
Abstract
Background
Cervical cancer ranks as the fourth most prevalent cancer among women globally, presenting a significant therapeutic challenge due to its resistance to cisplatin. Ephrin type-A receptor 2 (EPHA2) is prominently overexpressed in cervical cancer and plays a vital role in cisplatin resistance, although the underlying mechanisms remain incompletely elucidated. Mitochondrial dynamics, autophagy, and mitophagy are critical in mediating cisplatin resistance. Sesamol, a phytochemical compound, has exhibited promising anticancer properties. This study aims to investigate the regulatory role of EPHA2 in these pathways underlying cisplatin resistance and to investigate the potential of sesamol in overcoming this resistance and inhibiting cervical cancer progression.
Methods and result
In this study, we knocked down EPHA2 in the SiHa cell line and evaluated the resulting changes in molecular markers associated with mitochondrial dynamics, mitophagy, and autophagy. Our results indicated that EPHA2 knockdown (EPHA2-KD) led to enhanced mitochondrial fusion and reduced mitochondrial fission, mitophagy, and autophagy. Furthermore, we investigated the effect of EPHA2-KD and sesamol treatment on sensitising cervical cancer to cisplatin treatment. Our data revealed that EPHA2-KD and sesamol treatment significantly increases cellular sensitivity to cisplatin-induced cytotoxicity. Additionally, we demonstrated that sesamol effectively targets EPHA2, as evidenced by decreased EPHA2 expression levels following sesamol treatment.
Conclusion
In summary, targeting EPHA2 through knockdown or sesamol treatment enhances cisplatin sensitivity in cervical cancer by modulating mitochondrial dynamics, autophagy and mitophagy, suggesting promising therapeutic strategies to overcome chemoresistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cervical cancer is the fourth most common cancer among women worldwide [1]. Traditional treatments, including radiotherapy, chemotherapy, and surgery, face significant challenges as the disease progresses due to the increasing aggressiveness of cervical cancer cells. A major obstacle in effective treatment is overcoming both inherent and acquired drug resistance, which significantly hinders therapeutic success [2]. Despite the gravity of this issue, the intricate mechanisms through which cervical cancer cells orchestrate their resistance to chemotherapy remain insufficiently explored. The receptor tyrosine kinase EPHA2 is overexpressed in various cancers, including cervical cancer [3]. EPHA2 plays a crucial role in inducing chemoresistance. However, the mechanisms by which EPHA2 contributes to chemoresistance in cervical cancer are not well understood [4].
Mitochondria is a dynamic organelle essential for maintaining cellular homeostasis. Their dynamic behaviour is controlled by fission and fusion processes. Mitochondrial fission is when mitochondria divide or segregate into two separate mitochondrial organelles. Mitochondrial fission involves the recruitment of Drp1 to the outer mitochondrial membrane through receptors such as Fis1, MFF, and MIEF1/MIEF2, forming a constriction site where GTP hydrolysis drives mitochondrial division. Mitochondrial fusion is a cellular process where individual mitochondria merge their membranes, facilitated by specific proteins known as mitofusins (Mfn1 and Mfn2) and OPA1. Mitofusins are responsible for merging the outer membranes of mitochondria, while OPA1 acts on the inner mitochondrial membranes [5]. Irregularities in mitochondrial dynamics can cause tumour cells to develop both acquired and adaptive resistance to chemotherapeutic drugs, leading to significant variability in their response to chemotherapy [6].
Autophagy is a cellular process that degrades and recycles cellular components, including damaged organelles and proteins, to maintain cellular homeostasis. This mechanism is crucial for cell survival during stress, nutrient deprivation, or toxin exposure. Key molecules involved in autophagy include Beclin 1, ATG proteins (such as ATG12, ATG5, and ATG16), LC3, LAMP-2 A, ULK1, and VPS34, which collectively regulate the initiation, formation, and maturation of autophagosomes, as well as the degradation of cellular components within lysosomes. Autophagy protects cancer cells from chemotherapy, which makes them drug-resistant. It is reported that knocking down ATGs and Beclin 1 will inhibit autophagy and sensitise drug-resistant cancers [7].
