
Abstract
Purpose
The purpose of this review is to summarize recent updates regarding immune checkpoint inhibitor therapy in GBM patients including updates in brain immunology, clinical trials, mechanisms of resistance, and biomarkers of response.
Methods
PubMed was searched to identify recent relevant articles on immune checkpoint inhibitor therapy as it pertains to GBM. Clinicaltrials.gov was also searched to identify relevant clinical trials.
Results
The reported randomized phase 2 and 3 clinical trials of immune checkpoint inhibitors (alone or in combination with standard therapy) have not demonstrated a survival benefit to date in either newly diagnosed or recurrent GBM. A small randomized surgical study of neoadjuvant and adjuvant pembrolizumab suggested an increase in PFS and OS compared to adjuvant pembrolizumab only; further studies are needed to validate this finding.
Conclusions
Despite the positive impact of immune checkpoint inhibitors in many cancers, only a small subset of GBM patients respond to these agents. Further research is needed to identify biomarkers of response and therapies to rationally combine with immune checkpoint inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immune checkpoint inhibitors (ICIs) represent a class of agents that release the inhibitory brakes of T cells, thus activating the immune system to induce anti-tumor responses [1]. The ICIs approved by the United States Food and Drug Administration (FDA) block either cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or programmed cell death 1 (PD-1) or its ligands PD-L1/PD-L2. Many more ICIs are in development including agents that block other immune checkpoints such as lymphocyte-activation gene 3 (LAG3), T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), T-cell immunoglobulin- and mucin domain-3-containing molecule 3 (TIM3), B7H3, cluster of differentiation (CD)39, CD73, adenosine A2A receptor, and CD47. Despite the positive impact of ICIs on many cancers such as melanoma, most glioblastoma (GBM) patients do not respond to ICI therapy [2, 3]. Herein, we will review ICI therapy as it pertains to GBMs including basic brain immunology, mechanisms of resistance in GBM, reported clinical trials to date, and future directions.
Updates in brain immunology
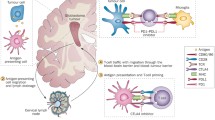
Evolutionarily, the central nervous system (CNS) have evolved to tightly regulate inflammatory and adaptive immune responses [4]. The brain is immunologically distinct compared to elsewhere in the body but is not as immunologically privileged as once thought and immune responses can indeed be elicited against antigens originating from the CNS [4,5,6]. While CNS immunobiology is still not fully elucidated, recent work has changed our understanding of CNS lymphatic drainage, CNS antigen presentation, and the role of the blood–brain barrier (BBB). Within the quiescent CNS, microglia serve as the primary immune cell as most peripheral immune cells such as naïve lymphocytes, circulating monocytes, and dendritic cells (DCs)—the classic antigen presenting cells (APCs)—are largely excluded [7]. The BBB, a highly selective semipermeable structure composed of tight junctions between endothelial cells, was felt to isolate the brain from the peripheral immune system. Evidence now shows that the meninges plays a role in CNS immune surveillance by providing a means to bypass the BBB. Although there is no obvious lymphoid tissue in the brain, there is indeed a glial-lymphatic (glymphatic) pathway including functional lymphatic vessels in the meninges that drain cerebrospinal fluid (CSF) containing solute and immune cells from the brain into deep cervical lymph nodes [4, 6]. Therefore, even in the quiescent CNS, brain-derived antigens can be carried through this glymphatic pathway where they are taken by up meningeal APCs which in turn interact with peripheral T cells.
Immune checkpoint inhibitor mechanism of action
Both CTLA-4 and PD-1 are inhibitory receptors that help modulate T cell immune responses [1]. Antigen presenting cells (APCs) ingests tumor-specific antigens and migrate to draining lymph nodes to present these antigens to naïve T cells via its T cell receptor (TCR). In order to complete T cell activation, another costimulatory interaction between B7 on APCs and CD28 on T cells is needed. This dual signaling induces T cell proliferation and cytokine release, leading to an immune response. In response to T cell activation, CTLA-4 is induced in T cells, binding B7 subtypes with greater affinity than CD28, thus leading to T cell downregulation and deactivation. Anti-CTLA-4 antibodies (e.g., ipilimumab) thus inhibit binding of B7 subtypes to CTLA-4 on T cells and amplify T cell immunity. PD-1 is expressed at later stages of T cell activation and on most activated immune cells such as macrophages, DCs, B cells, and T cells whereas PD-L1 and PD-L2 are expressed on hematopoietic and nonhematopoietic cells such as APCs and cancer cells. Engagement between PD-1 and its ligand suppresses T cell activity. Inflammatory signals in tumor tissue induce PD-L1 expression, thus allowing tumors to evade the immune system. ICIs that target PD-1 (e.g., cemiplimab, nivolumab, pembrolizumab) or PD-L1 (e.g., atezolizumab, avelumab, durvalumab) allow T cells to reactivate against tumor cells. ICI therapy, therefore, depends on a population of T cells recognizing tumor through APCs.
