
Abstract
Increasing evidence suggests that an inflammatory microenvironment promotes invasion by glioblastoma (GBM) cells. Together with p38 mitogen-activated protein kinase (MAPK) activation being regarded as promoting inflammation, we hypothesized that elevated inflammatory cytokine secretion and p38 MAPK activity contribute to expansion of GBMs. Here we report that IL-1β, IL-6, and IL-8 levels and p38 MAPK activity are elevated in human glioblastoma specimens and that p38 MAPK inhibitors attenuate the secretion of pro-inflammatory cytokines by microglia and glioblastoma cells. RNAi knockdown and immunoprecipitation experiments suggest that the p38α MAPK isoform drives inflammation in GBM cells. Importantly, p38 MAPK inhibition strongly reduced invasion of U251 glioblastoma cells in an inflammatory microenvironment, providing evidence for a p38 MAPK-regulated link between inflammation and invasiveness in GBM pathophysiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) cells have an extraordinary capacity to invade brain tissue beyond the surgical resection margin, accounting for the inability to treat this tumour effectively. An important factor that drives the invasion of transformed cells is the inflammatory microenvironment. Chronic inflammation in combination with oncogenic mutations contributes to up-regulation of inflammatory cytokines [1, 2], which support migration and invasion in carcinogenesis [3].
Glioblastomas are highly inflammatory in nature, because they are infiltrated by microglia [4] and surrounded by a pool of pro-inflammatory cytokines including tumour necrosis factor α (TNFα) and interleukin (IL) IL-1β. These cytokines are likely to promote the expansion of GBMs by upregulating other pro-tumourigenic mediators, for example IL-6 and IL-8 [5–7]. IL-8 amplifies the inflammatory microenvironment and also has chemotactic and angiogenic properties [8, 9]. IL-6 is mitogenic for tumour cells [10–13], and both TNFα and IL-1β induce expression of matrix metalloproteases (MMPs) to increase GBM invasion [14]. Thus, blocking the production and/or activity of these pro-inflammatory cytokines could be important in controlling GBM invasiveness.
p38 mitogen-activated protein kinase (MAPK) is critically important in the development of inflammation and activates a variety of transcription factors and kinases to up-regulate TNFα and IL-1β expression [15, 16]. Furthermore, amplification of local inflammation via TNFα and IL-1β-induced IL-6 and IL-8 expression is also p38 MAPK dependent [17–19]. Although p38 MAPK is upregulated in human glioblastoma [20] and is believed to be central to inflammation accompanying neurodegeneration [21] and possibly neuro-oncology [22, 23], the therapeutic potential of p38 MAPK inhibition in GBM has been primarily investigated independently of its function in inflammation [20, 24, 25].
Here we investigated whether p38 MAPK inhibition can reduce the formation of an inflammatory GBM microenvironment and attenuate the invasion of GBM cells. By using the prototypical p38 MAPK inhibitor SB203580 and a unique blood–brain barrier (BBB)-permeable and selective p38 MAPK inhibitor, compound MW01-2-069A-SRM (069A) [26], we demonstrated that p38 MAPK inhibition reduces the secretion of pro-inflammatory cytokines from microglia and malignant cells, resulting in reduced migration and invasion of glioblastoma cells.
Materials and methods
Reagents and cell culture
U251 human glioblastoma cells (American Type Culture Collection) were cultured in RPMI-1640 (Sigma–Aldrich) supplemented with 10 % (v/v) FBS (Bovogen Biolabs), 2 mM glutamine (Sigma–Aldrich) and antibiotic–antimycotic solution (Invitrogen) at 37 °C and 5 % CO2. Primary microglia were purified from 14 to 19 weeks old aborted foetuses collected after therapeutic termination after obtaining informed consent (approved by the ethics committee of the University of New South Wales, Australia; HREC 08284; [27]). For RNAi knockdowns, 1.5 μg shRNA plasmid targeting human p38α at position 1,336–1,356 (5′-ggagaagatgcttgtattgga-3′; SABiosciences) and scrambled shRNA plasmid (5′-ggaatctcattcgatgcatac-3′) were transfected into 1 × 106 U251 cells with 4 μL lipofectamine 2000 (Invitrogen) as described elsewhere [28]. Cells were treated with lipopolysaccharide (LPS, Sigma–Aldrich), IL-1β, or TNFα (R&D Systems), SB203580, U0126, SP600125 (Sigma–Aldrich) or DMW-01-069A-SRM (069A) [26].