Mitophagy, the selective degradation of mitochondria via autophagy, plays a crucial role in the complex landscape of chemoresistance. Initially, mitophagy supports normal cellular metabolism and helps suppress tumorigenesis. However, prolonged chemotherapy induces mitophagy, fostering tolerance in tumour cells and endowing them with stemness characteristics, ultimately leading to chemoresistance. In mitophagy, the E3 ubiquitin ligase Parkin is recruited and activated by the mitochondrial kinase Pink1 upon mitochondrial depolarization. This activation leads to the ubiquitination of mitochondrial proteins, triggering mitophagy. Receptor-mediated mitophagy selectively targets and removes damaged or dysfunctional mitochondria from the cell, with BNIP3L/NIX acting as a receptor by binding to autophagosomal proteins like LC3, recruiting the damaged mitochondria for degradation [8].
Cisplatin, or cis-diamminedichloroplatinum (II), is a chemotherapeutic agent for treating various cancers, including bladder, head and neck, lung, ovarian, and testicular. However, despite its effectiveness, cisplatin treatment often encounters issues with drug resistance and numerous undesirable side effects. The clinical community recognises the potential of combining cisplatin with other drugs to address drug resistance and minimise toxicity. Such combination therapies aim to optimise treatment outcomes while reducing the adverse reactions associated with cisplatin monotherapy [9].
Sesamol, a natural compound found in sesame seeds and oil, has attracted attention for its therapeutic properties, including antioxidant, anti-inflammatory, and anti-cancer effects. Research into sesamol’s role as a metabolic regulator positions it as a promising candidate to combat chemoresistance in cervical cancer and other malignancies [10, 11].
In this study, we aim to investigate the regulatory role of EPHA2 on mitochondrial dynamics, autophagy, and mitophagy. Additionally, we aim to investigate the impact of EPHA2-KD on sensitising cervical cancer to cisplatin treatment. Furthermore, we explore the modulatory effect of sesamol on EPHA2 expression and its potential role in regulating sensitivity to cisplatin treatment. This exploration opens doors to innovative therapeutic approaches, offering a glimpse into novel strategies to overcome drug resistance challenges in treating cervical cancer.
Materials and methods
Cell culture
The human SiHa cell line was obtained from NCCS Pune (National Centre for Cell Science). Cells were cultivated in T25 flasks containing Dulbecco’s Modified Eagle Medium (DMEM), enriched with 10% FBS (Gibco, USA), and maintained under standard conditions at 37 °C with a 5% CO2 atmosphere.
EPHA2-KD plasmid transfection
We used transfection of pcDNA-EPHA2 to mimic intracellular EPHA2 levels. We performed the transfection using Lipofectamine transfection reagent (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer’s protocol.
Western blot
For Western blotting, we lysed the cells using lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, and 1 µg/ml leupeptin), adding 1 mM PMSF immediately before use (Cell Signaling Technology). We separated the total proteins on SDS-PAGE gel and transferred them to a polyvinyl difluoride (PVDF) membrane using the Bio-Rad Mini Trans-Blot® system. After that, we processed the blots according to standard Western blotting protocols. We obtained the antibodies from Abcam (Drp1, Fis1, Mfn1, Mfn2) and Cell Signaling Technology (β-actin, Parkin, BNIP3, Pink1, EPHA2).
Semi quantitative reverse transcription PCR (RT PCR)
We extracted total RNA from the cells using the TRIzol method (QIAzol Lysis Reagent, QIAGEN) and quantified it with the NanoDrop™ One/OneC Microvolume UV-Vis Spectrophotometer (Thermo Scientific, Catalog number: ND-ONE-W). We converted the RNA samples to cDNA using the QuantiTect Reverse Transcription Kit (QIAGEN, Cat. No. / ID: 205311). Next, we amplified the cDNA from various cell samples using specific primers, ran the amplified products on a 1% agarose gel, and quantified them using ImageJ software. The primers used for semi-quantitative RT-PCR are listed in Table 1.
MTT
We seeded 5000 cells in a 96-well plate and, after 24 h, performed three different MTT assays. In the first assay, we treated the cells (including both control and EPHA2-KD cell lines) with various concentrations of cisplatin (5 µM, 10 µM, 20 µM, 50 µM, 80 µM, 100 µM). In the second MTT assay, we treated SiHa cells with different concentrations of sesamol (0.1 mM, 0.25 mM, 0.5 mM, 1 mM, 2.5 mM, 5 mM). In the third MTT assay, we treated SiHa cells with different concentrations of cisplatin (5 µM, 10 µM, 20 µM, 50 µM, 80 µM, 100 µM) along with 2 mM of sesamol. We incubated the cells with these treatments for 48 h. After incubation, we replaced the treatment media with MTT-containing media and incubated for 3–4 h. Next, we removed the MTT-containing media and added DMSO to solubilise the crystal formazan. Finally, we measured the absorbance at 570 nm. We obtained cisplatin (Product Number: D3371) and sesamol (Product Number: S0418) from TCI Chemicals.
