
Abstract
Over the past few decades, hollow-structured silica particles have attracted significant interest from material’s scientists due to their unique properties and morphologies related to biomedical applications. This interest has spurred the development of synthetic processes of silica particles and in this paper we report on the microwave-based synthesis of nano-silica hollow spheres (NSHS) with uniform shape and size. This synthetic process produces NSHS in more efficient and swifter manner than previously reported approaches. TEM, SEM, TGA, and BET analysis revealed that the particles produced are hollow, uniform, porous spheres with high water retention capabilities. To explore the drug delivery capability of these spheres, Ibuprofen, as the model drug, was loaded into the NSHS to explore the drug load capacity and the drug release action of these NSHS. The findings of this study signal that the reported microwave-based NSHS has interesting practical relevance as promising drug delivery vehicles.

Graphical abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past several decades, 3D nano/microhollow-structured particles composed of ceramic materials have drawn intense interest in various fields of research such as waste removal, ion exchange, catalysis, sensing, prosthetic materials, fillers, and biomedical applications (Wang et al. 2016; Lei et al. 2014; Prieto et al. 2016; Prates et al. 2019; Ramli 2017; Vanherck et al. 2010; Son et al. 2007). This interest is due to the unique properties associated with these structures, viz., low density, large specific surface area, porosity, monodispersity, well-confinement effect, and chemical stability. From their first reporting during the 1980s (Kowalski et al. 1984; Blankenship 1996), the synthesis of well-defined, monodispersed hollow-structured materials with tailored properties has remained a challenge to materials scientists.
Ceramic mesoporous silica nano/microparticles have been widely utilized in biomedical and biotechnology applications, as drug delivery carriers, bio-fillers, and even cosmetics (Du et al. 2016; Giret et al. 2015; Tan et al. 2014; Bharti et al. 2015; Adhikari et al. 2018). They are practically useful due to the high and well-documented functionalized surfaces, biocompatibility, and biodegradability. By combining the unique hollow structure of these particles with functional silica properties, 3D silica nano/microhollow particles become more attractive as vehicles for drug delivery applications in comparison with their standard solid counterparts due to the enhanced porosity, unique morphology, low density, and high confinement effect of these hollow silica structures. It is understood that the hollow nature of the particles can store a significant amount of a drug with the mesoporous nature of the shell controlling the manner in which the drug is released. Moreover, due to the confinement effect of the hollow-structured silica particles, biological payloads can be transported safely without aggregation of the nanoparticles caused by hydrophobic particles during the delivery into the cells.
To date, several synthetic methods to produce various 3D hollow-structured ceramic metal oxides, with desirable morphologies and structure, have been reported (Wang et al. 2013; Titirici et al. 2006; Zeng 2007; Hu et al. 2010). Sacrifacial templating is currently the most widely applied method for synthesizing hollow particles (Hu et al. 2010; Lou et al. 2008; Qian et al. 2007). In this method, a template is coated via controlled precipitation/deposition of a desired material precursor onto the surface layers of the template to generate core@shell composites. The template particles are then selectively etched (sacrificed) by thermal or chemical means, leaving behind the desired shell structure composed of the material precursor. Various materials have been used as sacrificial templates such as emulsion droplets, organic polymeric spheres, and inorganic nanoparticles (Hu et al. 2010; Lou et al. 2008; Qian et al. 2007; Xia et al. 2019). In earlier studies, we have reported on the fabrication of various ceramic oxides with hollow-structures using simple sugar (monosaccharaides) as sacrificial templates via sol-gel and hydrothermal methods (Abdelaal et al. 2014, 2020; Abdelaal and Harbrecht 2015; Abdelaal 2018a, b). These studies showed that utilizing monosaccharaides as sacrificial templates is an efficient and cost-effective method to achieve desired 3D nano/microhollow structures. However, these methods required precise conditions, or time-consuming, or multistep processes with several chemical additives, and the formed hollow structures lack well-defined shape and/or homogenous distribution (heterogeneity of samples). Consequently, improvement in the synthetic processes and chemicals agents is requiring to obtain the homogeneous hollow particles.
Recently, microwave-assisted chemical reactions have been widely applied widely in various synthetic procedures (Priecel and Sanchez 2019; Dąbrowska et al. 2018; Zhu and Chen 2014; Nüchter et al. 2004). Microwaves are advantageous in chemical reactions due to the high reaction efficiency, low energy consumption, and short reaction times. It was envisaged that based on these advantages, microwave combustion could be used to enhance the production of nano-silica hollow spheres (NSHS), via core@shell processes. Microwave combustion proved to be advantageous in this study in reducing the reaction time and energy for producing core@shell structures, while also maintaining high homogeneity. As with previous studies, the carbonaceous core was then selectively removed by thermal treatment, in air, leading to freestanding well-defined 3D NSHS.
