ 0.79) (Fig. 6). In addition, there was a significant (p < 0.001) increase in the expression of interleukin 6 (IL6) mRNA in the hippocampus of alcohol treated mice (1.76x) when compared with controls. There was no significant change in the expression IL6 in the prefrontal cortex. Overall, our results show the differential impact of alcohol on hippocampal and prefrontal astroglial cells. Increased levels of MCT1 and MCT4 in the hippocampus might mediate enhanced uptake and metabolism of acetate. Along similar lines, we observed an increase in the expression of another astrocyte enriched gene, Acss1 (Acyl-CoA Synthetase Short Chain Family Member 1), in the hippocampus. However, this was not statistically significant after correction for multiple comparisons.

Transcriptional changes in the hippocampus and prefrontal cortex of alcohol administered mice. The RNA was extracted from brain tissue. The expression of RNA was estimated in Control (n = 10) and Alcohol (n = 10) treated mice using qPCR. Data (mean ± SD) are presented as relative change to control. Two-way ANOVA was used to calculate significance between the groups, and the Benjamini Hochberg correction method was used to calculate the adjusted P value for multiple comparisons. Adjusted p ≤ 0.05 was considered significant
Discussion
The brain region-specific chemical and molecular profiles of neuro-metabolites during chronic alcohol use is of clinical importance to stage ethanol-associated emotional state, and also in exploring appropriate therapeutic interventions to prevent relapse. In this regard, we carried out neurometabolic analysis using 1H-[13C]-NMR spectroscopy in conjunction with an administration of 13C labeled substrates, [1,6-13C2]glucose and [2-13C]acetate, to assess the neuronal and astroglial metabolic activity in the brain after chronic alcohol administration. The finding of the study show chronic alcohol use is associated with cognitive impairments and decreased cerebral glucose metabolism in hippocampus and striatum. Furthermore, chronic alcohol use results in increased cerebral acetate oxidation in hippocampus and prefrontal cortex. These changes in neurometabolism were associated with gene expression changes in several genes.
Although there were some minor changes in the levels of NAA, aspartate and myo-inositol in the striatum, hippocampus and cortex respectively, the small differences did not sustain the adjustment of the p-value for the multiple comparisons. The findings of the current study indicate no significant perturbation in the cerebral metabolite homeostasis in hippocampus and prefrontal cortex (Table. S1). Several magnetic resonance spectroscopy (MRS) studies in alcoholism have reported changes in levels of NAA, Choline, and creatine in different brain regions like frontal, medial and temporal lobes, cerebellum and thalamus [15, 39,40,41]. On the contrary, some of the MRS studies have shown normalization of metabolite levels after abstinence from chronic alcohol use [39, 42]. Deviations from above mentioned alcohol-induced neurochemical profiles have also been reported; i.e., there were no changes in GABA levels by chronic alcohol use except in alcohol comorbid smokers [43]. Alcohol-induced increased [44] and decreased [45] levels of glutamine have been reported in anterior cingulate in alcohol-abuse patients when compared to controls. These discrepancies might be due to the differences in history of subject, dosage, duration and methods used.