Colony formation assay
We seeded 20,000 cells in a 60 mm Petri plate (control and EPHA2-KD cell lines). After the cells attained the desired morphology, we treated them with the appropriate drugs (Cisplatin-20 ± 1.04 µM, Cisplatin-22.14 ± 0.5 µM + Sesamol 2 mM, Sesamol 4.8 ± 0.5 mM). After 48 h, we replaced the drug-containing media with normal media, allowing the cells to grow for 7–10 days. After 10 days, we fixed and stained the cells with crystal violet. We counted the colonies by selecting random squares on the plates and plotted a bar diagram using the average colony count data.
Migration assay
We seeded the cells (both control and EPHA2-KD cell lines) in a 6-well plate. Once the cells form a monolayer, we create a cell-free gap. We then treated the cells with the appropriate drugs (Cisplatin-20 ± 1.04 µM, Cisplatin-22.14 ± 0.5 µM + Sesamol 2 mM, Sesamol 4.8 ± 0.5 mM). We imaged the closure of the gap using an inverted microscope every 6 h for 48 h. We calculated the percentage of wound confluence by measuring the gap length with ImageJ software at 0 h (At0) and 48 h (Btx) using the specified formula.
% wound confluence = (At0- Btx) / At0 * 100.
Mito tracker red staining
We seeded the cells (both control and EPHA2-KD cell lines) in a 6-well plate and treated them with MitoTracker Red (M7512, Invitrogen) after they attained the desired morphology. We incubated the cells for 30 min and then visualised them using a fluorescent microscope with 20X magnification.
Docking
We obtained the EPHA2 PDB structure through homology modelling using SWISS-PROT and further processed it with BIOVIA Discovery Studio 2022 (BIOVIA Discovery Studio, Dassault Systems, San Diego, CA) [12]. We retrieved the SDF structure of sesamol from the PubChem database [13]. We performed the docking using PyRx (https://pyrx.sourceforge.io/) and then visualised the results with Discovery Studio (https://discover.3ds.com/discovery-studio-visualizer).
Statistical analysis
The results are presented as mean values ± standard deviation (SD). We performed statistical analysis using Student’s t-test for comparisons between two groups and one-way analysis of variance (ANOVA) for comparisons among more than two groups, utilising GraphPad Prism version 7.0 software (San Diego, USA). A p-value of less than 0.05 was regarded as statistically significant.
Result
EPHA2 as a regulator of mitochondrial dynamics
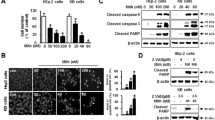
Mitochondrial dynamics play a vital role in cervical cancer progression; however, there have been no reports on the relationship between mitochondrial dynamics and EPHA2. To investigate the role of EPHA2 in mitochondrial dynamics, we first transiently transfected the SiHa cell line with an EPHA2 knockdown plasmid. The knockdown efficiency of EPHA2 protein in these cells was determined through western blot analysis and semi-quantitative RT-PCR (Fig. 1A and B). Fission and fusion are two crucial processes in mitochondrial dynamics. To examine the regulation of mitochondrial fission by EPHA2, we assessed the levels of Drp1, a key regulator of mitochondrial fission, and Fis1, a receptor for Drp1, at both the mRNA and protein levels in the EPHA2-KD cell line. EPHA2 knockdown decreased the Drp1 and Fis1 mRNA and protein levels (Fig. 1A and B). Additionally, we found that the expression level of MIEF1, a Drp1 receptor, was significantly reduced in the EPHA2-KD cell line compared to the control (Fig. 1B). These results indicate that EPHA2-KD reduces mitochondrial fission. To further analyse the potential role of EPHA2 in regulating mitochondrial fusion, we investigated the expression levels of Mfn1 and Mfn2, mitochondrial fusion regulators, in the EPHA2-KD cell line. EPHA2 knockdown increased the levels of Mfn1 and Mfn2 proteins. We also observed an increased expression level of Mfn1 mRNA in the EPHA2-KD cell line (Fig. 1A and B). These results indicate that EPHA2-KD enhances mitochondrial fusion. Evaluation of mitochondrial morphology using MitoTracker Red staining revealed a significant increase in mitochondrial fusion events within the EPHA2-KD cell line compared to the control (Fig. 1C). Collectively, the results suggest that EPHA2-KD increases mitochondrial fusion and decreases mitochondrial fission.
