
Abstract
Patients with glioblastomas, the most common primary tumors of the central nervous system, have poor prognoses because of uncontrolled tumor cell invasion and proliferation. β-Catenin plays an important role in tumor development. However, whether α-catenin expression contributes to β-catenin transactivation in glioma cells is largely unknown. We report here that α-catenin expression abrogates epidermal growth factor receptor (EGFR)-activation-induced β-catenin nuclear translocation in human glioblastoma cells, thereby attenuating β-catenin transactivation and the expression of its downstream genes CCND1 and c-myc. In addition, ectopic expression of α-catenin or depletion of β-catenin suppresses EGF-promoted glioblastoma cell migration, invasion, and proliferation. In contrast, α-catenin depletion promotes β-catenin nuclear translocation and transactivation, and tumor cell motility and growth. These findings reveal the importance of β-catenin regulation by α-catenin in cellular activities of glioblastoma cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas, the most common primary tumors that arise in the brain, include tumors derived from astrocytes, oligodendrocytes, and ependymal cells. The World Health Organization (WHO) classifies gliomas into four clinical grades on the basis of histology and prognosis [1]. Glioblastoma (GBM; WHO grade IV) is the most prevalent and malignant primary brain tumor in adults [1]. Despite the advances in surgical and clinical neuro-oncology, median survival time of patients with GBM is only approximately 1 year [2]. One of the most challenging clinical obstacles is the rapid growth and diffusion of GBM resulting from uncontrolled cell proliferation and invasion [1, 3], and the mechanisms underlying these malignant phenotypes remain to be defined.
The most frequent genetic alteration associated with GBM is amplification or mutation of the epidermal growth factor receptor (EGFR) gene, which occurs in about 50% of GBM [4]. Overexpression of EGFR promotes the migration, invasion, and proliferation of tumor cells and correlates with poor clinical prognosis [5–7]. EGFR activation regulates a wide range of cellular activities, including cell growth and differentiation, migration, metabolism, survival, and apoptosis [5, 8].
β-Catenin, a component of cell–cell adhesion structures, interacts with the cytoplasmic domain of E-cadherin and links E-cadherin to α-catenin, which in turn mediates anchorage of the E-cadherin complex to the cortical actin cytoskeleton [9]. In addition to its role in cell–cell adherens junctions, β-catenin is a key component of the Wnt/Wingless signaling pathway [10]. Activation of the Wnt pathway inhibits glycogen synthase kinase (GSK)-3β-dependent phosphorylation of β-catenin. Stabilized, hypophosphorylated β-catenin translocates to the nucleus and interacts with transcription factors of the T-cell factor/lymphoid enhancer factor-1 (TCF/LEF-1) family, leading to increased expression of genes such as c-myc and CCND1 (encoding for cyclin D1) [11–13]. Wnt/β-catenin signaling plays an important role in the development of a variety of human cancers [14, 15]. Our previous results showed that EGF stimulation results in β-catenin dissociation from α-catenin mediated by CK2 phosphorylation and increases of β-catenin nuclear translocation and transactivation, without altering β-catenin stability [16–18]. Although both E-cadherin and β-catenin have been intensively studied in terms of their roles in tumor development, α-catenin, which functions as a molecular switch that binds E-cadherin and β-catenin [19], has received less attention [20]. Loss or downregulation of α-catenin has been detected in cell lines derived from leukemia, colon and prostate cancers, and other primary human cancers [20]. The effect of α-catenin regulation on tumor development occurs at least partially through regulation of β-catenin [21, 22]. However, whether α-catenin expression contributes to β-catenin transactivation in glioma cells is largely unknown.
In this study, we demonstrate that α-catenin expression plays an instrumental role in EGF-induced β-catenin transactivation, thereby regulating GBM cell migration, invasion, and proliferation.
Results
α-Catenin inhibits EGF-induced β-catenin nuclear translocation
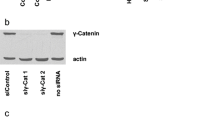
We previously showed that EGF stimulation results in nuclear translocation of β-catenin and an increase in β-catenin transactivation [16]. To examine the effect of α-catenin expression on EGF-induced regulation of β-catenin in glioma cells, we used EGF to treat EGFR-overexpressing U87/EGFR and U251/EGFR (data not shown) human GBM cells with or without expression of FLAG–α-catenin. Immunoblotting analysis of nuclear fractions of the cells showed that EGF-induced β-catenin nuclear translocation was largely inhibited by ectopic expression of α-catenin (Fig. 1a). In addition, depletion of α-catenin by expressing its short hairpin RNA (shRNA) significantly enhanced the level of nuclear β-catenin in the absence of EGF treatment, whereas the total expression levels of β-catenin were not obviously altered (Fig. 1b). Consistent with these findings, immunofluorescent studies revealed that depletion of α-catenin in U87/EGFR cells resulted in more nucleus-localized β-catenin in the absence of EGF treatment (Fig. 1c). These results indicate that α-catenin expression regulates β-catenin nuclear translocation.