Western blot analysis
Immunoblotting of cell lysates was performed as described elsewhere [17, 28]. Antibodies against total p38 MAPK (p38), activated p38 MAPK (p-p38), activated extracellular signal-regulated kinase (p-Erk), activated c-AMP response element binding (p-CREB), α-tubulin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were from Cell Signaling Technology and BD Transduction Laboratories.
Cytokine expression
U251 cells (1.5 × 106) were serum-starved for 2 h and treated with TNFα and IL-1β (10 ng/mL) for 24 h. Primary microglia (1.5 × 105) were treated with LPS (100 ng/mL) for 24 h. Test inhibitors (1–20 μM) were added 60 min before stimulus was applied and cell viability was monitored by MTT assay (MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). Cytokines secretion determined by ELISA was normalized to total protein concentration of cell lysates [17, 29].
Immunoprecipitation
U251 cells stimulated with TNFα or IL-1β (10 ng/mL, 15 min) were lysed with 500 μL RIPA lysis buffer and subjected to immunoprecipitation using p38 isoform-specific antibodies (Cell Signaling Technology) and the Catch and Release Immunoprecipitation kit (Millipore). Immunoprecipitates were analysed by immunoblotting for total and phosphorylated p38 MAPK as described elsewhere [17].
Wound migration assay
Conditioned media were prepared from 1.0 × 105 primary microglia treated with LPS (100 ng/mL) ± SB203580 (10 μM) or 069A (10 μM) for 24 h. Media from untreated microglia served as negative controls. Cytokines were quantified by ELISA. Migration experiments with U251 cells (1.5 × 105) and 500 μL microglia-conditioned medium were performed as described elsewhere [30]. Images (8 h) collected on an Olympus CellR microscope with Hamamatsu Orca ER camera were analysed by use of Image J software.
Transwell migration/invasion assay
U251 cells (3 × 104) starved for 24 h were seeded into Matrigel pre-coated invasion chambers (BD Biosciences). After 3 h, SB203580 or 069A (10 μM) was added and inserts were placed into media containing 10 % FBS. Alternatively, the inserts were placed into 500 μL conditioned media from microglia treated with LPS (100 ng/mL) ± SB203580 or 069A (10 μM). Invasion experiments were performed as described elsewhere [31]. Cells were counted in five independent fields (12 h) by use of the Nikon Eclipse TS100 microscope. Identical migration experiments were performed with non-Matrigel-treated inserts (BD Biosciences).
Protein extraction from patient samples
Human GBM specimens were obtained from the Steve and Lynette Waugh Brain Tumour Bank (Centre for Minimally Invasive Neurosurgery, Australia; approved by the Ethics Committee at St Vincent Hospital, Australia; SVH H03/037). Tumour tissues were flash frozen and stored in liquid nitrogen. For protein extraction, 100–300 mg tissue was homogenized in RIPA lysis buffer and the protein concentration quantified [32]. 50 µg of protein were used for quantification of cytokines by ELISA and 30 μg was used for immunoblotting.
Data analysis
Data analysis was performed with Prism 5 (GraphPad Software). Results are mean ± SEM from at least three independent experiments performed in duplicate. Statistical analysis was performed by one-way ANOVA, followed by the Newman–Keuls multiple comparison test (GraphPad Software). P values <0.05 were regarded as statistically significant. Immunoblots were analysed using Image J software and IC50 values were calculated by non-linear regression analysis with Excel.
Results
Expression of inflammatory mediators in human glioblastoma tissues
We analysed expression of IL-1β, IL-6, IL-8, and TNFα in primary glioblastomas (GBM, n = 15) and anaplastic astrocytomas (AA, n = 5) that had not been treated before the surgery, and compared the results with those from peripheral tissue of glioblastoma resections (Ctr; n = 3). Low IL-1β levels were observed in both control and AA tissues (3.54 ± 2.4 and 14.96 ± 4.6 pg/mL, respectively; Fig. 1a). In contrast, IL-1β was approximately tenfold higher in GBMs (119.2 ± 29.5 pg/mL) than in control tissues (P < 0.01). TNFα levels in GBM tissues did not reach statistical significance, but were two to threefold higher in GBMs (10.03 ± 3.5 pg/mL; Fig. 1b) than in control and AA (3.45 ± 0.8 and 2.93 ± 0.22 pg/mL, respectively). Control and AA resections contained small amounts of IL-6 (3.29 ± 1.6 and 16.81 ± 2.4 pg/mL, respectively; Fig. 1c), whereas IL-6 was significantly (P < 0.001) increased in GBMs (181.3 ± 53.5 pg/mL). Likewise, IL-8 levels in GBMs (21,808 ± 7,831 pg/mL) were 63-fold higher than in control and AA (58.48 ± 17.2 and 345.4 ± 101.1 pg/mL, respectively; P < 0.001; Fig. 1d). Hence, IL-6 and IL-8 levels in a subset of glioblastomas seem to be upregulated compared with non-malignant and lower-grade glioma tissues. These findings correlated with substantial amounts of p38 MAPK in several tumour tissues (Fig. 1e). Specifically, levels of p38 MAPK were detected in 7 out of 11 tumours analysed (63 %); phosphorylated (activated) p38 MAPK (p-p38) was found in 6 out of 11 tumours (54 %).