EPHA2 regulates mitophagy and autophagy
Autophagy plays a crucial role in the development and progression of cervical cancer. Mitophagy, a selective autophagic process, eliminates damaged or dysfunctional mitochondria. This process is vital in cervical cancer progression by enabling cancer cells to adapt to metabolic stress. However, no reports have been made on regulating mitophagy and autophagy by EPHA2. Beclin 1, ATG5, and LC3B are key activators of autophagy. To examine the regulation of EPHA2 on autophagy, we analysed the expression levels of Beclin 1, ATG5, and LC3B in the EPHA2-KD cell line. EPHA2 knockdown reduced the mRNA levels of Beclin 1, ATG5, and LC3B. The results indicate that EPHA2-KD reduces autophagy in cervical cancer (Fig. 2B). Additionally, to investigate the potential role of EPHA2 in regulating mitophagy, we examined the expression levels of Pink1, Parkin, and BNIP3, key activators of mitophagy (Fig. 2A). EPHA2 knockdown reduced the protein levels of Pink1, Parkin, and BNIP3, as well as the mRNA level of BNIP3 (Fig. 2A and B). This indicates that EPHA2-KD reduces mitophagy.
EPHA2 knockdown sensitises cervical cancer to cisplatin treatment
Mitochondrial dynamics, autophagy, and mitophagy play crucial roles in chemotherapy resistance to cisplatin in various cancers. Our previous findings revealed that EPHA2 regulates these processes. To investigate the sensitisation potential of EPHA2-KD towards cisplatin treatment, we performed an MTT assay for both control and EPHA2-KD cell lines. We found that the IC50 value for cisplatin was significantly reduced in the EPHA2-KD cell line (10.34 ± 1.25 µM) compared to the control (20 ± 1.04 µM), indicating that EPHA2-KD sensitises cells to cisplatin treatment (Fig. 3A). Increased cell proliferation and migration are hallmarks of cancer. We conducted colony formation and wound healing assays to explore the combined effects of EPHA2-KD and cisplatin on cancer cell migration and proliferation. The colony formation assay showed the following trend in average colony numbers: control > EPHA2-KD > cisplatin > EPHA2-KD + cisplatin (Fig. 3B). Similarly, the migration assay demonstrated a consistent pattern in migration potential: control > EPHA2-KD > cisplatin > EPHA2-KD + cisplatin (Fig. 3C). These results indicate that EPHA2-KD enhances the cisplatin inhibitory potential towards cancer cell migration and proliferation.
Sesamol sensitization for cisplatin treatment by regulating EPHA2 expression
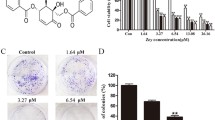
Sesamol, a plant-derived biomolecule with anticancer properties, was evaluated for its inhibitory effect on the SiHa cell line using an MTT assay, resulting in an IC50 value of 4.8 ± 0.5 mM (Fig. 4A). To explore an alternative to EPHA2 knockdown, we investigated sesamol’s sensitisation potential for cisplatin treatment. Western blot analysis showed that sesamol treatment at the IC50 concentration reduces EPHA2 expression (Fig. 4E). Docking analysis revealed a significant binding affinity of sesamol for EPHA2, with a binding energy of -9.34 kcal/mol. Within the EPHA2 protein’s binding pocket, sesamol forms two hydrogen bonds with Glycine 515 and Arginine 465, engages in an amide pi-stacking interaction with Leucine 402, and participates in Van der Waals interactions with Glutamate 403 (Fig. 4F). Our earlier results indicate that EPHA2-KD sensitises the SiHa cell line to cisplatin treatment. To further examine sesamol’s effect on cisplatin sensitivity, we co-administered cisplatin with sesamol (2 mM). This combination significantly reduced the IC50 value of cisplatin. While cisplatin alone had an IC50 value of 22.14 ± 0.5 µM, the combination with sesamol reduced it to 9.6 ± 0.43 µM. We also evaluated the impact of co-administering cisplatin and sesamol on cancer cell migration and proliferation (Fig. 4B). At their respective IC50 concentrations, sesamol (4.8 ± 0.5 mM) and cisplatin (20 ± 1.04 µM) exhibit almost equal inhibitory effects on cancer cell migration and proliferation, To achieve an equivalent inhibitory effect through combination therapy, sesamol needs to be administered at 2 mM and cisplatin at 9.6 ± 0.43 µM (Fig. 4C, D). These results indicated that sesamol treatment enhances the inhibitory potential of cisplatin for cancer cell migration and proliferation. In conclusion, sesamol effectively reduces EPHA2 expression, enhances cisplatin sensitivity, and inhibits cancer cell migration and proliferation in the SiHa cell line.