Our results show a prominent change in the neuronal (Fig. 3) and astroglial (Fig. 5) metabolic activity after chronic alcohol use in a brain region-specific manner. Brain tissues are high-energy-demanding to sustain various neurophysiological processes viz neurotransmission and generation of action potentials [46]. Consequently, reduced cerebral glucose oxidation attenuates the rate of ATP production with profound consequences on brain functions such as increased susceptibility to reactive oxygen species (ROS) induced neuronal damage [47]. Alcohol use induces ROS (especially at earlier time points) that preferentially damage membrane lipids including mitochondrial membrane lipids in neurons [48]. Earlier studies using similar approaches with 13C labeled substrates (glucose and or acetate) in mice have demonstrated reduced rates of neurotransmitter cycling flux and glucose oxidation in GABAergic and Glutamatergic neurons with no changes in astroglia metabolic activity in cortical and subcortical regions of mouse brain after acute alcohol exposure [17]. Our results are indicative of perturbation of neuronal (Fig. 3) and astroglial (Fig. 5) metabolic activity following chronic alcohol use. This could result in reduced metabolic activity in Glutamatergic and GABAergic neurons after chronic alcohol usage. Reduced hippocampal metabolic activity has been associated with cognitive impairments, and mistakes in verbal memory tests in humans [49,50,51]. Besides, lower glutamatergic activity has been observed in AD mice [30, 52, 53]. Current literature suggests that reduced glutamatergic transmission might be responsible for memory loss in AD patients [54, 55]. It has been shown that neurotransmitter cycling and neuronal glucose oxidation are stoichiometrically coupled [19, 56]. Hence, the finding of reduced glucose oxidation in glutamatergic and GABAergic neurons in alcohol-treated mice is indicative of decreased glutamate-glutamine and GABA-glutamine neurotransmitter cycling in the hippocampus of alcohol-treated mice.
The findings of the study further revealed an increased rate of acetate oxidation in the hippocampus and prefrontal cortex in alcohol treated mice (Fig. 5). The increased uptake of acetate has been reported in heavy drinkers [57]. Acetate is utilized specifically by astroglia, suggesting increased astroglial metabolic activity in alcohol-addicted mice. An increase in astroglial activity suggests astrogliosis response with neuroinflammation to chronic alcohol use. The brain can directly metabolize ethanol besides utilizing acetate produced by oxidation in the liver. Using [1,2-13C2]acetate and [2-13C]ethanol as labeled precursors, it has been shown that the brain can directly metabolize ethanol, and prior experience with chronic ethanol use enhances the utilization of ethanol in the brain, more specifically increased oxidation in the astroglial compartment [11].
Our results show increased expression of Gfap (Fig. 6) in hippocampus and prefrontal cortex suggestive of increased astroglia in alcohol administered mice. Additionally, several of the genes involved in uptake of monocarboxylates in astrocytes viz., MCT1 and MCT4 were upregulated in the hippocampus. These results are indicative of astrocyte dysregulation as both MCT1 and MCT4 are reported to be expressed in astrocytes [58]. There was no change in MCT2 expression levels in the hippocampus, a monocarboxylate transporter expressed in neurons [59,60,61]. The finding of increased expression of IL6 in the hippocampus suggests the presence of neuroinflammation. Alcohol-induced neuroinflammation has been reported earlier [62]. Furthermore, recent studies have reported increased expression of GFAP and MCT1 in reactive astrocytes [63, 64], which are more detrimental to neuronal health [65, 66]. Some reports have observed an enhanced risk of PFC neuronal damage as compared to the hippocampus, in response to alcohol [67]. Another possibility of this differential expression pattern in astrocytes in the hippocampus and PFC can be the potential astrocyte-mediated indirect support of energy requirements for hippocampal neurons. Such mechanisms might protect hippocampal neurons relatively more, as compared to PFC neurons. However, further investigations are required to probe the possibility of differential expression of MCTs in providing neuroprotection.
There are some limitations to the study. In this study, animals were infused with [1,6-13C2]glucose for two min, and the brain metabolism was arrested at 7 min. It may be possible that there is a slight drop in blood glucose level during 2 to 7 min. However, one may argue that a similar variation will occur in both groups of mice. In fact, the percent enrichment of [1-13C]glucose in the blood plasma was similar in both groups of mice. Hence, the impact of any variation in glucose level during the measurement on the neurometabolic flux will be nullified. Secondly, the neurometabolic measurement using single point infusion study assumes the build-up of 13C label into amino acids is linear during the 10 min of measurement. Previous in vivo measurements in rats and mice have indicated that GluC4 and GABAC2 labeling follows a sigmoidal curve that approached a steady state in 30–50 min depending on the anesthetics used [19, 26, 29, 68,69,70]. Therefore, 13C labeling of GluC4 and GABAC2 could be approximated to be linear during the first 20 min. Additionally, the label flown from GluC4/GABAC2 into GlnC4 via neurotransmitter cycling is not accounted for CMRGlc(Glu) and CMRGlc(GABA). Moreover, there may be some loss of 13C label as carbon dioxide. Hence, the rates of glucose oxidation in glutamatergic and GABAergic neurons may be underestimated. However, one may argue that the degree of underestimate in metabolic flux will be similar in alcohol-treated and control mice. Hence, the changes in the respective metabolic fluxes would remain the same, and will not affect the conclusion of the study. A more accurate measure of the impact of chronic alcohol on cerebral metabolic fluxes associated with different pathways could be achieved by analysis of 13C turnover of amino acids from [1,6-13C2]glucose [26, 29, 34]and [2-13C]acetate [27, 36, 71].
