
Abstract
Background
Epithelial ovarian cancer, especially high grade serous ovarian cancer (HGSOC) is by far, the most lethal gynecological malignancy with poor prognosis and high relapse rate. Despite of availability of several therapeutic interventions including poly-ADP ribose polymerase (PARP) inhibitors, HGSOC remains unmanageable and identification of early detection biomarkers and therapeutic targets for this lethal malady is highly warranted. Aberrant expression of protein kinase C iota (PKCί) is implicated in many cellular and physiological functions involved in tumorigenesis including cell proliferation and cell cycle deregulation.
Methods and results
Two high grade serous ovarian cancer cells SKOV3 and COV362 were employed in this study. PKCί was genetically knocked down or pharmacologically inhibited and several functional and biochemical assays were performed. We report that PKCί is overexpressed in HGSOC cells and patient tissue samples with a significant prognostic value. Pharmacological inhibition of PKCί by Na-aurothiomalate or its shRNA-mediated genetic knockdown suppressed HGSOC cell proliferation, EMT and induced apoptosis. Moreover, PKCί positively regulated GLUT1 and several other glycolytic genes including HK1, HK2, PGK1, ENO1 and LDHA to promote elevated glucose uptake and glycolysis in HGSOC cells. Mechanistically, PKCί drove glycolysis via PI3K/AKT/mTOR signalling. Na-aurothiomalate and highly selective, dual PI3K/mTOR inhibitor dactolisib could serve as novel anti-glycolytic drugs in HGSOC.
Conclusion
Taken together, our results indicate PKCί/PI3K/AKT/mTOR signalling cascade could be a novel therapeutic target in a lethal pathology like HGSOC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High grade serous ovarian cancer (HGSOC) is the deadliest gynecological malignancy which often displays high frequency of patient mortality [1]. Around 25–30% of all HGSOC are associated with germline mutations in BRCA1/2 genes and almost all cases of this lethal pathology harbor mutations in TP53 gene. Choice of treatment of HGSOC involves surgical debulking of tumor mass followed by chemotherapy using several drugs including cisplatin, carboplatin and paclitaxel [2]. Several drugs such as anti-angiogenesis molecules (bevacizumab, trebananib etc.), poly ADP-ribose polymerase (PARP) inhibitors (olaparib, niraparib etc.) have been developed against HGSOC and shown some promise to better manage patient health and survival outcome. Nonetheless, disease recurrence and therapy resistance hinder effective management of this lethal malady. Hence, identification of novel early detection biomarkers and therapeutic targets is highly warranted to manage this lethal pathology. Protein kinase C (PKC) family of serine/threonine kinases is categorized as classical (α, β, γ), novel (δ, ε, η, θ), atypical (ι, ζ) based on second messenger requirements and atypical PKCι is designated as classical oncogene in many human cancers [3, 4]. Emerging evidence suggests that aberrantly expressed PKCι modulates several signalling molecules and pathways including Yes-associated protein (YAP), vimentin, Rac1-MEK/ERK1/2 signaling axis and contributes to tumorigenesis of many major organs such as lung, pancreas and prostate [5,6,7]. Moreover, PKCι was shown to positively regulate glucose metabolism in non-small cell lung carcinoma in a Glut1/Myc signalling-dependent manner [8]. Therefore, several small molecule inhibitors including antirheumatoid agent aurothiomalate (ATM) have been developed against PKCι [9,10,11]. Although integrative genome analysis identified PKCι as a potential biomarker and oncogene in HGSOC and it was shown that PKCι positively drives HGSOC by regulating several signalling cascades such as YAP1, Cyclin E, Ect2 and MEK/ERK pathways [12,13,14,15], its role in regulating glycolysis and glucose metabolism in HGSOC remains unclear. Our results indicated that PKCι is overexpressed in HGSOC cells, tissue samples and its pharmacological inhibition by ATM or genetic knockdown hindered HGSOC cell glycolysis and glucose metabolism in a PI3K/AKT/mTOR-dependent manner. Taken together, we concluded that PKCι is a prognostic biomarker in EOC which positively drives glycolysis via PI3K/AKT/mTOR signalling and PKCι/PI3K/AKT/mTOR signalling axis is a novel therapeutic target in a lethal pathology like HGSOC (table 1.