GBM mechanisms of resistance to immune checkpoint inhibitors
GBMs exhibit a number of resistance mechanisms to ICI therapy [7, 8]. GBMs contain low numbers of tumor infiltrating lymphocytes (TILs) relative to other tumor types thus characterizing them as immunologically “cold” tumors. Reasons for this may include relatively limited access to the CNS, poor T-cell priming due to high tumor heterogeneity, and few suitable neoantigens [8]. In patients with GBM, T cells have been found to be sequestered in the bone marrow, to be less responsive to activation when circulating, and to demonstrate upregulation of markers of exhaustion when found intratumorally [9]. The GBM microenvironment is also highly immunosuppressive driven by multiple factors including infiltrating immunosuppressive cells such as tumor-associated macrophages (TAMs). In response to brain tumors and other inflammatory stimuli, brain stromal cells produce high levels of immunosuppressive cytokines such as transforming growth factor β (TGFβ) and interleukin-10 (IL-10) [4]. In addition, GBM patients are often treated with immunosuppressive drug such as steroids and temozolomide, which may limit the effectiveness of immunotherapy. Compared to tumors that are responsive to immunotherapy, GBMs have a lower expression of PD-L1 and lower prevalence of PD-1 expressing TILs [8].
Immune checkpoint inhibitor clinical trials in GBM
To date, none of the randomized phase 2 or randomized phase 3 clinical trials of PD-1 inhibitors in GBM patients, either alone or in combination with standard of care treatments, have demonstrated a clear overall survival (OS) benefit [10,11,12,13,14] (Table 1). CheckMate 298 examined the addition of nivolumab to upfront radiation and temozolomide in newly diagnosed O6-methylguanine-DNA-methyltransferase (MGMT) methylated GBM [13]. Although final results including progression-free survival (PFS) have not been published, an independent data monitoring committee determined in 2020 that, based on the number of events to date, the study would not meet its primary endpoint of OS in patients with no baseline corticosteroid use or in the overall randomized population. CheckMate 298 compared radiation with nivolumab or temozolomide in newly diagnosed MGMT unmethylated GBM and similarly did not meet the primary endpoint of OS at final analysis; final results including PFS are also pending publication [12]. CheckMate 143 randomized recurrent GBM patients to nivolumab or bevacizumab demonstrating longer PFS with bevacizumab 3.5 months versus nivolumab 1.5 months (HR 1.97; 95% CI 1.57–2.48; P < 0.001) and no difference in OS (HR 1.04; 95% CI 0.83–1.30; P = 0.76) [10]. A phase 2, randomized study comparing pembrolizumab with or without bevacizumab in patients with recurrent GBM demonstrated a PFS benefit from combination therapy 4.1 months over pembrolizumab alone 1.43 months (P = 0.0025) but no difference in OS (P = 0.87) [11]. Finally, there was no difference in PFS or OS in a randomized study of patients with recurrent GBM who received nivolumab with standard dose bevacizumab 10 mg/kg or low dose bevacizumab 3 mg/kg every 2 weeks [14]. Preliminary results have been modest at best from single arm studies of durvalumab in patients with newly diagnosed MGMT unmethylated GBM [15], recurrent bevacizumab naïve GBM [16,17,18], or recurrent bevacizumab refractory GBM [19].
Despite these negative results, lessons can be learned. In trials of recurrent GBM comparing nivolumab versus bevacizumab (CheckMate 143) [10] or pembrolizumab with or without bevacizumab [11], no baseline corticosteroid use was associated with longer survival in the ICI group. This may be due to the negative effects of corticosteroids on T cell function. Additionally, corticosteroid use may represent a patient population with larger tumor burden or more aggressive tumors that cannot tolerate the delay to response with ICI therapy. For the few responders to nivolumab (7.8%) in CheckMate 143, the median duration of response was numerically longer compared to the responders in the bevacizumab arm (11.1 months versus 5.3 months) to suggest that a select group of GBM patients could benefit from ICI therapy [10].
Several small trials have examined the impact of neoadjuvant PD-1 inhibitor on tumor tissue [21,22,23]. Cloughesy et al. randomized recurrent GBM patients undergoing surgery to a single dose of neoadjuvant pembrolizumab versus no neoadjuvant dose prior to surgery, followed by adjuvant pembrolizumab in both arms [22]. Although originally designed to examine correlative endpoints, the study unexpectedly demonstrated an improvement in PFS (3.3 versus 2.4 months) and OS (13.7 versus 7.5 months) with neoadjuvant pembrolizumab compared to no neoadjuvant pembrolizumab. In single arm surgical window of opportunities studies of neoadjuvant PD-1 inhibitor, patients enrolled in the De Groot et al. pembrolizumab study seemed to have better survival outcomes (median OS 20 months) compared to historical controls [21] whereas no clear survival benefit was seen in recurrent GBM patients enrolled in the Schalper et al. nivolumab study (median OS 7.3 months) [23]. Larger randomized studies will be needed to validate a possible survival benefit with neoadjuvant PD-1 inhibitor.