Since NSHS has high potential to be promising vehicles in biomedical applications, the hydration (water retention) ability of NSHS was tested to further understand the effect the porous and hollow nature of these particles would have on the loading capability of drug. It was observed that ibuprofen could be successfully loaded onto and into the NSHS and that the release of the drug is strongly linked to the structure of the NSHS. This has important implications for nanomedicine drug delivery.
Materials and methods
Materials
Fructose and Na2SiO3 solution (containing about 25% SiO2) were obtained from Merck (Darmstadt, Germany). Ibuprofen (USP grade), hexane, and phosphate-buffered saline (PBS) [BioPerformance, pH = 7.4] were obtained from Sigma-Aldrich. All chemicals were analytical grade and used without further purification. Deionized water (conductivity ~ 1.5 μscm−1) was used.
Fabrication of the nano-silica hollow spheres (NSHS)
In this study, nano-silica hollow spheres (NSHS) were synthesized by employing a microwave combustion method. In this method, fructose (simple sugar) was used as a sacrificed template, with sodium silicate (water glass) also acting as the silica precursor. The morphology of the NSHS was investigated and controlled by varying the degree of microwave irradiation and the concentration of sodium silicate used.
Typically, 3.6 g (20 mmol) of fructose was dissolved in 20 mL of distilled water followed by the addition of 0.5 mL water glass (equivalent to 9.83 mmol). The mixture was then sonicated (using Cole–Parmer, 500 W ultrasonic homogenizer, model wz-04711-30) for 5 min at room temperature. The homogenous mixture was then irradiated in a microwave (MARS Extraction and Digestion system, Model XP-1500, CEM Corp., Matthews, NC) at 80 °C for 2 h at a heating rate of 5 °C/min in a Teflon-lined vessel (Dupont, Wilimington, DE).
The formed samples (core@shell composite particles) were then centrifuged at 4000 rpm for 10 min and the obtained material was then washed several times with deionized water and air-dried at 60 °C for 5 h. The resulted core@shell composite was then calcined, in air, at 550 °C at a heating rate of 5 °C min−1 for 4 h to remove the carbonaceous core, leading to freestanding nano-silica hollow spheres (NSHS).
Characterization
The size and shape of the resultant NSHS was observed by scanning electron microscopy (SEM, Hitachi S-4800). Transmission electron microscopy (TEM) micrographs were taken on a JEOL 3010 high resolution in order to recognize the hollow structure of the core@shell composite particles and NSHS. The average distributions of the particle size were calculated using ImageJ program. X-ray powder diffraction (XRD) analysis data of the solid products were taken on an X’Pert MPD, PANalytical using Cu-Kα radiation. N2 adsorption/desorption isotherms, pore size distribution, and the specific surface area (SBET) were investigated using a Micromeritics ASAP 2020 gas sorptometer. Before the adsorption measurements, the sample was degased under vacuum at 200 °C overnight. The N2 adsorption/desorption isotherms were then recorded at 77 K over a wide range of relative pressures (P/Po) from 0.01 to 0.995. The specific surface area and the pore size distribution were calculated from the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. The FT-IR spectra of the solid products were taken on a Nicolet 6700, and thermal gravimetric analysis data (TGA) was taken on a METTLER TOLEDO-TGA/SDTA 851e apparatus. Prior to the measurement, the sample was dried in a vacuum oven at 60 °C overnight to eliminate as much water as possible. Then, the TGA was tested in a range of 30–700 °C at a heating rate of 10 °C/m. The drug release profiles were recorded using UV–Vis spectroscopy (Ocean Optics USB4000 spectrometer).
Water retention evaluation (hydration test)
The evaluation of the water retention capability of the NSHS was done using TGA analysis. Firstly, the as-synthesized NSHS sample was soaked in deionized water for 24 h. Thereafter, the recovered wet sample was then dried using a hair drier for 20 min and then air-dried overnight - at room temperature to eliminate the water adsorbed on the outer surface of NSHS. The TGA experiment was carried out under air in the range of 60–160 °C at a heating rate of 1 °C/ min.
Drug release test
A 100-mg porous NSHS was dispersed in 25 mL of ibuprofen-hexane solution with a concentration of 1.8 mg mL−1, and stirred for 24 h. The amount of ibuprofen absorbed by the sample was determined by UV spectrometry, resulting in 31 wt% with respect to the powdered starting material.
For in vitro ibuprofen release, 10 mg of ibuprofen containing mesoporous NSHS was dispersed into 20 mL of phosphate buffer solution. The mixtures were stirred at 150 rpm. The sample solutions were then centrifuged at 4000 rpm for 15 min. The clear supernatants were collected and analyzed by UV spectroscopy, and the release profiles were recorded.
Results and discussion
A novel, microwave-based method for the rapid synthesis of NSHS with well-defined size and shape is reported on. In this method, fructose is used as the source of the core carbonaceous sphere with sodium silicate acting as the silica shell precursor. The carbonaceous core is then oxidized in air to leave behind the NSHS. In order to control the morphology of the core@shell composites, varying microwave irradiation times and concentrations of sodium silicate were used. A graphic of a proposed mechanism of the formation of the NSHS is illustrated in Fig. 1.