Materials and methods
Cell culture, cell transfection and tissue sample collection
High grade serous ovarian cancer (HGSOC) cells SKOV3 was obtained from National Centre for Cell Science (NCCS), India. HGSOC cell COV362 was kindly gifted by Dr. Pallavi Agarwal, Amity University Noida. All cell lines were authenticated using STR profiling. Cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Brazil), 1x penicillin/streptomycin and maintained in humidified incubator containing 5% CO2 at 37 °C. We routinely checked the cells for mycoplasma contamination using MycoAlert Plus Mycoplasma detection kit (Lonza). Non-targeting control shRNA (SHC016), shRNAs against PRKCI (TRCN0000006037 and TRCN0000006039, Sigma), a plasmid ectopically expressing PKCι (PKCι-HA, Addgene Cat # 21252) were transfected in EOC cells using lipofectamine 3000 according to manufacturer’s protocol. Normal ovarian and tumor tissue samples were obtained from Rajiv Gandhi Cancer Institute & Research Centre, India. Ethical clearance was approved by the institutional review board. The clinicopathological data of the patient samples are as follows:
Cell proliferation, clonogenic and migration assays
HGSOC cells were plated in 96-well plate, subjected to treatment with varying concentrations of ATM, dactolisib or shRNAs directed against PKCι and MTT assay was performed to analyze cell proliferation. Clonogenic assay was performed by seeding cells in 6-well plate followed by aforementioned treatments. Colonies were fixed with 100% methanol, stained with 0.25% crystal violet and counted. Migration of EOC cells was analyzed by scratch assay. Briefly, EOC cells were plated in 24-well dishes to near confluency, a wound was created followed by treatment of 1.5 µM of ATM and 2 nM of Dactolisib and wound closure was monitored in a time-dependent manner. For cell proliferation, clonogenic and migration assays, HGSOC cells were treated with ATM and dactolisib for 72 h, 10 days and 48 h, respectively.
Western blotting, immunofluorescence and immunohistochemistry
Western blotting was performed as described previously [16]. Briefly, whole cell extract was prepared by lysing the cells in ice cold RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% triton X-100, 1% Na-deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors (Thermo Fisher, USA). 20 µg of total protein was resolved in SDS-PAGE, transferred to nitrocellulose membrane and the immunoblots were incubated with designated primary antibodies. Finally, the immunoblots were developed using ECL chemiluminescence substrate (Pierce, USA). For immunofluorescence assay, cells were grown in poly-lysine coated coverslips, fixed in 4% paraformaldehyde, washed thrice with 1X ice cold PBS, incubated with anti-PKCι primary antibodies overnight at 4 °C. Furthermore, the coverslips were incubated with anti-rabbit secondary antibody conjugated with Alexa Fluor-600, counterstained with DAPI and the images were analyzed in Leica fluorescent microscope fitted with 63X objective. Glass slides containing the sections of normal ovarian and tumor tissues were subjected to immunohistochemical assay. Briefly, slides were deparaffinized, rehydrated and antigens were retrieved by heating the slides in 100 mM Na-citrate buffer pH 6 followed by blocking and incubation with anti-PKCι antibody overnight at 4 °C. Moreover, the slides were incubated with anti-rabbit secondary antibody, dehydrated, stained with DAB substrate (Thermo Fisher, USA), counterstained with haematoxylin and mounted with DPX. The images were analyzed in Leica fluorescent microscope fitted with 20X objective. Tissue staining intensity and % of cells stained positive for PKCί were measured by semiquantitative H score as described previously [17]. Briefly, H score was obtained by the equation, 3 × the percentage of solid-staining nuclei (score at 3) + 2 × the percentage of opaque-staining nuclei (score at 2) + the percentage of translucent-staining nuclei (score at 1), yielding results in a range of 0 to 300.