These surgical trials provide insight into the immunological effects of PD-1 inhibitor on GBM tumor tissue. Neoadjuvant PD-1 blockade induced intratumoral inflammation as demonstrated by increases in interferon-gamma and repressed the cell-cycle-related transcriptional activity of tumor cells [22, 23]. Although de Groot et al. found no significant increase in the number of CD8+ cytotoxic T cells within pre-treated tumor tissue by cytometry by time of flight [21], Cloughesy et al. did find focal upregulation of PD-L1 and CD8+ T cell infiltrate by multiplex immunofluorescence [22]. De Groot et al. also found a marked infiltration of immunosuppressive macrophages in the tumor tissue of pre-treated patients, which may help mediate resistance to anti-PD-1 therapy [21].
Biomarkers of response
A small percentage of GBM patients experience prolonged responses to immune checkpoint blockade but it is not known what predicts response to ICI therapy in GBM. The most common biomarkers utilized across cancer to predict benefit from immunotherapy are high levels of microsatellite instability (MSI-H), mismatch repair deficiency (dMMR), and high tumor mutational burden (TMB-H). MMR deficiency is caused by mutation in one of the DNA mismatch repair genes: mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), mutS homolog 6 (MSH6), or postmeiotic segregation increased 2 (PMS2) [24, 25]. The majority of dMMR tumors and MSI-H tumors have a high TMB, but not all tumors with high TMB are dMMR or MSI-H. The FDA granted accelerated approval to pembrolizumab for adult and pediatric patients with unresectable or metastatic solid tumors (tissue agnostic) with dMMR in 2017 [26] or with high TMB (defined as ≥ 10 mutations/megabase per an FDA approved test) in 2020 [27]. These accelerated approvals were based on several trials including phase 2 KEYNOTE-158 study, which enrolled patients with advanced noncolorectal cancers classified as MSI-high by PCR or dMMR based on the immunohistochemical loss of at least one MMR protein [25]. Among the 13 patients (5.6%) enrolled with brain cancer (tumor histologies not specified), there were no responses, median PFS was 1.1 months and median OS was 5.6 months.
Most newly diagnosed GBMs have a low mutational burden [28], but there are subsets of GBM patients with high mutational burden (hypermutation) [29]. Studies suggest that hypermutation in gliomas can occur through two main pathways [29]. The first is via constitutional defects in DNA polymerase and mismatch repair genes. Responses to ICI therapy have been reported in GBM patients with germline dMMR such as childhood biallelic mismatch repair deficiency (bMMRD) [30]. bMMRD GBMs have significantly higher mutational load than sporadic gliomas and can harbor mean neoantigen loads 7 to 16 times higher than those in immunoresponsive melanomas, lung cancers, or microsatellite-unstable GI cancers [30]. The second and more common pathway is following temozolomide treatment in association with MMR defects. Hypermutation is reported in approximately 25% of temozolomide-treated gliomas and occurring more frequently in IDH mutant tumors. Retrospective analyses suggest that patients with post-temozolomide hypermutated gliomas may not benefit from PD-1 blockade [29, 31]. However, we await results from several clinical trials of PD-1 inhibitors in hypermutated gliomas for clarification; these include NCT02658279 (a pilot study of pembrolizumab in patients with recurrent malignant glioma with a hypermutator phenotype) [32], NCT03557359 (a phase 2 study of nivolumab for recurrent or progressive IDH mutant gliomas with prior exposure to alkylating agents) [33], and NCT03718767 (a phase 2 study of nivolumab in patients with IDH-mutant gliomas with and without hypermutator phenotype) [34]. A phase 2 Alliance trial is also now underway exploring the benefit of ipilimumab/nivolumab in recurrent GBM with elevated mutational burden based on recent pathology (NCT04145115) [35].
Even though dMMR, high MSI, and high TMB may help predict response to PD-1 blockade in some cancers, these biomarkers may not be generalizable across all cancer types, a reflection of fundamental differences in immune biology across different cancers [36]. For example, MMR inactivation is an early event in dMMR colorectal cancer but is a late and subclonal event in post-temozolomide gliomas [29]. In addition, hypermutation occurs prior to cancer treatment in smoking-associated non-small cell lung cancer and ultraviolet-associated melanoma as opposed to following treatment in gliomas. While the degree of microsatellite instability generally correlates with response in most cancers, GBM is one of the few tumor types where higher MSI may not predict response to ICI therapy [37]. Similarly, higher TMB is associated with longer overall survival after immunotherapy across several cancer types, except in glioma where higher TMB trended towards poorer survival [38, 39].