It may be possible that chronic ethanol exposure may affect liver pathology that will release different substrates into the blood, thus compensating for the glucose metabolism in the brain. In this regard, it is noteworthy that ketone bodies play an important role in brain energy metabolism in prolonged fasting and pathological conditions [72, 73]. Moreover, the metabolism of the branched-chain amino acids such as valine, leucine, and isoleucine has been shown to be linked with glutamate homeostasis [74]. Additionally, glutamine from blood is transported to brain, and labels glutamate [75]. Hence, due to compromised liver metabolism, BHB, branched-chain amino acids, and glutamine may be used as additional sources of energy in the ethanol-treated mice. However, the plasma level of these metabolites was found to be unperturbed in the alcohol-treated mice (Table S1). Hence, it is unlikely that ketone bodies and branched-chain amino acids compensate for the energy (ATP) deficiency associated with hypoglucose metabolism observed in the alcohol-treated mice (Table S4).
The findings of the study highlight that chronic alcohol use perturbs the neural circuitry in region specific as well as neural cell type specific manner. The reduction in neuronal metabolic activity and enhanced astroglial activity is seen with memory inflexibility in mice exposed with chronic alcohol. Further, neurometabolic analysis and molecular measurements will provide better insights in addiction etiology.
Data Availability
Data will be available on request.
Abbreviations
- Ala:
-
Alanine
- AlaC3 :
-
[3-13C]alanine
- Asp:
-
Aspartate
- AspC3 :
-
[3-13C]aspartate
- Cho:
-
Choline
- Cre:
-
Creatine
- CMRAce(Ox) :
-
Cerebral metabolic rate of acetate oxidation
- CMRGlc(Ox) :
-
Cerebral metabolic rate of glucose oxidation
- GABA:
-
γ-Aminobutyric acid
- GABAC2 :
-
[2-13C]GABA
- GABAC4 :
-
[4-13C]GABA
- Gln:
-
Glutamine
- GlnC4 :
-
[4-13C]Glutamine
- Glu:
-
Glutamate
- GluC3 :
-
[3-13C]Glutamate
- GluC4 :
-
[4-13C]Glutamate
- GPC:
-
Glycerophosphocholine
- LacC3 :
-
[3-13C]Lactate
- m-Ino:
-
myo-Inositol
- NAA:
-
N-Acetylaspartate
- NMR:
-
Nuclear magnetic resonance
- Tau:
-
Taurine
- TSP:
-
3-Trimethylsilyl[2,2,3,3-D4]-propionate
References
Nutt DJ, King LA, Phillips LD (2010) Drug harms in the UK: a multicriteria decision analysis. The Lancet 376:1558–1565
Koob GF (2015) The dark side of emotion: the addiction perspective. Eur J Pharmacol 753:73–87
Koob GF (2017) The Dark Side of Addiction: The Horsley Gantt to Joseph Brady Connection. J Nerv Ment Dis 205:270–272
Wise RA, Koob GF (2014) The development and maintenance of drug addiction. Neuropsychopharmacology 39:254–262
Fama R, Le Berre AP, Hardcastle C, Sassoon SA, Pfefferbaum A, Sullivan EV, Zahr NM (2019) Neurological, nutritional and alcohol consumption factors underlie cognitive and motor deficits in chronic alcoholism. Addict Biol 24:290–302
Bruijnen CJ, Dijkstra BA, Walvoort SJ, Markus W, VanDerNagel JE, Kessels RP, De Jong CA (2019) Prevalence of cognitive impairment in patients with substance use disorder. Drug Alcohol Rev 38:435–442
Le Berre AP, Fama R, Sullivan EV (2017) Executive functions, memory, and social cognitive deficits and recovery in chronic alcoholism: a critical review to inform future research. Alcohol Clin Exp Res 41:1432–1443
White AM, Matthews DB, Best PJ (2000) Ethanol, memory, and hippocampal function: a review of recent findings. Hippocampus 10:88–93