RNA isolation, qRT-PCR
2 µg of total RNA was isolated using Trizol reagent (Life Technologies, USA), reversed transcribed to cDNA using iScript cDNA synthesis kit (Bio-Rad, USA) and qRT-PCR analysis was performed using iTaq SYBR Green Supermix (Bio- Rad, USA) using Step One Plus qRT-PCR thermal cycler (Applied Biosystems).
Apoptosis assay
Apoptosis assay was performed as described earlier [16]. Briefly, treated HGSOC cells were trypsinized, stained with Annexin V-Alexa fluor 488 antibody and propidium iodide (Molecular Probes, Thermo Fisher Scientific, USA) according to manufacturer’s instructions. We analyzed the cells in BD Accuri (BD Biosciences) flow cytometer and the results were analyzed in BD Accuri C6 software.
Statistical analysis
Data were represented as mean of three independent experiments ± S.D. Statistical significance was analyzed using the student t-test for comparison between two groups (two-tailed) and results from more than two groups were analyzed by ANOVA. All statistical analyses were performed using Graph Pad Prism IX software.
Results
PKCι is a prognostic marker in high grade serous ovarian cancer (HGSOC)
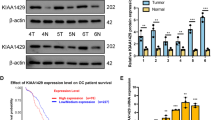
To address expression profile and copy number variations (CNVs) of PKCι in HGSOC, we analyzed 1949 human patient samples from the cancer genome atlas (TCGA) and observed that PRKCI gene which encodes PKCι protein, is amplified up to 24% in ovarian cancer tissue samples (Fig. 1A). Analysis from GEPIA2 online tool which links expression databases with TCGA and GTEx, also showed significant overexpression of PKCι in ovarian cancer tissue samples as compared to normal tissues (Fig. 1B). Elevated expression of PKCι was found to be associated with poor survival of HGSOC patients (Fig. 1C). Next, we assessed expression of PKCι in two HGSOC cell lines SKOV3 and COV362 by both Western blotting and qRT-PCR. We found that as compared to HeK293, a non-cancerous human epithelial cell, SKOV3 and COV362 expressed significantly higher PKCι at both mRNA and protein levels (Fig. 1D). Subcellular localization of PKCι in SKOV3 and COV362 by immunofluorescence assay showed that PKCι was mostly localized in the cytoplasm (Fig. 1E). Immunohistochemical analysis revealed that PKCι expression was significantly higher in HGSOC tissue samples as compared to normal ovarian tissue (Fig. 1F). Taken together, our results indicated that PKCι is overexpressed in HGSOC and it my serve as a novel prognostic biomarker in this lethal pathology.

PKCί is overexpressed and associated with poor prognosis in high grade serous ovarian cancer. (A). TCGA data analysis of genetic alteration of PRKCI gene in high grade serous ovarian cancer (HGSOC) is shown. (B) Differential expression box plot of PKCί in HGSOC obtained from TCGA and GEPIA is shown. (C) Disease-free survival of HGSOC patients overexpressing PKCί by Kaplan-Meier analysis is shown. (D) Expression of PKCί in designated cell lines was analyzed by Western blotting (left panel) and qRT-PCR (right panel). (E) Subcellular localization of PKCί in HGSOC cells COV362 and SKOV3 was analyzed by immunofluorescence technique. (F) Normal ovarian tissue and HGSOC patient tissue samples were analyzed for PKCί expression using immunohistochemistry (left panel). H-scoring was performed to quantify PKCί expression. Actin served as loading control in D. The graph represents average of at least three independent experiments with error bars representing SEM (***, P < 0.001; ****, P < 0.0001)
Pharmacological inhibition of PKCί suppresses cell proliferation, EMT and induces apoptosis in HGSOC cells