Discussion
Receptor tyrosine kinases (RTKs) regulate various cellular processes and downstream signalling pathways. EPHA2, a well-studied RTK, has been extensively researched in the context of cancer. It is often overexpressed in cervical cancer. Our study reveals that knocking down EPHA2 enhances mitochondrial fusion while reducing mitochondrial fission, mitophagy, and autophagy. Although cisplatin has been a cornerstone chemotherapy drug for numerous cancers over the decades, resistance to this drug remains a significant clinical challenge. EPHA2 is crucial in mediating chemoresistance, yet the underlying mechanisms are not fully understood. Given that mitochondrial dynamics, autophagy, and mitophagy are critical factors in cisplatin resistance, our findings indicate that EPHA2 knockdown sensitises the SiHa cell line to cisplatin treatment.
Mitochondrial fission causes cisplatin resistance under hypoxic conditions through reactive oxygen species in ovarian cancer cells [14]. Similarly, cisplatin-resistant gastric cancer cells show higher levels of mitophagy markers, mitochondrial fission, autophagy, and mitophagosomes compared to non-resistant cells [15]. In hepatocellular carcinoma, lowering MFF levels decreases Drp1 expression, making cells more susceptible to cisplatin [16]. Also, cisplatin-resistant ovarian and osteosarcoma cells exhibit a fragmented mitochondrial phenotype, characterised by increased levels of pro-fission proteins MFF, Fis1 and Drp1 [17]. Our research shows that knocking down EPHA2 reduces mitochondrial fission by lowering Drp1 and Fis1 expression, potentially increasing cisplatin sensitivity in the EPHA2-KD cell lines. Maintaining a balance between mitochondrial fission and fusion is crucial for cell function. Cisplatin-resistant ovarian and osteosarcoma cells exhibit decreased levels of fusion proteins Mfn1 and Opa1 [17]. We also examined the expression of mitochondrial fusion proteins Mfn1 and Mfn2 in EPHA2-KD cell lines and found their levels increased, suggesting that this could enhance cisplatin susceptibility. So, targeting mitochondrial dynamics through EPHA2 modulation offers a promising strategy to overcome cisplatin resistance in cervical cancer.
As indicated in existing literature, mitochondria undergo fragmentation to facilitate mitophagy, aiding in the removal of dysfunctional mitochondria by autophagosomes [18]. In hepatocellular carcinoma, inhibiting Drp1-mediated mitophagy by using Mdivi-1(Drp1 inhibitor) enhances sensitivity to cisplatin [19]. Similarly, cisplatin-resistant ovarian carcinoma and osteosarcoma cells exhibit increased mitochondrial fragmentation and elevated levels of the mitophagy receptor BNIP3. Silencing BNIP3 restores drug sensitivity in these resistant cells [17]. Additionally, research on cisplatin-resistant lung cancer cells demonstrates heightened BNIP3-dependent autophagy induction under hypoxic conditions, supporting cell survival and underscoring the role of the tumour microenvironment in drug resistance [20]. Similarly, other studies have also documented that parkin1-mediated mitophagy contributes to the induction of cisplatin resistance [21]. Our findings suggest that EPHA2 knockdown decreases mitophagy by reducing Pink1, Parkin, and BNIP3 expression levels, potentially enhancing cisplatin sensitivity in EPHA2-KD cell lines.
The relationship between mitochondrial dynamics and autophagy is crucial for maintaining cellular homeostasis. Studies indicate that salivary gland adenoid cystic cancer, which exhibits increased autophagy, shows resistance to cisplatin treatment. Additionally, the downregulation of Beclin 1, an autophagy regulator, enhances cisplatin-mediated apoptosis [22]. In lung adenocarcinoma cells (A549), acquired cisplatin resistance has been associated with elevated autophagy [23]. A recent resource database highlights genes implicated in cisplatin resistance in cancer, specifically those involved in autophagosome formation, like ATG5, ATG7, ATG12, ATG14, and BECN1 [24]. Similarly, increased levels of LC3A are associated with cisplatin resistance and poorer outcomes in ovarian clear cell carcinomas [25]. Our investigation of autophagy marker expression in EPHA2-KD cell lines revealed reduced mRNA levels of ATG5, Beclin 1, and LC3B, suggesting potential enhancement of cisplatin sensitivity.