Gilpin NW, Herman MA, Roberto M (2015) The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry 77:859–869
Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW Jr (2014) Addiction as a stress surfeit disorder. Neuropharmacology 76:370–382
Wang J, Du H, Jiang L, Ma X, de Graaf RA, Behar KL, Mason GF (2013) Oxidation of ethanol in the rat brain and effects associated with chronic ethanol exposure. Proc Natl Acad Sci U S A 110:14444–14449
Yang W, Singla R, Maheshwari O, Fontaine CJ, Gil-Mohapel J (2022) Alcohol use disorder: neurobiology and therapeutics. Biomedicines 10:1192
Chastain G (2006) Alcohol, neurotransmitter systems, and behavior. J Gen Psychol 133:329–335
Valenzuela CF (1997) Alcohol and neurotransmitter interactions. Alcohol Health Res World 21:144
Jagannathan NR, Desai NG, Raghunathan P (1996) Brain metabolite changes in alcoholism: an in vivo proton magnetic resonance spectroscopy (MRS) study. Magn Reson Imaging 14:553–557
Enculescu C, Kerr ED, Yeo KYB, Schenk G, Fortes MRS, Schulz BL (2019) Proteomics reveals profound metabolic changes in the alcohol use disorder brain. ACS Chem Neurosci 10:2364–2373
Tiwari V, Veeraiah P, Subramaniam V, Patel AB (2014) Differential effects of ethanol on regional glutamatergic and GABAergic neurotransmitter pathways in mouse brain. J Neurochem 128:628–640
Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG (2006) Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab 26:865–877
Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, Behar KL (2004) Glutamatergic neurotransmission and neuronal glucose oxidation are coupled during intense neuronal activation. J Cereb Blood Flow Metab 24:972–985
Gruetter R, Seaquist ER, Ugurbil K (2001) A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab 281:E100-112
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22:1523–1531
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858
Karisetty BC, Maitra S, Wahul AB, Musalamadugu A, Khandelwal N, Guntupalli S, Garikapati R, Jhansyrani T, Kumar A, Chakravarty S (2017) Differential effect of chronic stress on mouse hippocampal memory and affective behavior: Role of major ovarian hormones. Behav Brain Res 318:36–44
Khandelwal N, Dey SK, Chakravarty S, Kumar A (2019) miR-30 family miRNAs mediate the effect of chronic social defeat stress on hippocampal neurogenesis in mouse depression model. Front Mol Neurosci 12:188
Amalakanti S, Bhat UA, Mylavarapu MB, Khandelwal N, Sundarachary NV, Chakravarty S, Kumar A (2021) Gene expression analysis identifies cholesterol metabolism dysregulation in hippocampus of phenytoin-resistant pentylenetetrazol-kindled epileptic mice. Neuromolecular Med 23:485–490
Duarte JM, Gruetter R (2013) Glutamatergic and GABAergic energy metabolism measured in the rat brain by (13) C NMR spectroscopy at 14.1 T. J Neurochem 126:579–590
Lanz B, Xin L, Millet P, Gruetter R (2014) In vivo quantification of neuro-glial metabolism and glial glutamate concentration using 1H-[13C] MRS at 14.1T. J Neurochem 128:125–139
Mishra PK, Kumar A, Behar KL, Patel AB (2018) Subanesthetic ketamine reverses neuronal and astroglial metabolic activity deficits in a social defeat model of depression. J Neurochem 146:722–734
Tiwari V, Ambadipudi S, Patel AB (2013) Glutamatergic and GABAergic TCA cycle and neurotransmitter cycling fluxes in different regions of mouse brain. J Cereb Blood Flow Metab 33:1523–1531