Zhao et al. examined the immune and genomic correlates of response by retrospectively profiled 66 GBM patients treated with PD-1 inhibitors (pembrolizumab or nivolumab), including 17 long-term responders to PD-1 inhibitors [40]. Responsive tumors demonstrated enrichment of mitogen-activated protein kinase (MAPK) pathway alterations, namely BRAF and PTPN11 mutations, whereas non-responsive tumors were significantly enriched for PTEN mutations associated with immunosuppressive gene signatures.
Future directions
Many questions with respect to ICI therapy in GBM remain unanswered. A small percentage of GBM patients may experience prolonged responses to single agent PD-1 blockade, but further investigation into predictive biomarkers is warranted.
Given the limited benefit to date with single agent PD-1 blockade, combination therapies are now being pursued in GBM patients (Table 2) [2]. Upregulation of alternate immune checkpoints may represent a possible resistance mechanism to PD-1 therapy. Therefore, rational combinations include PD-1 inhibitors with other checkpoint inhibitors; trials are now underway exploring combinations of a PD-1 inhibitor with anti-LAG3 (NCT02658981 [41]) or with anti-TIGIT (NCT04826393 [42]). Targeting the “cold” tumor microenvironment represents another strategy for overcoming resistance to ICI therapy. For example, glioma cells produce indolamine 2,3-dioxygenase (IDO), which depletes tryptophan and in turn stimulates accumulation of T regulatory cells and inhibits T cell activity [4]. A trial is now underway combining nivolumab with an IDO inhibitor in GBM [43]. Other trials combine PD-1 inhibitor with therapeutic cancer vaccines (NCT04013672 [44], NCT03018288 [45], NCT03665545 [46], NCT04201873 [47], NCT02287428 [48], NCT03750071 [49]). Vaccines may help increase the number of infiltrating tumor-specific T cells, and in turn, ICI therapy may help enhance the anti-tumor activity of the vaccines. Similarly, ICI therapy may enhance the anti-tumor activity of chimeric antigen receptor T cell (CAR-T) therapy (NCT04003649 [50], NCT03726515 [51]) and oncolytic viral therapy (NCT04479241 [52], NCT03576612 [53], NCT02798406 [54]) thus providing a rationale for combining these immunotherapeutic approaches with ICI therapy. Finally, other studies explore combinations with specific targeted therapies such as vorinostat (NCT03426891 [55]), a histone deacetylases inhibitor that may help restore tumor immune recognition and synergize with ICI therapy, and the Akt inhibitor ipatasertib (NCT03673787 [56]), given the possible role of a hyperactive PI3K/Akt pathway may play into impaired antitumor responses.
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
Bagchi S, Yuan R, Engleman EG (2021) Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol 16:223–249. https://doi.org/10.1146/annurev-pathol-042020-042741
Khasraw M, Reardon DA, Weller M, Sampson JH (2020) PD-1 inhibitors: do they have a future in the treatment of glioblastoma? Clin Cancer Res 26:5287–5296. https://doi.org/10.1158/1078-0432.Ccr-20-1135
Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, Batchelor TT, Bindra RS, Chang SM, Chiocca EA, Cloughesy TF, DeGroot JF, Galanis E, Gilbert MR, Hegi ME, Horbinski C, Huang RY, Lassman AB, Le Rhun E, Lim M, Mehta MP, Mellinghoff IK, Minniti G, Nathanson D, Platten M, Preusser M, Roth P, Sanson M, Schiff D, Short SC, Taphoorn MJB, Tonn JC, Tsang J, Verhaak RGW, von Deimling A, Wick W, Zadeh G, Reardon DA, Aldape KD, van den Bent MJ (2020) Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 22:1073–1113. https://doi.org/10.1093/neuonc/noaa106
Sampson JH, Gunn MD, Fecci PE, Ashley DM (2020) Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer 20:12–25. https://doi.org/10.1038/s41568-019-0224-7
Dunn GP, Okada H (2015) Principles of immunology and its nuances in the central nervous system. Neuro-Oncology 17:vii3–vii8. https://doi.org/10.1093/neuonc/nov175
Rustenhoven J, Kipnis J (2019) Bypassing the blood–brain barrier. Science 366:1448. https://doi.org/10.1126/science.aay0479
Jackson CM, Choi J, Lim M (2019) Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol 20:1100–1109. https://doi.org/10.1038/s41590-019-0433-y