Graphic illustration of the microwave-based method for the synthesis of mesoporous NSHS
Unlike the conventional heating methods (Zheng et al. 2019; Guo et al. 2019; Darr et al. 2017), the electromagnetic energy of the microwaves assisted in the rapid decomposition of fructose into organic species and acids which final results in solid carbonaceous spheres with O-functionalities. Simultaneously, the silicic acid, which is generated due to the acidic hydrolysis of the sodium silicate, anchors to the fructose-derived carbonaceous spheres to generate core@shell composite spheres. Two key factors facilitate this process as follows: (I) the O-functionalities on the surface of the carbonaceous spheres; and (II) the silanol groups (-Si-OH) located on the surface of the silicic acid that provides strong binding sites with the fructose-derived carbonaceous spheres due to hydrogen bonding and electrostatic attraction. The synthesis of these core@shell structures are produced at lower reaction times and temperatures than previously reported resulting in a faster synthetic approach with controlled morphology. The carbonaceous core is selectively removed by heat treatment at 550 °C for 4 h, oxidation in air. This results in the densification and the cross-linking of the incorporated silicic acid to generate free-standing NSHS. Upon oxidation and removal of the carbonaceous core, the size of the resultant silica shell is reduced by about 55% when compared to the core@shell structure.
Figure 1 a–b shows the SEM images of the core@shell composite spheres generated through the microwave-based reaction and the NSHS generated after the heat treatment, respectively. SEM images reveal hollow structure with well-defined 3D spherical morphology and uniform size distribution with an average diameter of 190 nm. The inset image in Fig. 2b shows a broken wall of a hollow sphere (marked with an arrow) which provides further confirmation of the formation of hollow structure. The particle size distribution (Fig. 3 a–b) and SEM analysis (Fig. 2 a–b) clearly indicate that the average size of the core@shell composite spheres and NSHS are 420 nm and 190 nm, respectively. Although the morphology was well-maintained after sacrificing of the carbonaceous core, the size of the NSHS is reduced by about 55% in comparison with the original composite spheres after heat treatment, as observed from Figs. 2 and 3. The reason of this behavior is caused by calcination and removal of the interior carbonaceous core of the core@shell composite spheres.