Soni ND, Ramesh A, Roy D, Patel AB (2021) Brain energy metabolism in intracerebroventricularly administered streptozotocin mouse model of Alzheimer’s disease: A 1H-[13C]-NMR study. J Cereb Blood Flow Metab 41:2344–2355
Patel AB, Rothman DL, Cline GW, Behar KL (2001) Glutamine is the major precursor for GABA synthesis in rat neocortex in vivo following acute GABA-transaminase inhibition. Brain Res 919:207–220
Bagga P, Chugani AN, Varadarajan KS, Patel AB (2013) In vivo NMR studies of regional cerebral energetics in MPTP model of Parkinson’s disease: recovery of cerebral metabolism with acute levodopa treatment. J Neurochem 127:365–377
de Graaf RA, Brown PB, Mason GF, Rothman DL, Behar KL (2003) Detection of [1,6–13C2]-glucose metabolism in rat brain by in vivo 1H-[13C]-NMR spectroscopy. Magn Reson Med 49:37–46
Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL (2005) The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci U S A 102:5588–5593
Rae C, Fekete AD, Kashem MA, Nasrallah FA, Bröer S (2012) Metabolism, compartmentation, transport and production of acetate in the cortical brain tissue slice. Neurochem Res 37:2541–2553
Patel AB, de Graaf RA, Rothman DL, Behar KL, Mason GF (2010) Evaluation of cerebral acetate transport and metabolic rates in the rat brain in vivo using 1H-[13C]-NMR. J Cereb Blood Flow Metab 30:1200–1213
Waniewski RA, Martin DL (1998) Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci 18:5225–5233
Xin L, Mlynarik V, Lanz B, Frenkel H, Gruetter R (2010) 1H-[13C] NMR spectroscopy of the rat brain during infusion of [2-13C] acetate at 14.1 T. Magn Reson Med 64:334–340
Bendszus M, Weijers HG, Wiesbeck G, Warmuth-Metz M, Bartsch AJ, Engels S, Boning J, Solymosi L (2001) Sequential MR imaging and proton MR spectroscopy in patients who underwent recent detoxification for chronic alcoholism: correlation with clinical and neuropsychological data. AJNR Am J Neuroradiol 22:1926–1932
Zahr NM, Carr RA, Rohlfing T, Mayer D, Sullivan EV, Colrain IM, Pfefferbaum A (2016) Brain metabolite levels in recently sober individuals with alcohol use disorder: Relation to drinking variables and relapse. Psychiat Res Neuroim 250:42–49
Lee DW, Kim SY, Lee T, Nam YK, Ju A, Woo DC, You SJ, Han JS, Lee SH, Choi CB, Kim SS, Shin HC, Kim HY, Kim DJ, Rhim HS, Choe BY (2012) Ex vivo detection for chronic ethanol consumption-induced neurochemical changes in rats. Brain Res 1429:134–144
Durazzo TC, Gazdzinski S, Rothlind JC, Banys P, Meyerhoff DJ (2006) Brain metabolite concentrations and neurocognition during short-term recovery from alcohol dependence: preliminary evidence of the effects of concurrent chronic cigarette smoking. Alcohol Clin Exp Res 30:539–551
Mason GF, Petrakis IL, de Graaf RA, Gueorguieva R, Guidone E, Coric V, Epperson CN, Rothman DL, Krystal JH (2006) Cortical gamma-aminobutyric acid levels and the recovery from ethanol dependence: preliminary evidence of modification by cigarette smoking. Biol Psychiatry 59:85–93
Thoma R, Mullins P, Ruhl D, Monnig M, Yeo RA, Caprihan A, Bogenschutz M, Lysne P, Tonigan S, Kalyanam R, Gasparovic C (2011) Perturbation of the glutamate-glutamine system in alcohol dependence and remission. Neuropsychopharmacology 36:1359–1365
Prisciandaro JJ, Schacht JP, Prescot AP, Renshaw PF, Brown TR, Anton RF (2016) Associations between recent heavy drinking and dorsal anterior cingulate N-acetylaspartate and glutamate concentrations in non-treatment-seeking individuals with alcohol dependence. Alcohol Clin Exp Res 40:491–496