Next, to assess the inhibitory effect of PKCί in HGSOC cells COV362 and SKOV3, we pharmacologically inhibited it by a highly selective, potent small molecule inhibitor Na-aurothiomalate (ATM, Fig. 2A, top panel) for 72 h and examined the cytotoxic effect the inhibitor on HGSOC cells by MTT assay. It was revealed that ATM suppressed HGSOC cell proliferation in a dose-dependent manner and the IC50 value of ATM was 1.5 ± 0.14 µM and 1.47 ± 0.09 µM for COV362 and SKOV3, respectively (Fig. 2A, bottom panel). We validated these results by genetic knockdown of PKCί using shRNAs in COV362 and SKOV3 cells. In line with the previous observations, shRNA-mediated knockdown of PKCί significantly suppressed HGSOC cell proliferation as analyzed by MTT assay (Fig. 2B). Furthermore, treatment of 1.5 µM of ATM for 10 days significantly suppressed colony forming potential of HGSOC cells as analyzed by clonogenic assay (Fig. 2C). As inhibition of PKCί significantly suppressed HGSOC cell proliferation and ATM rendered its cytotoxic effect at micromolar range, we questioned if inhibition of PKCί by ATM could induce apoptosis in COV362 and SKOV3 cells. We treated the cells with 1.5 µM of ATM for 16 h, stained with Annexin V conjugated with Alexa fluor 488 antibody, propidium iodide and subjected the cells to flow cytometry. Indeed, pharmacological inhibition of PKCί significantly induced apoptosis in HGSOC cells COV362 and SKOV3 (Fig. 2D). Emerging evidences suggest that PKCί signalling contributes to epithelial-to-mesenchymal transition (EMT) and metastasis of several human cancers [3, 18]. To corroborate these conclusions in our HGSOC cellular model system, we treated COV362 and SKOV3 cells with 1.5 µM of ATM for 48 h and monitored migratory potentials of the cells by wound healing assay in a time-dependent manner. We found that ATM significantly inhibited wound closure in both cell lines (Fig. 2E). Moreover, 16 h of treatment of ATM significantly suppressed the expressions of EMT markers (N-cadherin and vimentin) in HGSOC cells at both mRNA and protein levels as analyzed by qRT-PCR and Western blotting (Fig. 2F). Taken together, our data indicated that PKCί positively regulated HGSOC cell proliferation, migration, EMT and pharmacological inhibition of PKCί induced apoptosis in COV362, SKOV3 cells.

Pharmacological inhibition of PKCί suppresses HGSOC cell proliferation, migration and induces apoptosis. (A) Chemical structure of PKCί inhibitor Na-aurothiomalate (ATM) is shown (top panel). COV362 and SKOV3 cells were treated with varying concentrations of ATM for 72 h and cell cytotoxicity was measured by MTT assay. (B) PKCί was knocked down using 2 shRNAs and cell proliferation was analyzed by MTT assay. (C) Colony forming potential of COV362 and SKOV3 cells upon ATM-mediated inhibition of PKCί was analyzed by clonogenic assay. Cells were treated with ATM for 10 days and clonogenic assay was performed. (D) HGSOC cells were treated with 1.5 µM of ATM for 16 h, stained with Annexin V-Alexa fluor 488 antibody, propidium iodide and apoptosis was measured by flow cytometry. (E) HGSOC cells were treated as described in D and wound healing assay was performed. (F) HGSOC cells were treated as described in D and expressions of designated EMT markers were measured by qRT-PCR (left panel) and Western blotting (right panel). Actin served as loading control in F. The graph represents average of at least three independent experiments with error bars representing SEM (*, P < 0.5; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001)
PKCί positively drives glycolysis via PI3K/AKT/mTOR signalling cascade