Expression of pro-inflammatory cytokines and p38 MAPK in human glioblastoma specimens. a–d Proteins (50 μg) extracted from three controls (Ctr), five anaplastic astrocytomas (AA), and 15 glioblastoma (GBM) specimens were analysed by ELISA for expression of IL-1β, TNFα, IL-6, and IL-8, as indicated. Each data point is an average of two independent experiments with duplicate samples (**P < 0.01, ***P < 0.001). e Proteins (30 μg) extracted from AA and GBM specimens were analysed by immunoblotting for expression of total p38 MAPK (p38), phosphorylated p38 MAPK (p-p38), and GAPDH as control for protein loading
p38 MAPK inhibitors reduce inflammation in microglia and GBM cells
To investigate whether p38 MAPK inhibition can reduce production of inflammatory cytokines in human GBM resections, we first examined cytokine secretion in LPS-stimulated primary human microglia ± p38 MAPK inhibitors SB203580 and 069A. LPS is widely used for studying inflammation associated with CNS disorders, including brain tumours [33–35]. Non-stimulated human microglia secrete only residual amounts of TNFα (122.3 ± 59 pg/mL), IL-1β (445.9 ± 190 pg/mL), IL-6 (406.0 ± 47 pg/mL), and IL-8 (20,931 ± 5,601 pg/mL), however LPS-activation resulted in a greater than threefold increase in TNFα, IL-1β, IL-6, and IL-8 production (data not shown). Both p38 MAPK inhibitors attenuated TNFα secretion, and 50 % inhibition was observed at 3 μM SB203580 (IC50 = 2.90 ± 0.02 μM) and 069A (IC50 = 2.65 ± 0.1 μM, Table 1). Furthermore, SB203580 and 069A reduced secretion of IL-1β, IL-6, and IL-8 with IC50 values in the low-μM range (Table 1), without reducing cell viability as judged by MTT assays (1–20 μM inhibitor, data not shown).
We next analysed whether GBM cells produce IL-6 and IL-8 upon inflammatory stimulation. IL-1β stimulation of U251 cells (Fig. 2a) resulted in 19.4 ± 6 (P < 0.05) and 432.5 ± 111-fold (P < 0.001) increases of IL-6 and IL-8 secretion, respectively. TNFα stimulated IL-6 and IL-8 secretion was increased 2.8 ± 0.4 (not significant) and 133.3 ± 44-fold (P < 0.001), respectively. Because IL-1β and TNFα activate Erk, p38 MAPK, and c-Jun N-terminal kinase (JNK) pathways in glioblastoma cells (Fig. 2b; [23, 36–38]) we examined whether inhibitors of these pathways attenuate IL-1β and TNFα-induced inflammation (Table 1). IL-6 secretion in IL-1β-stimulated U251 cells was attenuated by p38 MAPK inhibitors SB203580 and 069A (IC50 = 2.90 ± 0.4 and 4.60 ± 1.0 μM, respectively). Moreover, IL-1β-induced IL-8 was inhibited by SB203580 and 069A with IC50 values of 31.4 ± 4 and 25.8 ± 9 μM, respectively. Similarly, SB203580 inhibited TNFα-induced production of IL-6 (IC50 = 7.62 ± 0.8 μM), whereas 069A was less effective in reducing TNFα-induced IL-6 (IC50 = 22.04 ± 4.7 μM). Finally, TNFα-induced IL-8 secretion was potently inhibited by p38 MAPK inhibitors (IC50 = 2.41 ± 0.2 and 5.63 ± 1.0 μM for SB203580 and 069A, respectively). Inhibition of the p38 MAPK signalling pathway was routinely confirmed by immunoblotting of cell lysates against phoshorylated p38 MAPK and its direct targets CREB/ATF-1 (Fig. 2c, d). MTT assays revealed no cytotoxic effects of 1–20 μM SB203580 and 069A (data not shown). Finally, we tested the anti-inflammatory efficacy of inhibitors of Erk and JNK pathway (U0126 and SP600125, respectively). Both inhibitors attenuated secretion of IL-6 and IL-8 on stimulation with IL-1β (IC50 = 4.89–14.03 μM, Table 1), but failed to reduce TNFα-induced IL-6 (IC50 > 50 μM); indicating the lesser importance of the ERK and JNK pathways in the TNFα-mediated inflammatory response of GBM cells. In summary, p38 MAPK signalling seems to be a crucial pathway involved in the production of multiple inflammatory mediators from microglia and GBM cells.