Howarth C, Gleeson P, Attwell D (2012) Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab 32:1222–1232
Angelova PR, Abramov AY (2018) Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett 592:692–702
Jung ME, Metzger DB (2010) Alcohol withdrawal and brain injuries: beyond classical mechanisms. Molecules 15:4984–5011
Gardner S, Sohrabi S, Shen K, Rainey-Smith S, Weinborn M, Bates K, Shah T, Foster J, Lenzo N, Salvado O (2016) Cerebral glucose metabolism is associated with verbal not visual performance in community-dwelling older adults. J Alzheimers Dis 10:x–x
Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ (2008) Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging 29:676–692
Mosconi L, Tsui W-H, De Santi S, Li J, Rusinek H, Convit A, Li Y, Boppana M, De Leon M (2005) Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology 64:1860–1867
Patel AB, Tiwari V, Veeraiah P, Saba K (2018) Increased astroglial activity and reduced neuronal function across brain in AβPP-PS1 mouse model of Alzheimer’s disease. J Cereb Blood Flow Metab 38:1213–1226
Tiwari V, Patel AB (2012) Impaired glutamatergic and GABAergic function at early age in APPswe-PS1dE9 mice: implications for preclinical diagnosis of Alzheimer’s disease 1. J Alzheimers Dis 20(10):3233
Schallier A, Smolders I, Van Dam D, Loyens E, De Deyn PP, Michotte A, Michotte Y, Massie A (2011) Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer’s disease. J Alzheimers Dis 24:287–300
Chen KH, Reese EA, Kim HW, Rapoport SI, Rao JS (2011) Disturbed neurotransmitter transporter expression in Alzheimer’s disease brain. J Alzheimers Dis 26:755–766
Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG (1998) Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci U S A 95:316–321
Jiang L, Gulanski BI, De Feyter HM, Weinzimer SA, Pittman B, Guidone E, Koretski J, Harman S, Petrakis IL, Krystal JH, Mason GF (2013) Increased brain uptake and oxidation of acetate in heavy drinkers. J Clin Invest 123:1605–1614
Pierre K, Pellerin L (2005) Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem 94:1–14
Pellerin L, Halestrap AP, Pierre K (2005) Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res 79:55–64
Bergersen LH, Magistretti PJ, Pellerin L (2005) Selective postsynaptic co-localization of MCT2 with AMPA receptor GluR2/3 subunits at excitatory synapses exhibiting AMPA receptor trafficking. Cereb Cortex 15:361–370
Pierre K, Magistretti PJ, Pellerin L (2002) MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J Cereb Blood Flow Metab 22:586–595
Flores-Bastias O, Karahanian E (2018) Neuroinflammation produced by heavy alcohol intake is due to loops of interactions between Toll-like 4 and TNF receptors, peroxisome proliferator-activated receptors and the central melanocortin system: a novel hypothesis and new therapeutic avenues. Neuropharmacology 128:401–407
Nam M-H, Ko HY, Lee S, Park YM, Hyeon SJ, Won W, Kim SY, Jo HH, Chung J-I, Han Y-E; Lee G-H, Ju Y, Stein TD, Kong M, Lee L, Lee SE, Oh S-J, Chun J-H, Park KD, Ryu H, Yun M, Lee CJ (2021) Visualization of reactive astrocytes in living brain of Alzheimer’s disease patient. https://doi.org/10.1101/2021.04.13.439744
Jurga AM, Paleczna M, Kadluczka J, Kuter KZ (2021) Beyond the GFAP-astrocyte protein markers in the brain. Biomolecules 11:1361
Li T, Chen X, Zhang C, Zhang Y, Yao W (2019) An update on reactive astrocytes in chronic pain. J Neuroinflammation 16:1–13
Li K, Li J, Zheng J, Qin S (2019) Reactive astrocytes in neurodegenerative diseases. Aging Dis 10:664