Recent reports suggest that PKC signalling promotes glucose uptake and glycolysis to positively drive several cellular and physiological functions including cell proliferation [8, 19, 20]. We questioned if PKCί modulated elevated glucose transport and high propensity of glycolysis, hallmarks of cancer [21] in HGSOC cells that positively drove aberrant cell proliferation. We treated COV362 and SKOV3 cells with 1.5 µM of ATM and checked the expressions of glucose transporter GLUT1 and several other glycolytic genes including HK1, HK2, PGK1, ENO1 and LDHA by both qRT-PCR and Western blotting. It was evident that pharmacological inhibition of PKCί by ATM significantly suppressed expressions of these genes at both mRNA and protein levels-implying that PKCί indeed positively drives glycolysis in HGSOC (Fig. 3A-C). We confirmed this result by shRNA-mediated knockdown of PKCί in COV362 and SKOV3 cells as well (Fig. 3B-C). Furthermore, ATM-mediated inhibition or shRNA-mediated genetic knockdown of PKCί significantly suppressed glucose transport in HGSOC cells as analyzed by 2-deoxy glucose (2DG)-6-phosphate uptake assay (Fig. 3D). To corroborate these findings, we ectopically overexpressed PKCί from a plasmid (PKCί-HA) in COV362, SKOV3 cells and checked if overexpression of PKCί could rescue ATM or shRNA-mediated suppression of the observed glycolytic phenotypes. Indeed, overexpression of PKCί-HA in these two HGSOC cells significantly augmented expressions of GLUT1 and other glycolytic genes including HK1, HK2, PGK1, ENO1 and LDHA as analyzed by qRT-PCR and Western blotting (Fig. 3E). Next, we investigated the molecular basis of PKCί-mediated glucose uptake and glycolysis in HGSOC. Emerging evidence suggests that PI3K/AKT/mTOR pathway play a vital role in glucose metabolism of tumors and a regulatory association between PKCί and PI3K/AKT/mTOR signalling was established in osteosarcoma [15, 22, 23]. Therefore, we investigated if PKCί positively drove glucose uptake and glycolysis via PI3K/AKT/mTOR signalling in HGSOC. COV362 and SKOV3 cells were treated with 1.5 µM of ATM or shRNAs directed against PKCί and expression levels of components of PI3K/AKT/mTOR signalling pathway were analyzed by Western blotting. Indeed, catalytic inactivation or genetic knockdown of PKCί significantly suppressed expressions of phospho-AKT, phospho-mTOR (Fig. 3F), suggesting that PKCί positively regulated HGSOC cell proliferation, migration by modulating glucose uptake and glycolysis via PI3K/AKT/mTOR signalling.
Furthermore, to validate translational significance of PKCί/PI3K/AKT/mTOR signalling axis in regulating elevated glucose uptake and glycolysis in HGSOC, we inhibited PI3K/AKT/mTOR signalling by treating COV362 and SKOV3 cells with an imidazoquinolinone derivative dactolisib, a highly potent, dual inhibitor of PI3K/mTOR (Fig. 4A) and checked the expressions of GLUT1 and other glycolytic genes including HK1, HK2, PGK1, ENO1 and LDHA as analyzed by qRT-PCR and Western blotting. Treatment of dactolisib for 72 h was shown to be cytotoxic to HGSOC cells at nanomolar range with IC50 values were 1.96 ± 0.09 nM and 2.12 ± 0.13 nM for COV362 and SKOV3 respectively, as analyzed by MTT assay (Fig. 4B). Treating HGSOC cells with 2 nM dactolisib for 10 days significantly suppressed colony formation as measured by clonogenic assay (Fig. 4C) and importantly, expressions of GLUT1 and other glycolytic genes including HK1, HK2, PGK1, ENO1 and LDHA were also suppressed by treatment of dactolisib for 16 h as analyzed by qRT-PCR and Western blotting (Fig. 4D-E). Taken together, our results showed that PKCί positively regulated HGSOC cell proliferation, migration by modulating elevated glucose uptake and glucolysis via PI3K/AKT/mTOR signalling and pharmacological inhibition of PKCί/PI3K/AKT/mTOR signalling axis could serve as a novel therapeutic choice to manage a lethal pathology like HGSOC.