SEM images of the sample, (a) the core@shell composite spheres before heat treatment, and (b) the NSHS after heat treatment at 550 °C for 4 h in air. The inset image in (b) is that of a ruptured sphere after the heat treatment, confirming the hollow structure of the sphere

Average particle size distribution of the product, (a) the core@shell composite spheres before heat treatment, and (b) the NSHS after heat treatment at 550 °C for 4 h in air
Figure 4a provides clear evidence for the postulation of a spatial separation of the silica rich shells and the fructose-derived carbonaceous cores because of the abrupt contrast distinction in TEM micrograph of the spherical hybrid particle. Figure 4b illustrates the morphology of NSHS. The clear contrast between the dark wall and the bright inner indicates the hollow structure of the silica spheres with wall thickness of 16 nm on average. These TEM images also further validate the 55% size shrinkage after the heat treatment, as concluded from SEM analysis and particle size distributions Figs. 2 and 3, respectively.

TEM images of (a) the core@shell composite spheres before heat treatment, and (b) the NSHS after heat treatment at 550 °C for 4 h in air
The effect of the concentration of sodium silicate on particle size and morphology (Fig. S1 and S2, see the Supporting Information) indicates that the amount of sodium silicate added has a substantial impact on the morphology of the formed solid particles. By simply changing the amount of sodium silicate to a higher concentration (1 mL, equivalent to 19.67 mmol), a spongy like structure was observed (Fig. S2, the Supporting Information). Additionally, it observed that when decreasing the concentration of the added sodium silicate to 0.1 mL (equivalent to 1.97 mmol), no hollow spheres were observed (Fig. S3, the Supporting Information), revealing a concentration limit for the synthesis of NSHS.
Another relevant factor is the influence of the microwave irradiation time. When the time is increased to 3 h, the growth of the core@shell precursor continued and it was observed that the morphology after this heat treatment was sponge-like in structure (Fig. S4, the Supporting Information). Whereas, decreasing the irradiation time to 1 h is not sufficient to ensure well-formed spheres. Control of both irradiation time and concentration of sodium silicate are essential to producing spheres with controlled morphology and yield.
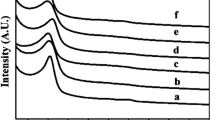
The powder X-ray diffraction pattern of the NSHS after the heat treatment shows one broad peak centered at about 22° (Fig. 5). This is a characteristic for amorphous silica (Martinez et al. 2006). The inset XRD pattern of the core@shell composite indicates the amorphous nature of the hybrid composite spheres before heat treatment.

XRD patterns of the NSHS after thermal treatment at 550 °C for 4 h in air. The inset is that of core@shell composite before thermal treatment
Figure 6 a and b display the IR spectra of the sample before and after heat treatment, respectively. A comparison between both the IR spectrum reveals the disappearance of the peaks around 2930 and 1705 C-H stretching and C=O group (Abdelaal 2015), respectively, after heat treatment. In addition, the appearance of peaks around 468, 805, and 1102 cm−1 which correspond to the three vibration bands of the Si-O-Si bond is observed. The peak at about 3400 to 3450 cm−1 in the hollow-structured sample (after heat treatment) is ascribed to the hydroxyl groups on the surface of silica (Ryu and Tomozawa 2006). The existence of an absorption peak around 1640 cm−1 in the IR spectrum of the NSHS (after heat treatment) is due to the stretching and deformation vibration of adsorbed water molecules (Abdelaal et al. 2014). IR spectra are a clear evidence of the complete removal of the carbonaceous core and the formation of SiO2 after the heat treatment.

FT-IR spectra of (a) the core@shell composite spheres before heat treatment, and (b) the NSHS after heat treatment at 550 °C for 4 h in air
Figure 7 shows the thermogravimetric analysis (TGA) of the hybrid core@shell composite precursor. As shown in Fig. 7, a marginal weight loss of about 3%, below 100 °C, occurs due to the evaporation of the physically adsorbed H2O. Then, at about 300 °C, the pyrolysis of the carbonaceous core of the solid composite as well as the condensation of the terminal silanol groups occurs (Fowler et al. 2001), resulting in a total mass decrease of 86% at about 500 °C. The TGA analysis has two significant implications. Firstly, it indicates the complete removal of the carbonaceous core at about 500 °C. Secondly, it shows the thermal stability of the as-synthesized NSHS up to at least 700 °C.