Mitochondrial dynamics and mitophagy are intricately linked processes. Based on the literature, we investigated their relationship in EPHA2-KD cell lines. It is reported that Parkin plays a crucial role by ubiquitinating mitochondrial fusion proteins Mfn1 and Mfn2, marking damaged mitochondria for degradation and initiating mitophagy [26]. Reduced levels of Parkin due to EPHA2-KD may impair this ubiquitination process, enhancing mitochondrial fusion. BNIP3 directly interacts with the Opa1 oligomer to inhibit its activity and promote mitochondrial fission [27]. BNIP3 induces Drp1 translocation in cardiac myocytes, promoting mitochondrial fission [29]. Reduced levels of BNIP3 in EPHA2-knockdown cells may hinder mitochondrial fission by inhibiting the translocation of Drp1 and directly interacting with the Opa1 oligomer to modulate its activity. BNIP3 also facilitates Parkin translocation and promotes mitophagy [28]; hence, reduced BNIP3 levels in EPHA2-KD cells decrease ubiquitin-mediated mitophagy. In conclusion, EPHA2 knockdown disrupts mitophagy by reducing Parkin and BNIP3 levels, enhancing mitochondrial fusion and decreasing mitochondrial fission.

EPHA2 regulates mitochondrial dynamics. (A) Western blot showing the protein expression of mitochondrial dynamics molecules in the EPHA2-KD cells and their control cells. (B) mRNA expression of mitochondrial dynamics molecules in the EPHA2-KD cells and their control cells. Both protein and mRNA expression bands were quantified and normalized with internal control. Significance was shown as *p < 0.05, **p < 0.01, and ***p < 0.001. (C) Mito tracker red staining for the EPHA2-KD and its control was taken at 20X magnification

EPHA2 knockdown decreases autophagy and mitophagy. (A) Western blot showing the protein expression of autophagy and mitophagy molecule in the EPHA2-KD cells and their control cells, (B) mRNA expression of autophagy and mitophagy molecule in the EPHA2-KD cells and their control. Both protein and mRNA expressions band were quantified and normalized with internal control. Significance was shown as *p < 0.05, **p < 0.01 and ***p < 0.001

EPHA2-KD sensitises cervical cancer for Cisplatin treatment. (A) MTT shows the % cell viability of EPHA2-KD cells and their control cells against the different concentrations of cisplatin treatment. (B) Colony formation assay showing the number of colonies formed in EPHA2-KD cells and their control cells after cisplatin treatment. (C) Wound healing assay showing the wound confluence in EPHA2-KD cells and their control cells after cisplatin treatment. Significance was shown as *p < 0.05, **p < 0.01, and ***p < 0.001

Sesamol sensitises cervical cancer to cisplatin treatment by inhibiting EPHA2 expression. (A) MTT assay showing the % cell viability of the SiHa cell line against the different concentrations of sesamol treatment. (B) MTT assay showing the % cell viability in the SiHa cell line for different concentrations of cisplatin, with and without sesamol administration. (C) Colony formation assay showing the number of colonies formed after cisplatin, sesamol, and sesamol + cisplatin treatment compared to their control. (D) The wound healing assay showed the wound confluence after cisplatin, sesamol, and sesamol + cisplatin treatment compared to their control. (E): Docking interaction between the sesamol and EPHA2 molecule. (F) Western blot showing the expression level of EPHA2 after sesamol treatment. Protein expression bands were quantified and normalized with internal control. Significance was shown as *p < 0.05, **p < 0.01, and ***p < 0.001
Seeking an alternative to EPHA2-KD, we explored sesamol, a plant-derived bioactive molecule with known anticancer activity. Similar to EPHA2-KD, sesamol sensitises the SiHa cell line for Cisplatin treatment. Combining plant-derived bioactive molecules with cisplatin monotherapy can effectively mitigate both chemoresistance and the associated side effects [29]. A combination therapy involving sesamol and cisplatin enhances the inhibitory effects of cisplatin on cancer cell migration and proliferation. Sesamol treatment at the IC50 value reduces EPHA2 expression in SiHa cell lines, suggesting that sesamol sensitises SiHa cells to cisplatin treatment by inhibiting EPHA2 expression. The docking analysis indicates that sesamol exhibits a binding affinity towards EPHA2 by a notable binding energy of -9.34 kcal/mol. In the binding pocket, sesamol engages in two hydrogen bonds with residues Glycine 515 and Arginine 465 within the EPHA2 protein. This is pivotal for stabilising the protein-ligand complex by affecting affinity and selectivity. Furthermore, sesamol demonstrates an amide pi stacking interaction with leucine 402, evidencing a pronounced affinity relative to other interactions. Additionally, pi-alkyl interactions facilitate the conformational stability of sesamol and its integration into the EPHA2 binding pocket. Moreover, weak Van der Waals forces contribute to the cohesive interaction between sesamol and EPHA2. So, Sesamol emerges as a promising alternative to EPHA2-KD, effectively sensitising SiHa cells to cisplatin treatment through inhibition of EPHA2 expression and strong binding affinity to the protein, suggesting its potential as a critical component in combating chemoresistance and enhancing therapeutic outcomes.