PKCί positively regulates glucose uptake and glycolysis in HGSOC. A-C. COV362 and SKOV3 cells were either treated with 1.5 µM of ATM or transfected with shRNAs directed against PKCί and expressions of Glut1 and indicated glycolytic enzymes were analyzed by qRT-PCR and Western blotting. D. COV362 and SKOV3 cells were treated as described in A and 2-deoxy-glucose-6P uptake was measured. E. PKCί was ectopically overexpressed from a plasmid (PKCί-HA) in designated HGSOC cells and expressions of Glut1 and indicated glycolytic enzymes were analyzed by qRT-PCR and Western blotting. F. HGSOC cells were treated as described in A and whole cell lysates were analyzed by Western blotting using indicated antibodies. Actin served as loading control in F. The graph represents average of at least three independent experiments with error bars representing SEM (*, P < 0.5; **, P < 0.01; ***, P < 0.001, ****, P < 0.0001)

Pharmacological inhibition of PI3K/AKT/mTOR signalling suppresses glycolysis in HGSOC. (A) Chemical structure of highly selective, dual PI3K/mTOR inhibitor dactolisib is shown. (B) COV362 and SKOV3 cells were treated with varying concentrations of dactolisib for 72 h and cell cytotoxicity was measured by MTT assay. (C) HGSOC cells were treated with dactolisib for 10 days and colony forming potential of COV362 and SKOV3 cells upon dactolisib-mediated inhibition of PI3K/mTOR was analyzed by clonogenic assay. (D-E) COV362 and SKOV3 cell were treated with 2 nM of dactolisib for 16 h and expressions of Glut1 and indicated glycolytic enzymes were analyzed by qRT-PCR and Western blotting. (F) Cartoon signifying the positive role of PKCί/PI3KAKT/mTOR signalling cascade in glucose uptake and glycolysis in HGSOC is shown
Discussion
High grade serous ovarian cancer (HGSOC) is the deadliest gynecological cancer worldwide. Although application of several drugs including carboplatin, paclitaxel and development of several novel drugs including PARP inhibitors such as olaparib initially improve patient outcome, HGSOC remains as leading cause of cancer-related death due to chemoresistance and high rate of disease relapse in females.
Tumor cells reprogram energy metabolism and heavily rely on aerobic glycolysis for energy production even in the presence of oxygen, and this aerobic glycolysis is a known modulator of tumorigenesis and tumor development. The tumor microenvironment (TME) is composed of tumor cells, various immune cells, cytokines, and extracellular matrix, among other factors, and is a complex niche supporting the survival and development of tumor cells and through which they interact and co-evolve with other tumor cells. In addition, lactic acid, a metabolite of glycolysis, can also accumulate in the TME, leading to reduced extracellular pH and immunosuppression, and affecting the TME [24]. Like many other tumors, HGSOC cells prefer aerobic glycolysis as an energy metabolism pathway. Via elevated glucose uptake and glycolysis, tumor cells convert glucose into lactic acid to greatly produce energy and promote tumor progression [25]. Alterations of glycolytic activity, modulation of key rate-limiting enzymes such as hexokinases, signalling molecules and pathways including PI3K/AKT, HIF1α play a critical role in tumor cell growth, proliferation and therapy response. Therefore, discovery of novel glycolytic modulators and understanding their molecular basis might shed light on identifying improved opportunities for better management of HGSOC.