α-Catenin inhibits EGF-induced β-catenin nuclear translocation. a Nuclear fractions (left panel) or total cell lysates (right panel) were prepared from U87/EGFR cells with or without FLAG–α-catenin expression. Cells were treated with EGF (100 ng/ml) for 8 h. Immunoblotting analysis was performed with the indicated antibodies. b Nuclear fractions (left panel) or total cell lysate (right panel) were prepared from U87/EGFR cells expressing a control shRNA or α-catenin shRNA. The cells were treated with EGF (100 ng/ml) for 8 h. Immunoblotting analysis was performed with the indicated antibodies. (c) U87/EGFR cells were stably expressed with control shRNA or α-catenin shRNA. Immunofluorescent staining was performed with a β-catenin antibody
α-Catenin expression regulates EGF-induced β-catenin transactivation
β-Catenin interacts with transcription factors of the TCF/LEF-1 family, leading to increased expression of genes such as CCND1 and c-myc [11]. To investigate the effect of α-catenin on β-catenin transactivation in response to EGFR activation, we first examined α-catenin regulation of TCF/LEF-1 transcriptional activity in U87/EGFR cells. TCF/LEF-1 luciferase reporter analysis showed that expression of FLAG–α-catenin or depletion of β-catenin expression significantly suppressed EGF-induced β-catenin transactivation (Fig. 2a). In contrast, depletion of α-catenin largely enhanced the β-catenin transactivation, which was further increased by EGF treatment (Fig. 2a). Immunoblotting analyses showed that expression of FLAG–α-catenin or depletion of β-catenin inhibited EGF-induced expression of cyclin D1 and c-Myc, whereas α-catenin depletion enhanced the expression of these proteins (Fig. 2b). These results indicate that α-catenin expression regulates EGF-induced β-catenin transactivation and expression of its downstream genes CCND1 and c-myc.

α-Catenin expression regulates EGF-induced β-catenin transactivation. a U87/EGFR cells were stably expressed with control shRNA, FLAG–α-catenin, α-catenin shRNA, or β-catenin shRNA. Luciferase activity was measured 8 h after treatment of cells with EGF (100 ng/ml) or no treatment. The relative levels of luciferase activity were normalized to the levels of untreated cells and to the levels of luciferase activity in the Renilla control plasmid. Data represent mean ± standard deviation of three independent experiments. b U87/EGFR cells were stably expressed with control shRNA, FLAG–α-catenin, α-catenin shRNA, or β-catenin shRNA. Immunoblotting analyses were performed with the indicated antibodies
β-Catenin regulation by α-catenin expression plays an important role in GBM cell proliferation and migration
β-Catenin transactivation has been closely related to tumor development. We next examined the role of β-catenin regulation by α-catenin in GBM cell proliferation by plating equal numbers of U87/EGFR cells, U87/EGFR cells expressing FLAG–α-catenin, or U87/EGFR cells with depletion of α-catenin or β-catenin. As shown in Fig. 3a, depletion of β-catenin or overexpression of α-catenin inhibited tumor cell proliferation. In contrast, depletion of α-catenin enhanced tumor cell proliferation.

β-Catenin regulation by α-catenin expression plays an important role in GBM cell proliferation and migration. a U87/EGFR cells (1 × 104) expressing control shRNA, FLAG–α-catenin, α-catenin shRNA, or β-catenin shRNA were seeded and counted at the indicated time. b U87/EGFR cells, which were stably expressed with control shRNA, FLAG–α-catenin, α-catenin shRNA, or β-catenin shRNA, were wounded by scraping with a micropipette tip and treated with EGF (100 ng/ml) or untreated for 16 h. Representative microphotographs are shown. c The number of migrated cells for each group was counted from six different fields under microscope. Results represent mean ± standard deviation of three independent experiments
Stimulation of EGFR-overexpressing cancer cells with EGF promotes cell migration [16, 23]. To examine the effect of regulation of expression of α-catenin and β-catenin on tumor cell migration, we conducted a monolayer wound healing assay. Compared with parental U87/EGFR cells, U87/EGFR cells with stable expression of FLAG–α-catenin or depletion of β-catenin migrated into the wound much more slowly (Fig. 3b, upper and middle panels). In addition, EGF-enhanced cell migration was inhibited by overexpression of α-catenin or by β-catenin depletion (Fig. 3b, bottom panel). In contrast, depletion of α-catenin increased tumor cell migration, which was further enhanced by EGF treatment (Fig. 3b). Given that we measured cell migration at 16 h and that cell proliferation did not differ significantly among the cell lines (Fig. 3a), the differences in cell migration were likely not caused by the differences in cell proliferation rates. These results indicated that α-catenin regulates β-catenin in EGF-induced tumor cell migration.