Conclusion
In conclusion, our study underscores the pivotal role of EPHA2, a prominent receptor tyrosine kinase, in mediating cisplatin resistance through modulation of mitochondrial dynamics, autophagy and mitophagy in cervical cancer. By demonstrating that EPHA2 knockdown enhances mitochondrial fusion while reducing fission and mitophagy autophagy, we provide mechanistic insights into how targeting EPHA2 can sensitise cervical cancer cells to cisplatin treatment. Moreover, our findings highlight sesamol as a promising alternative to EPHA2-KD, showing its efficacy in enhancing cisplatin sensitivity through downregulating EPHA2 expression. These results propose a novel therapeutic strategy for overcoming chemoresistance and improving treatment outcomes in cervical cancer. Further research into the clinical applicability and implications of EPHA2 modulation and sesamol combination therapy is warranted to advance these findings towards clinical translation.
Data availability
No datasets were generated or analysed during the current study.
References
Fowler JR, Maani EV, Dunton CJ, Gasalberti DP, Jack BW (2024) Cervical cancer. StatPearls
Burmeister CA, Khan SF, Schäfer G, Mbatani N, Adams T, Moodley J, Prince S (2022) Cervical cancer therapies: current challenges and future perspectives. Tumour Virus Res 13:200238
Zhao X, Liu J, Jin D et al (2022) EphA2 promotes the development of cervical cancer through the CXCL11/PD-L1 pathway. J Oncol 2022:1–17
Veiga RN, De Azevedo ALK, De Oliveira JC, Gradia DF (2024) Targeting EphA2: a promising strategy to overcome chemoresistance and drug resistance in cancer. J Mol Med 102:479–493
Van Der Bliek AM, Shen Q, Kawajiri S (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5:a011072–a011072
Jin P, Jiang J, Zhou L, Huang Z, Nice EC, Huang C, Fu L (2022) Mitochondrial adaptation in cancer drug resistance: prevalence, mechanisms, and management. J Hematol Oncol 15:97
Chang H, Zou Z (2020) Targeting autophagy to overcome drug resistance: further developments. J Hematol Oncol 13:159
Guan Y, Wang Y, Li B, Shen K, Li Q, Ni Y, Huang L (2021) Mitophagy in carcinogenesis, drug resistance and anticancer therapeutics. Cancer Cell Int 21:350
Dasari S, Bernard Tchounwou P (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740:364–378
Majdalawieh AF, Mansour ZR (2019) Sesamol, a major lignan in sesame seeds (Sesamum indicum): anti-cancer properties and mechanisms of action. Eur J Pharmacol 855:75–89
Kumar A, Bajaj P, Singh B et al (2024) Sesamol as a potent anticancer compound: from chemistry to cellular interactions. Naunyn-Schmiedeberg’s Arch Pharmacol. https://doi.org/10.1007/s00210-023-02919-2
Bairoch A (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28:45–48
Kim S, Thiessen PA, Bolton EE et al (2016) PubChem substance and compound databases. Nucleic Acids Res 44:D1202–D1213
Han Y, Kim B, Cho U, Park IS, Kim SI, Dhanasekaran DN, Tsang BK, Song YS (2019) Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 38:7089–7105
Xiao Y-Y, Xiao J-X, Wang X-Y, Wang T, Qu X-H, Jiang L-P, Tou F-F, Chen Z-P, Han X-J (2022) Metformin-induced AMPK activation promotes cisplatin resistance through PINK1/Parkin dependent mitophagy in gastric cancer. Front Oncol 12:956190
Li X, Wu Q, Ma F, Zhang X, Cai L, Yang X (2022) Mitochondrial fission factor promotes cisplatin resistancein hepatocellular carcinoma. Acta Biochim Biophys Sin (Shanghai) 54:301–310
Vianello C, Cocetta V, Catanzaro D et al (2022) Cisplatin resistance can be curtailed by blunting Bnip3-mediated mitochondrial autophagy. Cell Death Dis 13:398