PKCί belongs to the family of atypical PKC which are independent of requirement of Ca++ and do not require diacylglycerol (DAG) or phorbol esters for activation [4]. Overexpression and aberrant activation of PKCί is involved in tumorigenesis of many organs including lung and its oncogenic role majorly relies on post-translational modification of other proteins [3]. For example, PKCί-dependent phosphorylation and nuclear translocation of YAP1 promotes ovarian cancer [26]. Recent reports suggest possible involvement of PKCί in glucose uptake and glycolysis. Dejure et al. demonstrated that PKCί directly activates Glut1 via FoxO1/c-Myc signalling and promotes glycolysis [27]. Liu et al. showed that overexpression of PKCί elevates Glut1 expression via c-Myc activation and promotes glycolysis to drive non-small cell lung cancer [8]. In line with the previous results, our studies found that PKCί is significantly overexpressed in HGSOC cells and tumor tissue samples as compared to normal control and high expression of PKCί positively correlates with poor patient survival-indicating a prognostic value of this classic oncogene in HGSOC (Fig. 1). We have pharmacologically inhibited PKCί using Na-aurothiomalate and showed that its inactivation suppressed HGSOC cell proliferation, migration, EMT and induced apoptosis (Fig. 2). Most importantly, Na-aurothiomalate-mediated inhibition of PKCί significantly suppressed expressions of Glut1 and other major glycolytic enzymes such as HK1, HK2, PGK1, ENO1 and LDHA. shRNA-mediated genetic knockdown of PKCί also attested above mentioned observations in HGSOC cells COV362 and SKOV3 (Fig. 3). Importantly, ectopic overexpression of PKCί in these cells rescued Na-aurothiomalate or shPKCί-mediated glycolytic defect in HGSOC cells, implying that indeed, PKCί positively drives glucose uptake and glycolysis in HGSOC. Mechanistically, we demonstrated that PKCί drives glycolysis in a PI3K/AKT/mTOR signalling in COV362 and SKOV3 cells. Dual inhibition of PI3K and mTOR using highly potent small molecule dactolisib hindered glycolytic flux, suppressed proliferation of COV362 and SKOV3 cells (Fig. 4). These observations are in line with the notion of deep involvement of PI3K/AKT/mTOR signalling in glucose metabolism of cancer cells [22]. However, several caveats still remain in this study which should be clearly addressed in future. For example, a clear understanding of the molecular mechanism of putative inhibition of cellular glycolysis by dactolisib should be elucidated. Several assays including 2-NBDG uptake assay should be performed to monitor PKCί-regulated glucose uptake and glycolysis in the cells. In future, an attempt to design highly potent, small molecule inhibitors of PKCί/PI3K/AKT/mTOR signalling cascade should be made to inhibit glycolysis in HGSOC.
Conclusion
Taken together, our results indicate that PKCί is a driver of glycolysis in HGSOC and PKCί/PI3K/AKT/mTOR signalling cascade (Fig. 4F) could serve as a novel therapeutic target in HGSOC.
Data availability
No datasets were generated or analysed during the current study.
References
Desai A, Xu J, Aysola K, Qin Y, Okoli C, Hariprasad R, Chinemerem U, Gates C, Reddy A, Danner O, Franklin G, Ngozi A, Cantuaria G, Singh K, Grizzle W, Landen C, Partridge EE, Rice VM, Reddy ESP, Rao VN (2014) Epithelial ovarian cancer: an overview. World J Transl Med 3:1
Pignata S, Cecere SC, Du Bois A, Harter P, Heitz F (2017) Treatment of recurrent ovarian cancer. Ann Oncol 28:viii51–viii56
Fields AP, Regala RP (2007) Protein kinase C iota: human oncogene, prognostic marker and therapeutic target. Pharmacol Res 55:487–497
Tyagi K, Roy A (2021) Evaluating the current status of protein kinase C (PKC)-protein kinase D (PKD) signalling axis as a novel therapeutic target in ovarian cancer, Biochim. Biophys. Acta - Rev Cancer, 1875
Kim KH, Chung C, Kim JM, Lee D, Cho SY, Lee TH, Cho HJ, Yeo MK (2019) Clinical significance of atypical protein kinase C (PKCι and PKCζ) and its relationship with yes-associated protein in lung adenocarcinoma. BMC Cancer
Ratnayake WS, Apostolatos CA, Breedy S, Dennison CL, Hill R, Acevedo-Duncan M (2021) Atypical PKCs activate Vimentin to facilitate prostate cancer cell motility and invasion. Cell Adh Migr 15:37–57
Scotti ML, Bamlet WR, Smyrk TC, Fields AP, Murray NR (2010) Protein Kinase C Iota is Required for Pancreatic Cancer Cell Transformed Growth and Tumorigenesis, Cancer Res., 70 2064