Chuntova P, Chow F, Watchmaker PB, Galvez M, Heimberger AB, Newell EW, Diaz A, DePinho RA, Li MO, Wherry EJ, Mitchell D, Terabe M, Wainwright DA, Berzofsky JA, Herold-Mende C, Heath JR, Lim M, Margolin KA, Chiocca EA, Kasahara N, Ellingson BM, Brown CE, Chen Y, Fecci PE, Reardon DA, Dunn GP, Liau LM, Costello JF, Wick W, Cloughesy T, Timmer WC, Wen PY, Prins RM, Platten M, Okada H (2021) Unique challenges for glioblastoma immunotherapy—discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro-Oncology 23:356–375. https://doi.org/10.1093/neuonc/noaa277
Rahman M, Sawyer WG, Lindhorst S, Deleyrolle LP, Harrison JK, Karachi A, Dastmalchi F, Flores-Toro J, Mitchell DA, Lim M, Gilbert MR, Reardon DA (2020) Adult immuno-oncology: using past failures to inform the future. Neuro-Oncology 22:1249–1261. https://doi.org/10.1093/neuonc/noaa116
Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr O, Phuphanich S, Sepulveda JM, De Souza P, Sahebjam S, Carleton M, Tatsuoka K, Taitt C, Zwirtes R, Sampson J, Weller M (2020) Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol 6:1003–1010. https://doi.org/10.1001/jamaoncol.2020.1024
Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J, Nghiemphu L, Kaley T, Colman H, McCluskey C, Gaffey S, Smith TR, Cote DJ, Severgnini M, Yearley JH, Zhao Q, Blumenschein WM, Duda DG, Muzikansky A, Jain RK, Wen PY, Reardon DA (2021) Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res 27:1048–1057. https://doi.org/10.1158/1078-0432.Ccr-20-2500
Squibb BM (2019) Bristol-Myers Squibb announces phase 3 CheckMate -498 study did not meet primary endpoint of overall survival with opdivo (nivolumab) plus radiation in patients with newly diagnosed MGMT-unmethylated glioblastoma multiforme. Princeton, NJ. https://news.bms.com/news/corporatefinancial/2019/Bristol-Myers-Squibb-Announces-Phase-3-CheckMate--498-Study-Did-Not-Meet-Primary-Endpointof-Overall-Survival-with-Opdivo-nivolumab-Plus-Radiation-in-Patients-with-Newly-Diagnosed-MGMTUnmethylated-Glioblastoma-Multiforme/default.aspx
Squibb BM (2020) Bristol Myers Squibb announces update on phase 3 CheckMate -548 trial evaluating patients with newly diagnosed MGMT-methylated glioblastoma multiforme. Princeton, NJ. https://news.bms.com/news/details/2020/Bristol-Myers-Squibb-Announces-Update-on-Phase-3-CheckMate–548-Trial-Evaluating-Patients-with-Newly-Diagnosed-MGMT-Methylated-Glioblastoma-Multiforme/default.aspx
Ahluwalia MS, Rauf Y, Li H, Wen PY, Peereboom DM, Reardon DA (2021) Randomized phase 2 study of nivolumab (nivo) plus either standard or reduced dose bevacizumab (BEV) in recurrent glioblastoma (rGBM). J Clin Oncol 39:2015–2015. https://doi.org/10.1200/JCO.2021.39.15_suppl.2015
Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn G, Lim M, Cloughesy TF, Gan HK, Park AJ, Schwarzenberger P, Ricciardi T, Macri MJ, Ryan A, Venhaus RR (2019) Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J Clin Oncol 37:2032–2032. https://doi.org/10.1200/JCO.2019.37.15_suppl.2032
Reardon D, Kaley T, Dietrich J, Lim M, Dunn G, Gan H, Cloughesy T, Clarke J, Park A, Macri M, Ryan A, Ricciardi T, Reddy V, Venhaus R (2016) ATIM-04. Phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab [DUR]) in patients with glioblastoma (GBM): results for cohort B (DUR monotherapy), bevacizumab (BEV) naïve patients with recurrent GBM. Neuro-Oncology. https://doi.org/10.1093/neuonc/now212.069
Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, Cloughesy TF, Gan HK, Park AJ, Schwarzenberger P, Ricciardi T, Macri MJ, Ryan A, Venhaus RR (2017) Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J Clin Oncol 35:2042–2042. https://doi.org/10.1200/JCO.2017.35.15_suppl.2042
Reardon D, Kaley T, Dietrich J, Clarke J, Dunn G, Lim M, Cloughesy T, Gan H, Park A, Schwarzenberger P, Ricciardi T, Macri M, Ryan A, Venhaus R (2018) ATIM-38. Phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab, DURVA) + bevacizumab (BEV) in BEV-naïve patients with recurrent glioblastoma (GBM). Neuro-Oncology. https://doi.org/10.1093/neuonc/noy148.033
Reardon D, Kaley T, Dietrich J, Clarke JL, Dunn GP, Lim M, Cloughesy T, Gan HK, Park A, Schwarzenberger P, Ricciardi T, Macri M, Ryan A, Venhaus R (2017) ATIM-12. Phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab [DUR]) in patients with bevacizumab (BEV)-refractory recurrent glioblastoma (GBM). Neuro-Oncology. https://doi.org/10.1093/neuonc/nox168.108