U251 glioblastoma cells produce inflammatory cytokines, and kinase inhibitors SB203580 and 069A inhibit p38 MAPK signalling in glioblastoma cells. a U251 glioma cells were stimulated with IL-1β and TNFα (10 ng/mL) and secretion of IL-6 and IL-8 (pg/mL) was determined by ELISA. Results are means from 3–5 independent experiments with duplicate samples (*P < 0.05; ***P < 0.001). b U251 cells were stimulated with IL-1β or TNFα (10 ng/mL) for 15 min. Cell lysates were analysed for the amount of total p38 MAPK (p38), phosphorylated p38 MAPK (p-p38), and phosphorylated Erk (p-Erk1/2). c U251 cells were pre-treated ± SB203580 or 069A (1 and 10 μM) for 60 min and stimulated with TNFα (10 ng/mL) for 15 min. Cell lysates were analysed for the amount of total p38 MAPK (p38) and phosphorylated p38 MAPK (p-p38). Western blots are representatives of multiple independent experiments. d U251 cells were pre-treated ± SB203580 (10 μM) for 60 min and stimulated with TNFα (10 ng/mL) for 60 min. Cell lysates were analysed for the amount of phosphorylated CREB and ATF-1 (p-CREB/p-ATF-1) and α-tubulin as control for protein loading
p38α is an abundant p38 MAPK isoform activated in U251 GBM cells
p38 MAPK isoforms have been characterized in microglia [21] and several cancer cells [39, 40], but not in GBM cells. Therefore, we examined expression of p38 MAPK isoforms in U251 cells (Fig. 3a). p38α is the most prominent isoform (lanes 1 and 2), with substantial amounts of p38β MAPK, and only residual levels of p38γ and p38δ MAPKs (lanes 3–8). Next, we examined which p38 MAPK isoforms could be involved in the inflammatory response. Because p38 MAPK isoform-specific phospho-antibodies (e.g. p-p38α) were not available, immunoprecipitates of p38 MAPK isoforms from U251 cells ± TNFα or IL-1β were analysed for activated p38 MAPK (p-p38 MAPK). These studies only revealed activation of the p38α isoform (Fig. 3b, lanes 4 and 5); phosphorylation of p38β, p38γ, or p38δ was not detectable (data not shown).

Expression of p38 MAPK isoforms in U251 cells. a Cell lysates from U251 cells were analysed for the amount of p38α (lanes 1 and 2), p38β (lanes 3 and 4), p38γ (lanes 5 and 6), and p38δ (lanes 7 and 8). Amounts of α-tubulin in cell lysates are shown. b U251 cells were stimulated ± TNFα or IL-1β (10 ng/mL) for 15 min. Cell lysates were immunoprecipitated with anti-p38α (p38α Ab). Whole-cell lysates (lanes 1 and 2) and immunoprecipitates (IP) (lanes 3–5) were analysed for the amount of total (total p38) and phosphorylated (p-p38) p38 MAPK. Western blots are representative of three independent experiments. c, d U251 cells were transfected with RNAi targeting p38α MAPK (lanes 4–6) and control (scrambled, lanes 1–3). 72 h after transfection, cells were stimulated ± IL-1β or TNFα (10 ng/mL) for 15 min. Cell lysates were analysed for the amount of total p38α (p38α), phosphorylated p38 MAPK (p-p38), and α-tubulin (α-tub). Western blots of three independent experiments were quantified and the ratios p38α/α-tub, p-p38/α-tub, and p-p38/p-38α are given. IL-6 secretion in U251 cells transfected ± p38α RNAi and treated ± TNFα or IL-1β (10 ng/mL) was determined by ELISA
To substantiate a potentially central function of p38α MAPK in inflammation driven by GBM cells, we determined IL-6 secretion in U251 cells upon RNAi knockdown of p38α MAPK (Fig. 3c, d). p38α MAPK expression was reduced by 50.97 ± 1.5 % (Fig. 3c, compare lanes 1–3 with 4–6) and correlated with 53.58 ± 12.6 % (lanes 5 and 6) reduction of IL-1β and TNFα-induced p38 MAPK phosphorylation. Most importantly, p38α depletion significantly reduced (46.73 ± 6 %, P < 0.01) IL-6 secretion in IL-1β-stimulated cells (Fig. 3d). p38α MAPK knockdown was associated with a slight (15.32 ± 2 %), but statistically not significant, reduction of TNFα-induced IL-6 secretion. Hence, p38α MAPK strongly contributes to IL-1β-induced secretion of IL-6 in U251 cells.