Thermogravimetric analysis for the core@shell composite sample
Figure 8 displays the N2 adsorption-desorption isotherm for the as-synthesized NSHS. Typically, it is categorized as type IV isotherm, which reveals the mesoporous nature of the wall of the NSHS based on IUPAC sorting (Sing et al. 1985). Using the BET method, the surface area of the NSHS was calculated to be 330 m2g−1. The inset figure displays the pore size distribution of the NSHS, which was calculated using Barrett–Joyner–Halenda (BJH) method from the desorption curve. The pore size on average was noted to be 3.8 nm and the distribution is generally in a range of 2–100 nm. This may attributed to the existence of mesoporous channels and inner voids.

Nitrogen adsorption-desorption isotherm of the NSHS. The inset is representative of the BJH pore size distribution of the sample
Herein, we evaluated the water retention ability of the as-synthesized NSHS as the accessibility of water into the mesoporous walls and the inner voids of the NSHS could affect the loading capability of any biological payload in aqueous solutions. As shown in Fig. 9, thermogravimetric analysis (TGA) indicates that NSHS can harbor up to 19 wt% of water molecules (equivalent to 12.1 mmol g−1) which indicates the excellent hydration ability of the as-synthesized NSHS. The implication of this is that the NSHS has a high potential confinement effect. The TGA figure displays a weight loss between 60 and 70 °C which is ascribed to the fast evaporation of the free water. From 70 °C to 96 °C the water adsorbed onto the mesopores and the inner void through capillary effect was evaporated. Above 96 °C, no weight loss was observed and nearly all the free and adsorbed water had evaporated.

TGA curve of the NSHS after immersed in deionized water for 24 h
We tested the capability of the as-synthesized NSHS as a drug carrier for nanomedicine applications. In order to explore the drug loading capacity of the NSHS, the drug ibuprofen was loaded onto the NSHS. The observed maximum loading amount of the drug, evaluated by UV analysis, is ca. 31% which indicates that these as-synthesized materials have a larger loading capacity when compared with some previous studies (Zhou et al. 2007; Mitran et al. 2018). This reveals that the well-defined porosity and morphology and the high surface area are of the reported NSHS probably has a relevant impact on the loading mechanism. Figure 10 shows the release profile of the NSHS and their conventional silica solid counterparts prepared according to earlier studies (Zawrah et al. 2009). It was observed that the ibuprofen release profile from the ibuprofen-NSHS system (Fig. 11), in phosphate buffer solution (pH = 7.4) at 37 °C, had taken place through two stages as follows: (I) about 80% of the drug was released in 10 h; (II) the rest of the drug released slowly till the drug release was complete at a release time of 40 h. This observation has important implications with regard to the drug release mechanism of the ibuprofen-NSHS system as presented in Fig. 11: (I) The ibuprofen loaded on the outer surface of the NSHS was released in the first 10 h (the first release stage); (II) the ibuprofen payload in the pore channels and the inner void was slowly released; and it was completely released after 40 h (the second release stage); this reveals that the open-ended pore channels act as gates that have a control over the release of the ibuprofen payload.