Reardon DA, Kim T-M, Frenel J-S, Santoro A, Lopez J, Subramaniam DS, Siu LL, Rodon J, Tamura K, Saraf S, Morosky A, Stein K, Soria J-C (2016) ATIM-35. Results of the phase IB KEYNOTE-028 multi-cohort trial of pembrolizumab monotherapy in patients with recurrent PD-L1-positive glioblastoma multiforme (GBM). Neuro-Oncology 18:25–26. https://doi.org/10.1093/neuonc/now212.100
de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, Weathers SP, Loghin M, Kamiya Matsouka C, Yung WKA, Mandel J, Wu J, Yuan Y, Zhou S, Fuller GN, Huse J, Rao G, Weinberg JS, Prabhu SS, McCutcheon IE, Lang FF, Ferguson SD, Sawaya R, Colen R, Yadav SS, Blando J, Vence L, Allison J, Sharma P, Heimberger AB (2020) Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro-Oncology 22:539–549. https://doi.org/10.1093/neuonc/noz185
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, Du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O’Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY, Prins RM (2019) Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25:477–486. https://doi.org/10.1038/s41591-018-0337-7
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, Inogés S, de Andrea C, López-Diaz de Cerio A, Tejada S, Berraondo P, Villarroel-Espindola F, Choi J, Gúrpide A, Giraldez M, Goicoechea I, Gallego Perez-Larraya J, Sanmamed MF, Perez-Gracia JL, Melero I (2019) Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 25:470–476. https://doi.org/10.1038/s41591-018-0339-5
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372:2509–2520. https://doi.org/10.1056/NEJMoa1500596
Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord J-P, Geva R, Gottfried M, Penel N, Hansen AR, Piha-Paul SA, Doi T, Gao B, Chung HC, Lopez-Martin J, Bang Y-J, Frommer RS, Shah M, Ghori R, Joe AK, Pruitt SK, Diaz LA Jr (2020) Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol 38:1–10. https://doi.org/10.1200/JCO.19.02105
Marcus L, Lemery SJ, Keegan P, Pazdur R (2019) FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res 25:3753. https://doi.org/10.1158/1078-0432.CCR-18-4070
FDA approves pembrolizumab for adults and children with TMB-H solid tumors [Internet]. Silver Spring (MD): US Food and Drug Administration; 2020 [cited 2021 Aug 1]. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van ’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. https://doi.org/10.1038/nature12477
Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, Cortes-Ciriano I, Birzu C, Geduldig JE, Pelton K, Lim-Fat MJ, Pal S, Ferrer-Luna R, Ramkissoon SH, Dubois F, Bellamy C, Currimjee N, Bonardi J, Qian K, Ho P, Malinowski S, Taquet L, Jones RE, Shetty A, Chow KH, Sharaf R, Pavlick D, Albacker LA, Younan N, Baldini C, Verreault M, Giry M, Guillerm E, Ammari S, Beuvon F, Mokhtari K, Alentorn A, Dehais C, Houillier C, Laigle-Donadey F, Psimaras D, Lee EQ, Nayak L, McFaline-Figueroa JR, Carpentier A, Cornu P, Capelle L, Mathon B, Barnholtz-Sloan JS, Chakravarti A, Bi WL, Chiocca EA, Fehnel KP, Alexandrescu S, Chi SN, Haas-Kogan D, Batchelor TT, Frampton GM, Alexander BM, Huang RY, Ligon AH, Coulet F, Delattre JY, Hoang-Xuan K, Meredith DM, Santagata S, Duval A, Sanson M, Cherniack AD, Wen PY, Reardon DA, Marabelle A, Park PJ, Idbaih A, Beroukhim R, Bandopadhayay P, Bielle F, Ligon KL (2020) Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580:517–523. https://doi.org/10.1038/s41586-020-2209-9
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, Afzal S, Yalon M, Rechavi G, Magimairajan V, Walsh MF, Constantini S, Dvir R, Elhasid R, Reddy A, Osborn M, Sullivan M, Hansford J, Dodgshun A, Klauber-Demore N, Peterson L, Patel S, Lindhorst S, Atkinson J, Cohen Z, Laframboise R, Dirks P, Taylor M, Malkin D, Albrecht S, Dudley RW, Jabado N, Hawkins CE, Shlien A, Tabori U (2016) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34:2206–2211. https://doi.org/10.1200/jco.2016.66.6552
Ahmad H, Fadul CE, Schiff D, Purow B (2019) Checkpoint inhibitor failure in hypermutated and mismatch repair-mutated recurrent high-grade gliomas. Neurooncol Pract 6:424–427. https://doi.org/10.1093/nop/npz016
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT02658279, pembrolizumab (MK-3475) in patients with recurrent malignant glioma with a hypermutator phenotype [cited 2021 AUG 1]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02658279
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT03557359, nivolumab for recurrent or progressive IDH mutant gliomas [cited 2021 AUG 1]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03557359