p38 MAPK inhibitors reduce U251 glioblastoma cell migration and invasion in the presence of inflammatory cytokines
To determine whether p38 MAPK could contribute to the motility of GBM cells, we assayed U251 cells ± SB203580 and 069A (10 μM) by using Transwell migration and invasion chambers and with FBS (10 %) as chemoattractant. Quantification of migrating/invading cells from multiple experiments confirmed that motility of U251 cells treated with SB203580 and 069A was not significantly reduced (Fig. 4a, b). Immunoblotting confirmed inhibition of p38 MAPK phosphorylation (data not shown).

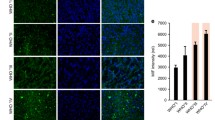
p38 MAPK inhibitors attenuate U251 glioblastoma cell migration and invasion in the presence of inflammatory cytokines. a, b Transwell migration/invasion was determined and quantified for U251 cells (see “Materials and methods” section). Serum-starved U251 cells seeded into un-coated (a) and Matrigel-precoated (b) Transwell inserts were treated with SB203580 or 069A (10 μM) and exposed to medium containing FBS (10 %) for 12 h. Migration/invasion was quantified in five independent fields. c, d Transwell migration/invasion was determined as described in the “Materials and methods” section. U251 cells were exposed to conditioned media from microglia stimulated ± lipopolysaccharide (100 ng/mL, LPS) in the presence or absence of p38 MAPK inhibitors SB203580 (10 μM, LPS + SB) or 069A (10 μM, LPS + 069A). Conditioned media (MG–CM) from untreated microglia served as control (Ctr). Images (12 h) shown in c are representative of four independent experiments. In the wound migration assay (d) U251 cells were exposed to conditioned media from microglia (±LPS, SB, 069A). Images were taken 8 h after wounding and migration was determined. Results are means from 3 or 4 independent experiments (*P < 0.05, **P < 0.01 for LPS vs Ctr, LPS + SB vs LPS and LPS + 069A vs LPS)
Next, we addressed the potential of p38 MAPK inhibitors to reduce U251 cell motility in the presence of inflammation. To mimic the inflammatory microenvironment, we prepared conditioned media (MG–CM) from LPS-stimulated human primary microglia ± p38 MAPK inhibitors. Cytokine quantification confirmed greater than threefold increased secretion of IL-1β, TNFα, IL-6, and IL-8 in LPS-stimulated microglia (data not shown), which corresponds to the increase of these cytokines in GBM tissues (three to tenfold increase, Fig. 1). Pre-treatment of LPS-activated microglia with SB203580 and 069A (10 μM) reduced TNFα (69.25 ± 15.2 and 54.76 ± 24.3 %), IL-6 (55.22 ± 28.0 and 59.11 ± 20 %), and IL-8 (65.8 ± 20.7 and 53.59 ± 33.0 %) secretion. These different MG–CMs were then used to investigate U251 cell motility in migration and invasion assays with Transwell chambers (Fig. 4d). U251 cell migration in the presence of media from LPS-activated microglia was increased by 51.48 ± 7.2 % (P < 0.05). Importantly, migration of U251 cells was reduced (63.8 ± 6 and 56.1 ± 13 %) to basal levels in the presence of SB203580 and 069A, respectively (LPS + SB, LPS + 069A, Fig. 4d, P < 0.05 for LPS vs LPS + SB and LPS vs LPS + 069A in Transwell migration assay). Similarly, the strongly increased number of GBM cells invading the Transwell chamber upon incubation with MG–CM from LPS-treated microglia indicates a more invasive phenotype of U251 cells when exposed to an inflammatory environment (Fig. 4c; for quantification see Fig. 4d). Importantly, invasion of U251 cells was significantly reduced (69.8 ± 11 and 49.1 ± 6 %; P < 0.01 for LPS vs LPS + SB and LPS vs LPS + 069A in Transwell invasion assay) when cells were exposed to MG–CM obtained from activated microglia pre-treated with SB203580 and 069A, respectively.