β-Catenin regulation by α-catenin expression plays an important role in EGF-induced GBM cell invasion
β-Catenin transactivation is involved in tumor cell invasion [16, 17]. To examine the effect of α-catenin expression on tumor cell invasion, we performed Matrigel Transwell invasion assays to measure the ability of tumor cells to invade through the Matrigel in 16 h, during which time cell proliferation did not differ significantly among the different cell lines (Fig. 3a). As shown in Fig. 4a, overexpression of α-catenin or depletion of β-catenin, which reduced basal levels of cell invasion, greatly inhibited EGF-induced U87/EGFR cell invasion. In contrast, α-catenin depletion enhanced the ability of U87/EGFR cells to invade, which was further increased by EGF treatment (Fig. 4a). This observation was further validated by quantitative colorimetric analysis (Fig. 4b). These findings indicate that α-catenin is a negative regulator for EGF-induced tumor cell invasion.

β-Catenin regulation by α-catenin expression plays an important role in EGF-induced GBM cell invasion. a U87/EGFR cells expressing a control shRNA, FLAG-tagged α-catenin, α-catenin shRNA, or β-catenin shRNA were plated on the top surface of the Matrigel in the absence or presence of EGF (100 ng/ml). Sixteen hours after plating, cells that migrated to the opposite side of the insert were stained with crystal violet. Representative microphotographs are shown. b The membranes with invaded cells were dissolved in 4% deoxycholic acid and read colorimetrically at 590 nm for quantification of invasion. Data represent mean ± standard deviation of three independent experiments
Discussion
Overexpression or activating mutation of EGFR frequently occurs in glioblastoma [2]. EGFR activation promotes tumor cell migration and invasion [5, 24]. We demonstrate here that β-catenin transactivation induced by EGFR activation can be regulated by α-catenin and that this regulation affects GBM cell motility and proliferation. Ectopic expression of α-catenin led to inhibition of nuclear β-catenin translocation and transactivation, whereas depletion of α-catenin enhanced both basal and EGF-induced nuclear translocation and transactivation of β-catenin. Consequently, the expression of the β-catenin downstream genes CCND1 and c-myc, as well as tumor cell migration, invasion, and proliferation, were modulated. These results underscore an instrumental role of α-catenin in EGF-induced β-catenin transactivation and GBM cell migration, invasion, and proliferation.
Activation of β-catenin by Wnt-dependent signaling or activating mutations of Wnt components has been observed in human cancers; however, Wnt-independent signaling is also involved in the regulation of β-catenin transactivation and tumorigenesis [25, 26]. β-Catenin-TCF/LEF-1 signaling can be activated by EGF, hepatocyte growth factor, and insulin-like growth factor I and II [16, 27, 28]. EGF-induced β-catenin nuclear accumulation and transactivation does not accompany a detectable change in β-catenin’s half-life or phosphorylation level by GSK-3β [16, 17], suggesting that the activation of β-catenin is regulated by a noncanonical pathway. Our previous studies showed that EGFR activation results in β-catenin transactivation by phosphorylation of β-catenin Ser552 by AKT and α-catenin Ser641 by CK2, which promotes dissociation of β-catenin from E-cadherin and α-catenin, respectively [17, 18]. These findings highlight a critical role of posttranslational modifications of β-catenin and α-catenin in EGF-induced β-catenin transactivation.
In addition to the regulation of β-catenin by posttranslational modifications, we demonstrate here that β-catenin transactivation can be regulated by expression levels of its interacting component α-catenin. Given that loss or downregulation of α-catenin has been detected in cancer cells, and that metastatic cancers are often found to have downregulated levels of α-catenin [20–22], inhibition of β-catenin by regulation of α-catenin expression could be an alternative therapeutic approach to treat cancers with high β-catenin activity.