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT03718767, nivolumab in patients with IDH-mutant gliomas with and without hypermutator phenotype [cited 2021 AUG 1]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03718767
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT04145115, a study testing the effect of immunotherapy (ipilimumab and nivolumab) in patients with recurrent glioblastoma with elevated mutational burden [cited 2021 AUG 1]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04145115
Ott M, Prins RM, Heimberger AB (2021) The immune landscape of common CNS malignancies: implications for immunotherapy. Nat Rev Clin Oncol. https://doi.org/10.1038/s41571-021-00518-9
Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, Ramaswamy AT, Durham JN, Bartlett B, Ma X, Srivastava R, Middha S, Zehir A, Hechtman JF, Morris LGT, Weinhold N, Riaz N, Le DT, Diaz LA, Chan TA (2019) Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364:485. https://doi.org/10.1126/science.aau0447
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69. https://doi.org/10.1126/science.aaa4971
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, Kaley TJ, Kendall SM, Motzer RJ, Hakimi AA, Voss MH, Russo P, Rosenberg J, Iyer G, Bochner BH, Bajorin DF, Al-Ahmadie HA, Chaft JE, Rudin CM, Riely GJ, Baxi S, Ho AL, Wong RJ, Pfister DG, Wolchok JD, Barker CA, Gutin PH, Brennan CW, Tabar V, Mellinghoff IK, DeAngelis LM, Ariyan CE, Lee N, Tap WD, Gounder MM, D’Angelo SP, Saltz L, Stadler ZK, Scher HI, Baselga J, Razavi P, Klebanoff CA, Yaeger R, Segal NH, Ku GY, DeMatteo RP, Ladanyi M, Rizvi NA, Berger MF, Riaz N, Solit DB, Chan TA, Morris LGT (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51:202–206. https://doi.org/10.1038/s41588-018-0312-8
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, Filip I, Orenbuch R, Goetz M, Yamaguchi JT, Cloney M, Horbinski C, Lukas RV, Raizer J, Rae AI, Yuan J, Canoll P, Bruce JN, Saenger YM, Sims P, Iwamoto FM, Sonabend AM, Rabadan R (2019) Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 25:462–469. https://doi.org/10.1038/s41591-019-0349-y
Lim M, Ye X, Piotrowski AF, Desai AS, Ahluwalia MS, Walbert T, Fisher JD, Desideri S, Nabors LB, Wen PY, Grossman SA (2020) Updated safety phase I trial of anti-LAG-3 alone and in combination with anti-PD-1 in patients with recurrent GBM. J Clin Oncol 38:2512–2512. https://doi.org/10.1200/JCO.2020.38.15_suppl.2512
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT04826393, ASP8374 + cemiplimab in recurrent glioma [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04826393
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT04047706, nivolumab, BMS-986205, and radiation therapy with or without temozolomide in treating patients with newly diagnosed glioblastoma [cited 2021 AUG 12]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04047706
Ahluwalia MS, Peereboom DM, Ciolfi M, Schilero C, Hobbs B, Ciesielski MJ, Fenstermaker RA (2020) Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. J Clin Oncol. https://doi.org/10.1200/JCO.2020.38.15_suppl.TPS2581
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT03018288, radiation therapy plus temozolomide and pembrolizumab with and without HSPPC-96 in newly diagnosed glioblastoma (GBM) [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03018288
Dutoit V, Marinari E, Dietrich P-Y, Migliorini D (2020) CTIM-08. Combination of the IMA950/POLY-ICLC multipeptide vaccine with pembrolizumab in relapsing glioblastoma patients. Neuro-Oncology. https://doi.org/10.1093/neuonc/noaa215.142
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT04201873, pembrolizumab and a vaccine (ATL-DC) for the treatment of surgically accessible recurrent glioblastoma [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04201873
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, Shukla SA, Hu Z, Li L, Le PM, Allesøe RL, Richman AR, Kowalczyk MS, Abdelrahman S, Geduldig JE, Charbonneau S, Pelton K, Iorgulescu JB, Elagina L, Zhang W, Olive O, McCluskey C, Olsen LR, Stevens J, Lane WJ, Salazar AM, Daley H, Wen PY, Chiocca EA, Harden M, Lennon NJ, Gabriel S, Getz G, Lander ES, Regev A, Ritz J, Neuberg D, Rodig SJ, Ligon KL, Suvà ML, Wucherpfennig KW, Hacohen N, Fritsch EF, Livak KJ, Ott PA, Wu CJ, Reardon DA (2019) Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565:234–239. https://doi.org/10.1038/s41586-018-0792-9