Ibuprofen release profile of NSHS

Graphic illustration of the structure and the release processes of ibuprofen-NSHS system
Conclusions
In conclusion, a novel and cost-effective method to develop NSHS via a microwave-assisted process, using fructose (as a sacrificial template) and water glass (as silica precursor) has been developed. This approach resulted in core@shell structures, which are produced at lower reaction times and temperatures than previously reported on. The microwave irradiation time and concentration of water glass have a significant effect on the morphology of the core@shell structures and ultimately the NSHS produced (by thermal decomposition of the core). TEM and SEM analysis revealed the synthesis of uniform 3D spherical particles with hollow interiors (final product, NSHS); while BET analysis revealed that the porous spheres have a high surface area. Additionally, TGA analysis revealed the high water retention capabilities of the as-synthesized hollow spheres. The drug delivery potential of these NSHS was investigated using ibuprofen and it was found that ibuprofen could be loaded successfully at 31% load capacity, and due to the porous nature of the NSHS, the drug was released in a slow and sustained manner. The as-synthesized NSHS has high potential as promising carriers in drug delivery applications.
References
Abdelaal HM (2015) Facile hydrothermal fabrication of nano-oxide hollow spheres using monosaccharides as sacrificial templates. ChemistryOpen 4:72–75
Abdelaal HM (2018a) One-pot path for the synthesis of hollow zirconia sub-microspheres using hydrothermal approach. Matt Lett 212:218–220
Abdelaal HM (2018b) Sonochemical fabrication of 3d chromium (iii) oxide hollow spheres using fructose as sacrificial template. Interceram- Int Ceram Rev 67:20–25
Abdelaal HM, Harbrecht B (2015) Fabrication of hollow spheres of metal oxide using fructose derived-carbonaceous spheres as sacrificial templates. C R Chimie 18:379–384
Abdelaal HM, Zawrah MF, Harbrecht B (2014) Facile one-pot fabrication of hollow porous silica nanoparticles. Chem Eur J 20:673–677
Abdelaal HM, Tawfik A, Shaikjee A (2020) A simple approach to synthesis uniform 3D hollow yttrium oxide spheres using a hydrothermal scheme. Mater Chem Phys 242:122530
Adhikari C, Mishra A, Nayak D, Chakraborty A (2018) J. Drug Deliv. Sci Tec 45:303–314
Bharti C, Nagaich U, Pal AK, Gulati N (2015) Mesoporous silica nanoparticles in target drug delivery system: a review. Int J Pharm Investig 5:124–133
Blankenship RM (1996) US Patent 5494971
Dąbrowska S, Chudoba T, Wojnarowicz J, Łojkowski W (2018) Current trends in the development of microwave reactors for the synthesis of nanomaterials in laboratories and industries: a review. Crystals 8:379
Darr JA, Zhang J, Makwana NM, Weng X (2017) Continuous hydrothermal synthesis of inorganic nanoparticles: applications and future directions. Chem Rev 117:11125–11238
Du X, Li X, Xiong L, Zhang X, Kleitz F, Qiao SZ (2016) Mesoporous silica nanoparticles with organo-bridged silsesquioxane framework as innovative platforms for bioimaging and therapeutic agent delivery. Biomaterials 91:90–127
Fowler CE, Khushalani D, Mann S (2001) Interfacial synthesis of hollow microspheres of mesostructured silica. Chem Commun:2028–2029
Giret S, Man MWC, Carcel C (2015) Mesoporous-silica-functionalized nanoparticles for drug delivery. Chem Eur J 21:13850–13865
Guo C, Wang P, Liao S, Si H, Chen S, Fan G, Wang Z (2019) Morphology-controllable hydrothermal synthesis of zirconia with the assistance of a rosin-based surfactant. Appl Sci 9:4145
Hu CY, Xu YJ, Duo SW, Li WK, Xiang JH, Li MS, Zhang RF (2010) Preparation of inorganic hollow spheres based on different methods. J Chin Chem Soc 57:1091–1098
Kowalski A, Vogel M, Blankenship RM (1984) US Patent:4427836
Lei C, Han F, Sun Q, Li W-C, Lu A-H (2014) Confined nanospace pyrolysis for the fabrication of coaxial Fe3O4@C hollow particles with a penetrated mesochannel as a superior anode for Li-ion batteries. Chem Eur J 20:139–145