Materials and methods
Cell lines, antibodies, and reagents
EGFR-overexpressed U87/EGFR cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine calf serum (HyClone, Logan, UT). Cell cultures were made quiescent by growing them to confluence and then replacing the medium with fresh medium containing 0.5% serum for 1 day. Monoclonal antibodies for α-catenin (G-11) and β-catenin (E-5) and polyclonal antibodies for c-Myc and cyclin D1 were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies for FLAG and tubulin and EGF were purchased from Sigma (St. Louis, MO). Puromycin and G418 were purchased from EMD Biosciences (San Diego, CA). Hoechst 33342 and Texas Red-conjugated anti-rabbit antibody were from Molecular Probes (Eugene, OR). HyFect transfection reagents were from Denville Scientific (Metuchen, NJ).
DNA constructs
Polymerase chain reaction–amplified human α-catenin was cloned into pEGFP N1-FLAG vector. pGIPZ control was generated with a control oligonucleotide, GCTGTTGACAGTGAGCGAGCTTCTAACACCGGAGGTCTTTAGTGAAGCCACAGATGTAAAGACCTCCGGTGTTAGAAGCGTGCCTACTGCCTCGGA. pGIPZ-α-catenin was generated with the following oligonucleotides: GCTGTTGACAGTGAGCGAGATGGTATCTTGAAGTTGAGGTAGTGAAGCCACAGATGTACCTCAACTTCAAGATACCATCGTGCCTACTGCCTCGGA and GCTGTTGACAGTGAGCGAGACTTAGGAATCCAGTATAAATAGTGAAGCCACAGATGTATTTATACTGGATTCCTAAGTCGTGCCTACTGCCTCGGA. pGIPZ-β-catenin was generated with the following oligonucleotides: GCTGTTGACAGTGAGCG AGTAGCTGATATTGATGGACAGTAGTGAAGCCACAGATGTACTGTCCATCAATATCAGCTACGTGCCTACTGCCTCGGA and GCTGTTGACAGTGAGCGAGGTGCTATCTGTCTGCTCTAGTAGTGAAGCCACAGATGTACTAGAGCAGACAGATAGCACCGTGCCTACTGCCTCGGA.
Transfection
Cells were plated at density of 4 × 105 per 60-mm dish, 18 h prior to transfection. Transfection was performed using either calcium phosphate or HyFect reagents (Denville Scientific) according to the vendor’s instructions. Transfected cultures were selected with hygromycin (200 μg/ml) or puromycin (5 μg/ml) for 10–14 days at 37°C. At that time, antibiotic-resistant colonies were picked, pooled, and expanded for further analysis under selective conditions.
Subcellular fractionation
Nuclei were isolated using the Nuclear Extract Kit from Active Motif North America (Carlsbad, CA) and the ProteoExtract Subcellular Proteome Extraction Kit from Calbiochem (San Diego, CA), according to the manufacturers’ instructions.
Immunoblotting analysis
Extraction of proteins from cultured cells using a modified buffer was followed by immunoblotting with corresponding antibodies, as described previously [29]. Each experiment was repeated at least three times.
Immunofluorescence analysis
Cells were fixed and incubated with primary antibodies, Alexa Fluor dye-conjugated secondary antibodies, and Hoechst 33342 according to standard protocols. Cells were examined using a deconvolution microscope (Zeiss, Thornwood, NY) with a 63-Å oil-immersion objective. Axio Vision software from Zeiss was used to deconvolute Z-series images.
Luciferase reporter gene assay
Transcriptional activities of TCF/LEF-1 in U87/EGFR cells were measured as described previously [17]. EGF (100 ng/ml) was added 8 h before harvesting. Each experiment was performed at least three times.
In vitro invasion assay
Cell invasion was assessed by invasion of cells through Matrigel-coated Transwell inserts (Greiner Bio-One North America Inc., Monroe, NC) during a 16-h time frame, as described previously [17]. The membranes with invaded cells were dissolved in 4% deoxycholic acid and read colorimetrically at 590 nm for quantification of invasion.
Cell migration assay
In the wound healing assay, cells were plated at 60% confluence in 10% serum-DMEM. Twelve hours after seeding, the monolayers were wounded by scoring with a sterile plastic 200-μl micropipette tip, washed, and fed with DMEM. Sixteen hours after wounding, cells were photographed using a low-magnification phase-contrast microscope, as described previously [24]. Migration of cells was quantified as the number of migrated cells. Data represent mean ± standard deviation of three independent experiments.