Wick W, Wick A, Chinot OL, Van Den Bent MJ, De Vos FYFL, Mansour M, Podola L, Lubenau H, Platten M (2020) Oral DNA vaccination targeting VEGFR2 combined with anti-PDL1 avelumab in patients with progressive glioblastoma: Safety run-in results—NCT03750071. J Clin Oncol 38:3001–3001. https://doi.org/10.1200/JCO.2020.38.15_suppl.3001
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT04003649, IL13Ralpha2-targeted chimeric antigen receptor (CAR) T cells with or without nivolumab and ipilimumab in treating patients with recurrent or refractory glioblastoma [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04003649
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT03726515, CART-EGFRvIII + pembrolizumab in GBM [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03726515
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT04479241, LUMINOS-101: PVSRIPO and pembrolizumab in patients with recurrent glioblastoma [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04479241
ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT03576612, GMCI, nivolumab, and radiation therapy in treating patients with newly diagnosed high-grade gliomas (GMCI) [cited 2021 SEP 24]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03576612
Zadeh G, Daras M, Cloughesy TF, Colman H, Kumthekar PU, Chen CC, Aiken R, Groves MD, Ong S, Ramakrishna R, Vogelbaum MA, Khagi S, Kaley T, Melear JM, Peereboom DM, Rodriguez A, Yankelevich M, Nair SG, Puduvalli VK, Nassiri F, Sonabend AM, Agensky L, Ewald B, Levisetti M, Lang FF (2020) LTBK-04. Phase 2 multicenter study of the oncolytic adenovirus DNX-2401 (tasadenoturev) in combination with pembrolizumab for recurrent glioblastoma; captive study (KEYNOTE-192). Neuro-Oncology. https://doi.org/10.1093/neuonc/noaa215.989
Sahebjam S, Forsyth P, Tran N, Mokhtari S, Arrington J, Jaglal M, Etame A, Liu J, Wicklund M, Gatewood T, Macaulay R, Robinson T, Yu M (2018) ATIM-08. A phase I trial of pembrolizumab and vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma (NCT03426891). Neuro-Oncology. https://doi.org/10.1093/neuonc/noy148.005
Tiu C, Biondo A, Welsh LC, Jones TL, Zachariou A, Prout T, Turner AJ, Daly R, Vivanco I, Yap C, Jenkins B, Crespo M, Riisnaes R, Carreira S, Gurel B, Tunariu N, Minchom A, Banerji U, de Bono JS, Lopez JS (2021) Abstract CT120: results of the glioblastoma multiforme (GBM) cohort of phase 1 trial Ice-CAP (NCT03673787): preliminary evidence of antitumour activity of Ipatasertib (Ipa) and Atezolizumab (A) in patients (pts) with PTEN loss. Cancer Res. https://doi.org/10.1158/1538-7445.AM2021-CT120
Brown CE, Aguilar B, Starr R, Yang X, Chang W-C, Weng L, Chang B, Sarkissian A, Brito A, Sanchez JF, Ostberg JR, D’Apuzzo M, Badie B, Barish ME, Forman SJ (2018) Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther 26:31–44. https://doi.org/10.1016/j.ymthe.2017.10.002
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot JM, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaa0984
Reardon DA, Brem S, Desai AS, Bagley SJ, Kurz SC, De La Fuente MI, Nagpal S, Welch MR, Hormigo A, Carroll N, Bartra SK, Campbell P, Bhatt K, Lowy I, Boyer J, Kraynyak K, Morrow MP, McMullan T, Weiner DB, Skolnik J (2020) INO-5401 and INO-9012 delivered intramuscularly (IM) with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma (GBM): Interim results. J Clin Oncol 38:2514–2514. https://doi.org/10.1200/JCO.2020.38.15_suppl.2514
Funding
None.
Author information
Authors and Affiliations
Contributions
I, as the sole author, contributed to the conception and design of this review. Material preparation, data collection and analysis were performed by myself. The first draft as well as all versions of the manuscript were written by myself. I have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Royalties from Wolters Kluwer (Up to Date); honorarium from Prime Oncology; honorarium from CONTINUUM: Lifelong Learning in Neurology; honorarium from Medlink; honorarium from Medscape.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lee, E.Q. Immune checkpoint inhibitors in GBM. J Neurooncol 155, 1–11 (2021). https://doi.org/10.1007/s11060-021-03859-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-021-03859-8