To provide additional evidence that p38 MAPK inhibition reduces the ability of U251 to migrate, we performed wound closure assays [31]. For U251 cells exposed to MG–CM from LPS-activated microglia (LPS) migration increased (178.0 ± 7 %, P < 0.05) compared with controls (Ctr) (Fig. 4d). When U251 cells were exposed to MG–CM from LPS-activated microglia pre-treated with p38 MAPK inhibitors (LPS + SB, LPS + 069A), migration of GBM cells was significantly (P < 0.05) reduced by 10 μM SB203580 or 069A (37.06 ± 11.5 and 39.58 ± 5.9 %, respectively). In summary, p38 MAPK inhibition is likely to reduce cytokine production from microglia cells, thereby reducing the inflammatory microenvironment and invasiveness of GBM cells.
Discussion
This study provided insight into the involvement of p38 MAPK in the inflammation and invasiveness of GBM cells. Elevated levels of IL-1β, IL-6, and IL-8 in human glioblastoma specimens suggest that inflammatory cytokines contribute to the progression of GBM. Importantly, expression and activity of p38 MAPK is also elevated in GBM tissues, and p38 MAPK inhibitors SB203580 and 069A, and p38 RNAi knockdown, strongly suppressed inflammatory responses in both the malignant and non-malignant cells that are believed to constitute glioblastomas.
Inflammatory cytokines with tumour-promoting activity [41] have been assessed in blood samples of GBM patients [42]. We found elevated TNFα and IL-1β levels in a subset of GBM tissues, supporting previous studies correlating these cytokines with glioblastoma [43, 44]. TNFα was expressed in low quantities, which is in agreement with results showing that GBM cells down-regulate production of TNFα by activated microglia [45]. However, TNFα, even at low concentrations, is a potent extracellular signal for exacerbation of the inflammatory microenvironment [46]. Along these lines, we demonstrated that TNFα and IL-1β can potentially activate the production of IL-6 and IL-8 by malignant cells. Our results showing increased IL-6 and IL-8 protein levels in GBMs support a number of studies demonstrating elevated mRNA levels encoding for these mediators [10, 11, 47]. Importantly, when examining the IL-6 and IL-8 expression analysis within the GBM samples, we observed several data points standing out an order of magnitude higher than the average value. The underlying mechanism of this observation is currently under investigation, but it is highly plausible that the increased expression of pro-inflammatory cytokines results from oncogenes such as EGFRvIII, which is known to up-regulate IL-6 and IL-8 [2, 13]. Thus, given the increased expression of inflammatory mediators and their malignancy-promoting activities, GBM therapy is likely to benefit from anti-inflammatory agents.
Our results imply p38 MAPK is important in the development of an inflammatory microenvironment that contributes to GBM invasiveness [3]. p38 MAPK drives the lysophosphatidic acid-stimulated migratory response of GBM cells [25] and invasion of GBM spheroids [20]. Although, in our work, inhibition of p38 MAPK in U251 cells resulted in minimally altered motility, significant anti-migratory and anti-invasive activity of p38 MAPK inhibitors was observed through inhibition of the inflammatory microenvironment generated by activated microglia. An attractive hypothesis that might contribute to explaining the anti-invasive efficacy of p38 MAPK inhibitors involves TNFα and IL-1β-induced activity of MMPs [14, 24, 48]. Because p38 MAPK regulates the release of MMPs from microglia and glioblastoma cells [24, 49], it is likely that p38 MAPK inhibitors attenuate GBM cell invasiveness by controlling the activity of inflammation-dependent MMPs. Further investigation is in progress to investigate the underlying mechanism.
In summary, this study provides in-vitro evidence of a link between inflammation and invasiveness in GBM pathophysiology, and initial insight supporting a function of p38 MAPK in this detrimental liaison. The invasiveness of GBM cells was efficiently attenuated by p38 MAPK inhibitors in a microglia-derived inflammatory microenvironment, emphasising possible involvement of p38 MAPK inhibitors as anti-invasive agents. As p38 MAPK inhibitors also sensitize the arrested (non-moving) GBM cells to cytotoxic therapy [20], further studies must be conducted to clarify whether targeting the activity of p38 MAPK can contribute to therapeutic benefit in GBM therapy.