References
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21(21):2683–2710. doi:10.1101/gad.1596707
Heimberger AB, Suki D, Yang D, Shi WM, Aldape K (2005) The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med 3:38. doi:10.1186/1479-5876-3-38
Demuth T, Berens ME (2004) Molecular mechanisms of glioma cell migration and invasion. J Neurooncol 70(2):217–228
Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD (1997) Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 57(18):4130–4140
Bianco R, Gelardi T, Damiano V, Ciardiello F, Tortora G (2007) Rational bases for the development of EGFR inhibitors for cancer treatment. Int J Biochem Cell Biol 39(7–8):1416–1431. doi:10.1016/j.biocel.2007.05.008
Gullick WJ (1991) Prevalence of aberrant expression of the epidermal growth-factor receptor in human cancers. Br Med Bull 47(1):87–98
Voldborg BR, Damstrup L, Spang-Thomsen M, Poulsen HS (1997) Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol 8(12):1197–1206
Chang LF, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410(6824):37–40
Perez-Moreno M, Fuchs E (2006) Catenins: keeping cells from getting their signals crossed. Dev Cell 11(5):601–612. doi:10.1016/j.devcel.2006.10.010
Huang H, He X (2008) Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol 20(2):119–125. doi:10.1016/j.ceb.2008.01.009
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281(5382):1509–1512
Nelson WJ, Nusse R (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303(5663):1483–1487
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 96(10):5522–5527
Polakis P (2000) Wnt signaling and cancer. Genes Dev 14(15):1837–1851
Chenn A (2008) Wnt/β-catenin signaling in cerebral cortical development. Organogenesis 4(2):76–80
Lu ZM, Ghosh S, Wang ZY, Hunter T (2003) Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 4(6):499–515
Fang DX, Hawke D, Zheng YH, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu ZM (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282(15):11221–11229. doi:10.1074/jbc.M611871200
Ji HT, Wang J, Nika H, Hawke D, Keezer S, Ge QY, Fang BL, Fang XX, Fang DX, Litchfield DW, Aldape K, Lu ZM (2009) EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-catenin from beta-catenin and transactivation of beta-catenin. Molecular Cell 36(4):547–559. doi:10.1016/j.molcel.2009.09.034
Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI (2005) Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 123(5):903–915. doi:10.1016/j.cell.2005.09.021
Benjamin JM, Nelson WJ (2008) Bench to bedside and back again: molecular mechanisms of alpha-catenin function and roles in tumorigenesis. Semin Cancer Biol 18(1):53–64. doi:10.1016/j.semcancer.2007.08.003
Giannini AL, Vivanco M, Kypta RM (2000) Alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J Biol Chem 275(29):21883–21888. doi:10.1074/jbc.M001929200
Hwang SG, Yu SS, Ryu JH, Jeon HB, Yoo YJ, Eom SH, Chun JS (2005) Regulation of beta-catenin signaling and maintenance of chondrocyte differentiation by ubiquitin-independent proteasomal degradation of alpha-catenin. J Biol Chem 280(13):12758–12765. doi:10.1074/jbc.M413367200
Muller T, Bain G, Wang X, Papkoff J (2002) Regulation of epithelial cell migration and tumor formation by beta-catenin signaling. Exp Cell Res 280(1):119–133. doi:10.1006/excr.2002.5630
Lu ZM, Jiang GQ, Blume-Jensen P, Hunter T (2001) Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol 21(12):4016–4031
Giles RH, van Es JH, Clevers H (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochimica Et Biophysica Acta-Reviews on Cancer 1653(1):1–24. doi:10.1016/s0304-419x(03)00005-2
Lu ZM, Hunter T (2004) Wnt-independent beta-catenin transactivation in tumor development. Cell Cycle 3(5):571–573
Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, Capeau J (2001) Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3 beta inhibition and Ras activation. Oncogene 20(2):252–259
Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L (2001) IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene 20(36):4942–4950
Lu ZM, Liu D, Hornia A, Devonish W, Pagano M, Foster DA (1998) Activation of protein kinase C triggers its ubiquitination and degradation. Mol Cell Biol 18(2):839–845
Acknowledgments
This study was supported by National Cancer Institute grants 5R01CA109035 (Z.L.) and CA-16672 (Cancer Center Support Grant), American Cancer Society Research Scholar Award RSG-09-277-01-CSM (Z.L.), an institutional research grant from The University of Texas MD Anderson Cancer Center (Z.L.), and the A. Lavoy Moore Endowment Fund (H.J.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ji, H., Wang, J., Fang, B. et al. α-Catenin inhibits glioma cell migration, invasion, and proliferation by suppression of β-catenin transactivation. J Neurooncol 103, 445–451 (2011). https://doi.org/10.1007/s11060-010-0413-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-010-0413-4