Lou XW, Archer LA, Yang Z (2008) Hollow micro−/nanostructures: synthesis and applications. Adv Mater 20:3987–4019
Martinez J, Sanchez SAP, Ortega-Zarzosa G, Ruiz F, Chumakov Y (2006) Rietveld refinement of amorphous SiO2 prepared via sol–gel method. Mater Lett 60:3526–3529
Mitran R, Matei C, Berger D, Bajenaru L, Moisescu MG (2018) Controlling drug release from mesoporous silica through an amorphous, nanoconfined 1-tetradecanol layer. Eur J Pharm Biopharm 127:318–325
Nüchter M, Ondruschka B, Bonrath W, Gum A (2004) Microwave assisted synthesis – a critical technology overview. Green Chem 6:128–141
Prates ARM, Meunier F, Dodin M, Franco RM, Farrusseng D, Tuel A (2019) Hydrogenation size-selective Pt/hollow beta catalysts. Chem Eur J 25:2972–2977
Priecel P, Sanchez JAL (2019) Advantages and limitations of microwave reactors: from chemical synthesis to the catalytic valorization of biobased chemicals. ACS Sustain Chem Eng 7(1):3–21
Prieto G, Tüysüz H, Duyckaerts N, Knossalla J, Wang G-H, Schüth F (2016) Hollow nano- and microstructures as catalysts. Chem Rev 116:14056–14119
Qian H, Lin G, Zhang Y, Gunawan P, Xu R (2007) A new approach to synthesize uniform metal oxide hollow nanospheres via controlled precipitation. Nanotechnology 18:355602
Ramli RA (2017) Hollow polymer particles: a review. RSC Adv 7:52632–52650
Ryu S-R, Tomozawa M (2006) Fictive temperature measurement of amorphous SiO2 films by IR method. J Non-Cryst Solids 352:3929–3935
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, Siemieniewska T (1985) Reporting physisorption data for gas/solid system with special reference to the determination of surface area and porosity. Pure Appl Chem 57:603–619
Son SJ, Bai X, Lee SB (2007) Inorganic hollow nanoparticles and nanotubes in nanomedicine: part 1. Drug/gene delivery applications. Drug Discov Today 12:650–656
Tan SY, Ang CY, Li P, Yap QM, Zhao Y (2014) Drug encapsulation and release by mesoporous silica nanoparticles: the effect of surface functional groups. Chem Eur J 20:11276–11282
Titirici MM, Antonietti M, Thomas A (2006) A generalized synthesis of metal oxide hollow spheres using a hydrothermal approach. Chem Mater 18:3808–3812
Vanherck K, Aerts A, Martensa J, Vankelecom I (2010) Hollow filler based mixed matrix membranes. Chem Commun 46:2492–2494
Wang X, Yang YJ, Ma Y, Yao J-N (2013) Controlled synthesis of multi-shelled transition metal oxide hollow structures through one-pot solution route. Chin Chem Lett 24:1–6
Wang X, Feng J, Bai Y, Zhang Q, Yin Y (2016) Synthesis, properties, and applications of hollow micro−/nanostructures. Chem Rev 116:10983–11060
Xia Y, Na X, Wu J, Ma G (2019) The horizon of the emulsion particulate strategy: engineering hollow particles for biomedical applications. Adv Mater 31:1801159
Zawrah MF, El-Kheshen AA, AbdelaaL HM (2009) Facile and economic synthesis of silica nanoparticles. J Ovonic Res 5:129–133
Zeng HC (2007) Ostwald ripening: a synthetic approach for hollow nanomaterials. Curr Nanosci 3:177–181
Zheng J, Zhang T, Zeng H, Guo W, Zhaho B, Sun Y, Li Y, Jiang L (2019) Multishelled hollow structures of yttrium oxide for the highly selective and ultrasensitive detection of methanol. Small 15:1804688
Zhou J, Wu W, Caruntu D, Yu MH, Martin A, Chen JF, O'Connor CJ, Zhou WL (2007) Synthesis of porous magnetic hollow silica nanospheres for nanomedicine application. J Phys Chem C 111:17473–17477
Zhu YJ, Chen F (2014) Microwave-assisted preparation of inorganic nanostructures in liquid phase. Chem Rev 114:6462–6555
Acknowledgments
Prof. Dr. M. Salama for valuable and fruitful discussion.
Funding
This work was supported by the Ceramics Laboratory and the National Research Centre (NRC), Egypt.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1746 kb)
Rights and permissions
About this article

Cite this article
Abdelaal, H.M., Shaikjee, A. Microwave-based fast synthesis of clear-cut hollow spheres with mesoporous wall of silica nanoparticles as excellent drug delivery vehicles. J Nanopart Res 22, 171 (2020). https://doi.org/10.1007/s11051-020-04918-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-020-04918-3