Fowler A-K, Thompson J, Chen L, Dagda M, Dertien J, Dossou KSS, Moaddel R, Bergeson SE, Kruman II (2014) Differential sensitivity of prefrontal cortex and hippocampus to alcohol-induced toxicity. PLoS ONE 9:e106945
de Graaf RA, Mason GF, Patel AB, Rothman DL, Behar KL (2004) Regional glucose metabolism and glutamatergic neurotransmission in rat brain in vivo. Proc Natl Acad Sci U S A 101:12700–12705
Lai M, Gruetter R, Lanz B (2017) Progress towards in vivo brain 13C-MRS in mice: Metabolic flux analysis in small tissue volumes. Anal Biochem 529:229–244
McNair LM, Mason GF, Chowdhury GM, Jiang L, Ma X, Rothman DL, Waagepetersen HS, Behar KL (2022) Rates of pyruvate carboxylase, glutamate and GABA neurotransmitter cycling, and glucose oxidation in multiple brain regions of the awake rat using a combination of [2-13C]/[1-13C]glucose infusion and 1H-[13C]NMR ex vivo. J Cereb Blood Flow Metab 42:1507–1523
Sonnay S, Poirot J, Just N, Clerc AC, Gruetter R, Rainer G, Duarte JMN (2018) Astrocytic and neuronal oxidative metabolism are coupled to the rate of glutamate-glutamine cycle in the tree shrew visual cortex. Glia 66:477–491
Jensen NJ, Wodschow HZ, Nilsson M, Rungby J (2020) Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int J Mol Sci 21(22):8767
Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr (1967) Brain metabolism during fasting. J Clin Invest 46:1589–1595
Yudkoff M, Daikhin Y, Lin ZP, Nissim I, Stern J, Pleasure D, Nissim I (1994) Interrelationships of leucine and glutamate metabolism in cultured astrocytes. J Neurochem 62:1192–1202
Bagga P, Behar KL, Mason GF, De Feyter HM, Rothman DL, Patel AB (2014) Characterization of cerebral glutamine uptake from blood in the mouse brain: implications for metabolic modeling of 13C NMR data. J Cereb Blood Flow Metab 34:1666–1672
Acknowledgements
This research was supported by the Department of Biotechnology, India [BT/PR27426/MED/122/140/2018 to AK]. UAB acknowledge Council of Scientific and Industrial Research (CSIR), India and department of Biotechnology, India for fellowship. Dr. Robin de Graff of Yale University for providing the POCE pulse sequence. Mr. Jedy Jose, CCMB, Animal House, is duly acknowledged for his constant support in maintaining the quality of animals used for the study.
Funding
This study was supported by Department of Biotechnology grant no: BT/PR27426/MED/122/140/2018.
Author information
Authors and Affiliations
Contributions
UAB, ABP and AK designed the experiments. UAB, SAK, SC performed the experiments, UAB, ABP, AK analyzed the data. UAB, ABP, AK wrote the manuscript. ABP and AK managed the resources and supervised whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of Interests
The authors have no relevant financial or non-financial interests to disclose.
Ethical Approval
All experimental procedures involving mice were approved by the Institutional Animal Ethics Committee (IAEC) of CSIR-CCMB, Hyderabad, and conducted in accordance with the guidelines established by the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forests, Government of India.
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bhat, U.A., Kumar, S.A., Chakravarty, S. et al. Differential Effects of Chronic Ethanol Use on Mouse Neuronal and Astroglial Metabolic Activity. Neurochem Res 48, 2580–2594 (2023). https://doi.org/10.1007/s11064-023-03922-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03922-y