References
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger P, Tan P, Sah DW, Cavenee WK, Furnari FB (2011) EGFRvIII promotes glioma angiogenesis and growth through the NFκB, interleukin-8 pathway. Oncogene. doi:10.1038/onc.2011.563
Solinas G, Marchesi F, Garlanda C, Mantovani A, Allavena P (2010) Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev 29:243–248
Hussain SF, Yang D, Suki D, Aldape K, Grimm E et al (2006) The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol 8:261–279
Yamaguchi S, Tanabe K, Takai S, Matsushima-Nishiwaki R, Adachi S et al (2009) Involvement of Rho-kinase in tumor necrosis factor-α-induced interleukin-6 release from C6 glioma cells. Neurochem Int 55:438–445
Griffin BD, Moynagh PN (2006) Persistent interleukin-1β signaling causes long term activation of NFκB in a promoter-specific manner in human glial cells. J Biol Chem 281:10316–10326
Radeff-Huang J, Seasholtz TM, Chang JW, Smith JM, Walsh CT et al (2007) Tumor necrosis factor-α-stimulated cell proliferation is mediated through sphingosine kinase-dependent Akt activation and cyclin D expression. J Biol Chem 282:863–870
Brat DJ, Bellail AC, Van Meir EG (2005) The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol 7:122–133
de la Iglesia N, Konopka G, Lim KL, Nutt CL, Bromberg JF et al (2008) Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci 28:5870–5878
Tchirkov A, Rolhion C, Bertrand S, Doré JF, Dubost JJ et al (2001) IL-6 gene amplification and expression in human glioblastomas. Br J Cancer 85:518–522
Tchirkov A, Khalil T, Chautard E, Mokhtari K, Véronèse L et al (2007) Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer 96:474–476
Weissenberger J, Loeffler S, Kappeler A, Kopf M, Lukes A et al (2004) IL-6 is required for glioma development in a mouse model. Oncogene 23:3308–3316
Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW et al (2010) Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 24:1731–1745
Sarkar S, Yong VW (2009) Inflammatory cytokine modulation of matrix metalloproteinase expression and invasiveness of glioma cells in a 3-dimensional collagen matrix. J Neurooncol 91:157–164
Schieven GL (2005) The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem 5:921–928
Kumar S, Boehm J, Lee JC (2003) p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2:717–726
Munoz L, Ramsay EE, Manetsch M, Ge Q, Peifer C et al (2010) Novel p38 MAPK inhibitor ML3403 has potent anti-inflammatory activity in airway smooth muscle. Eur J Pharmacol 635:212–218
Yoshino Y, Aoyagi M, Tamaki M, Duan L, Morimoto T et al (2006) Activation of p38 MAPK and/or JNK contributes to increased levels of VEGF secretion in human malignant glioma cells. Int J Oncol 29:981–987
Kim YJ, Hwang SY, Oh ES, Oh S, Han IO (2006) IL-1β, an immediate early protein secreted by activated microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK and NF-kappaB pathways. J Neurosci Res 84:1037–1046
Demuth T, Reavie LB, Rennert JL, Nakada M, Nakada S et al (2007) MAP-ing glioma invasion: mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol Cancer Ther 6:1212–1222
Munoz L, Ammit AJ (2010) Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58:561–568
Dziembowska M, Danilkiewicz M, Wesolowska A, Zupanska A, Chouaib S et al (2007) Cross-talk between Smad and p38 MAPK signalling in transforming growth factor β signal transduction in human glioblastoma cells. Biochem Biophys Res Commun 354:1101–1106
Tanabe K, Matsushima-Nishiwaki R, Yamaguchi S, Iida H, Dohi S et al (2010) Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J Neuroinflammation 7:16
Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H et al (2009) Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA 106:12530–12535
Malchinkhuu E, Sato K, Horiuchi Y, Mogi C, Ohwada S et al (2005) Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 24:6676–6688
Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM et al (2007) A novel p38 MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J Neuroinflammation 4:21
Guillemin GJ, Smythe G, Takikawa O, Brew BJ (2005) Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 49:15–23
Vila de Muga S, Timpson P, Cubells L, Evans R, Hayes TE et al (2009) Annexin A6 inhibits Ras signalling in breast cancer cells. Oncogene 28:363–377