Twig G, Shirihai OS (2011) The interplay between mitochondrial dynamics and Mitophagy. Antioxid Redox Signal 14:1939–1951
Ma M, Lin X, Liu H, Zhang R, Chen R (2020) Suppression of DRP1–mediated mitophagy increases the apoptosis of hepatocellular carcinoma cells in the setting of chemotherapy. Oncol Rep. https://doi.org/10.3892/or.2020.7476
El-Guindy DM, Ibrahim FMk ADA, El-Horany HE-S, Sabry NM, Elkholy RA, Mansour W, Helal DS (2023) Hypoxia-induced autophagy in triple negative breast cancer: association with prognostic variables, patients’ survival and response to neoadjuvant chemotherapy. Virchows Arch 482:823–837
Li Z, Wang Y, Wu L, Dong Y, Zhang J, Chen F, Xie W, Huang J, Lu N (2019) Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkin–mediated mitophagy. Oncol Rep. https://doi.org/10.3892/or.2019.7345
Ma B, Liang L, Liao G, Liang Y, Liu H, Zheng G, Su Y (2013) Inhibition of autophagy enhances cisplatin cytotoxicity in human adenoid cystic carcinoma cells of salivary glands. J Oral Pathol Med 42:774–780
Wu T, Wang M-C, Jing L, Liu Z-Y, Guo H, Liu Y, Bai Y-Y, Cheng Y-Z, Nan K-J, Liang X (2015) Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des Devel Ther 9:6421–6431
Huang D, Savage SR, Calinawan AP, Lin C, Zhang B, Wang P, Starr TK, Birrer MJ, Paulovich AG (2021) A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 40:6395–6405
Miyamoto M, Takano M, Aoyama T, Soyama H, Yoshikawa T, Tsuda H, Furuya K (2017) Inhibition of autophagy protein LC3A as a therapeutic target in ovarian clear cell carcinomas. J Gynecol Oncol 28:e33
Gegg ME, Cooper JM, Chau K-Y, Rojo M, Schapira AHV, Taanman J-W (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19:4861–4870
Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnauné-Pelloquin L (2010) The BH3‐only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep 11:459–465
Lee Y, Lee H-Y, Hanna RA, Gustafsson ÅB (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301:H1924–H1931
Dasari S, Njiki S, Mbemi A, Yedjou CG, Tchounwou PB (2022) Pharmacological effects of cisplatin combination with natural products in cancer chemotherapy. IJMS 23:1532
Acknowledgements
Mubthasima P P is thankful to CSIR, New Delhi, India for the award of fellowship (31/005(0563)/2019-EMR-I). The authors thank DST -Science and Engineering Research Board (DST-SERB) and CSIR-CFTRI for funding. All the authors also acknowledge Central Instruments Facility and Services department (CIFS), Academy of Scientific and Innovative Research (AcSIR) and Director, CSIR-CFTRI, Mysuru, for constant support to carry out this work.
Funding
DST -Science and Engineering Research Board (DST-SERB) - ECR/2018/000894.
Author information
Authors and Affiliations
Contributions
M P P: Conceptualization, methodology, Validation, Data Curation, Writing—Original Draft. A K: Conceptualization, Methodology, Project administration, Funding acquisition, Resource, Investigation, Writing-Review and Editing. SAS: Technical expertise, Resource, Writing—Review and Editing. All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
It is not applicable.
Conflict of interests
The authors declare that they have no financial interests.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mubthasima, P.P., Singh, S.A. & Kannan, A. Sesamol-mediated targeting of EPHA2 sensitises cervical cancer for cisplatin treatment by regulating mitochondrial dynamics, autophagy, and mitophagy. Mol Biol Rep 51, 949 (2024). https://doi.org/10.1007/s11033-024-09875-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09875-x