Liu L, Lei B, Wang L, Chang C, Yang H, Liu J, Huang G, Xie W (2019) Protein kinase C-iota-mediated glycolysis promotes non-small-cell lung cancer progression. Onco Targets Ther 12:5835
Kwiatkowski J, Baburajendran N, Poulsen A, Liu B, Tee DHY, Wong YX, Poh ZY, Ong EHQ, Dinie N, Cherian J, Jansson AE, Hill J, Keller TH, Hung AW (2019) Fragment-based Discovery of a small-molecule protein kinase C-iota inhibitor binding post-kinase domain residues, ACS Med. Chem Lett 10:318–323
Mansfield AS, Fields AP, Jatoi A, Qi Y, Adjei AA, Erlichman C, Molina JR (2013) Phase I dose escalation study of the protein kinase C iota inhibitor aurothiomalate for advanced non-small cell lung cancer, ovarian cancer, and pancreatic cancer. Https://Doi 31:2551–2551. https://doi.org/10.1200/Jco.2013.31.15_suppl.2551
Regala RP, Thompson EA, Fields AP (2008) Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res 68:5888–5895
Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, Cadungog MG, O’Brien-Jenkins A, Massobrio M, Roby KF, Katsaros D, Gimotty P, Butzow R, Weber BL, Coukos G (2006) Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res 66:4627–4635
Wang Y, Hill KS, Fields AP (2013) PKCι maintains a tumor-initiating cell phenotype that is required for ovarian tumorigenesis. Mol Cancer Res 11:1624–1635
Wang Y, Justilien V, Brennan KI, Jamieson L, Murray NR, Fields AP (2017) PKCι regulates nuclear YAP1 localization and ovarian cancer tumorigenesis, Oncogene
Nanos-Webb A, Bui T, Karakas C, Zhang D, Carey JPW, Mills GB, Hunt KK, Keyomarsi K (2016) PKC iota promotes ovarian tumor progression through deregulation of cyclin E, Oncogene, 35 2428
Tyagi K, Roy A, Mandal S (2023) Pharmacological inhibition of protein kinase D suppresses epithelial ovarian cancer via MAPK/ERK1/2/Runx2 signalling axis. Cell Signal, 110
Kumar V, Yu J, Phan V, Tudor IC, Peterson A, Uppal H (2017) Androgen receptor immunohistochemistry as a Companion Diagnostic Approach to predict clinical response to Enzalutamide in Triple-negative breast Cancer. 1–19. https://doi.org/10.1200/PO.17.00075
Murray NR, Kalari KR, Fields AP (2011) Protein kinase Cι expression and oncogenic signaling mechanisms in cancer. J Cell Physiol
Leung SWS, Shi Y (2021) 432 The glycolytic process in endothelial cells and its implications, Acta Pharmacol. Sin. 43 (2021) 251–259
Pan D, Xu L, Guo M (2022) The role of protein kinase C in diabetic microvascular complications. Front Endocrinol (Lausanne), 13
Kroemer G, Pouyssegur J (2008) Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 13:472–482
Hoxhaj G, Manning BD (2020) The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 20:74
Qu L, Li G, Xia D, Hongdu B, Xu C, Lin X, Chen Y (2016) PRKCI negatively regulates autophagy via PIK3CA/AKT-MTOR signaling, Biochem. Biophys Res Commun 470:306–312
Zhou D, Duan Z, Li Z, Ge F, Wei R, Kong L (2022) The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front Pharmacol, 13
Li C, Liu FY, Shen Y, Tian Y, Han FJ (2023) Research progress on the mechanism of glycolysis in ovarian cancer. Front Immunol, 14
Wang Y, Justilien V, Brennan KI, Jamieson L, Murray NR, Fields AP (2017) PKCι regulates nuclear YAP1 localization and ovarian cancer tumorigenesis. Oncogene 36:534–545
Dejure FR, Eilers M (2017) MYC and tumor metabolism: chicken and egg. EMBO J 36:3409–3420
Funding
This work was supported by the Ramalingaswami Re-entry fellowship Grant (BT/HRD/35/02/2006), Department of Biotechnology, Govt. of India to AR.
Author information
Authors and Affiliations
Contributions
K.T. and A.R. wrote the main manuscript text. K.T. prepared all figures. S.M. reviewed the manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tyagi, K., Roy, A. & Mandal, S. Protein kinase C iota promotes glycolysis via PI3K/AKT/mTOR signalling in high grade serous ovarian cancer. Mol Biol Rep 51, 983 (2024). https://doi.org/10.1007/s11033-024-09918-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09918-3