Munoz L, Selig R, Yeung YT, Peifer C, Laufer S (2010) Fluorescence polarisation binding assay to develop inhibitors of inactive p38α mitogen-activated protein kinase. Anal Biochem 401:125–133
Hagel M, George EL, Kim A, Tamimi R, Opitz SL et al (2002) The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol Cell Biol 22:901–915
Bryce NS, Clark ES, Leysath LJ, Currie JD, Webb DJ et al (2005) Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol 15:1276–1285
Peterson G (1977) A simplification of the protein assay method by Lowry et al. which is more generally applicable. Anal Biochem 83:346–356
Hu W, Ralay Ranaivo H, Roy SM, Behanna HA, Wing LK et al (2007) Development of a novel therapeutic suppressor of brain proinflammatory cytokine up-regulation that attenuates synaptic dysfunction and behavioral deficits. Bioorg Med Chem Lett 17:414–418
Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS (2011) HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci 31:1081–1092
Thuringer D, Hammann A, Benikhlef N, Fourmaux E, Bouchot A et al (2011) Transactivation of the epidermal growth factor receptor by heat shock protein 90 via Toll-like receptor 4 contributes to the migration of glioblastoma cells. J Biol Chem 286:3418–3428
Paugh BS, Bryan L, Paugh SW, Wilczynska KM, Alvarez SM et al (2009) Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J Biol Chem 284:3408–3417
Kesanakurti D, Chetty C, Bhoopathi P, Lakka SS, Gorantla B et al (2011) Suppression of MMP-2 attenuates TNF-α induced NF-κB activation and leads to JNK mediated cell death in glioma. PLoS One 6:e19341
Tanabe K, Kozawa O, Iida H (2011) Midazolam suppresses interleukin-1β-induced interleukin-6 release from rat glial cells. J Neuroinflammation 8:68
Junttila MR, Ala-aho R, Jokilehto T, Peltonen J, Kallajoki M et al (2007) p38α and p38δ mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 26:5267–5279
Chen L, Mayer JA, Krisko TI, Speers CW, Wang T et al (2009) Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res 69:8853–8861
Balkwill F, Mantovani A (2010) Cancer and Inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther 87:401–406
Reynes G, Vila V, Martin M, Parada A, Fleitas T et al (2011) Circulating markers of angiogenesis, inflammation, and coagulation in patients with glioblastoma. J Neurooncol 102:35–41
Sharma V, Dixit D, Koul N, Mehta VS, Sen E (2011) Ras regulates interleukin-1β-induced HIF-1α transcriptional activity in glioblastoma. J Mol Med 89:123–136
Maruno M, Kovach J, Kelly PJ, Yanagihara T (1997) Distribution of endogeneous tumour necrosis factor α in gliomas. J Clin Pathol 50:559–562
Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE (2008) Astrocytic regulation of human monocytic/microglial activation. J Immunol 181:5425–5432
Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ et al (2003) Tumor necrosis factor α induces angiogenic factor up-regulation in malignant glioma cells. Cancer Res 63:4181–4187
Hong TM, Teng LJ, Shun CT, Peng MC, Tsai JC (2009) Induced interleukin-8 expression in gliomas by tumor-associated macrohages. J Neurooncol 93:289–301
Rao JS (2003) Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3:489–501
Park MJ, Park IC, Hur JH, Kim MS, Lee HC et al (2002) Modulation of phorbol ester-induced regulation of matrix metalloproteinases and tissue inhibitors of metalloproteinases by SB203580, a specific inhibitor of p38 mitogen-activated protein kinase. J Neurosurg 97:112–118
Acknowledgments
This study was supported by grants from the Cure for Life Foundation, Alzheimer’s Australia, and The JR and JO Wicking Foundations (managed by ANZ Trustees) to LM; the National Health and Medical Research Council of Australia (510293, 510294) and the University of Sydney (2010-02681) to TG; the Australian Research Council (DP1094232) to NB, and the National Health and Medical Research Council of Australia (1009914) to GG. The authors acknowledge scientific and technical input and support from the Australian Microscopy and Microanalysis Research Facility (AMMRF) at the University of Sydney.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Yeung, Y.T., Bryce, N.S., Adams, S. et al. p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human U251 glioblastoma cells. J Neurooncol 109, 35–44 (2012). https://doi.org/10.1007/s11060-012-0875-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-012-0875-7