
Abstract
Circular RNAs (circRNAs) are a unique family of endogenous RNAs devoid of 3′ poly-A tails and 5′ end caps. These single-stranded circRNAs, found in the cytoplasm, are synthesized via back-splicing mechanisms, merging introns, exons, or both, resulting in covalently closed circular loops. They are profusely expressed across the eukaryotic transcriptome and offer heightened stability against exonuclease RNase R compared to linear RNA counterparts. This review endeavors to provide a comprehensive overview of circRNAs’ characteristics, biogenesis, and mechanisms of action. Furthermore, aimed to shed light on the potential of circRNAs as significant biomarkers in various cancer types. It has been performed an exhaustive literature review, drawing on recent studies and findings related to circRNA characteristics, synthesis, function, evaluation techniques, and their associations with oncogenesis. CircRNAs are intricately associated with tumor progression and development. Their multifaceted roles encompass gene regulation through the sponging of proteins and microRNAs, controlling transcription and splicing, interacting with RNA binding proteins (RBPs), and facilitating gene translation. Due to these varied roles, circRNAs have become a focal point in tumor pathology investigations, given their promising potential as both biomarkers and therapeutic agents. CircRNAs, due to their unique biogenesis and multifunctionality, hold immense promise in the realm of oncology. Their stability, widespread expression, and intricate involvement in gene regulation underscore their prospective utility as reliable biomarkers and therapeutic targets in cancer. As our understanding of circRNAs deepens, advanced techniques for their detection, evaluation, and manipulation will likely emerge. These advancements might catalyze the translation of circRNA-based diagnostics and therapeutics into clinical practice, potentially revolutionizing cancer care and prognosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Circular RNA (circRNA) is a diverse class of non-coding RNA (ncRNA) known to regulate both post-transcriptional and transcriptional gene expression [1]. The concept of ‘circular RNA’ was first introduced in 1976 by Sanger et al. who identified this unique structure in viroids [2]. Unlike linear RNAs, circRNAs lack polyadenylation and are formed through a 3-to-5 splicing mechanism, connecting the donor and acceptor sequences [2,3,4,5,6,7,8]. Studies estimate that up to one-fifth of activated human genes can potentially generate circRNAs, which can vary in length from a few hundred to thousands of nucleotides [3,4,5]. The absence of free ends in circRNAs makes them more stable than their linear counterparts, rendering them resistant to typical RNA degradation processes [6]. These molecules also exhibit evolutionary conservation, with specific expression patterns in tissues and developmental stages among eukaryotes [3, 7, 8]. Advances in whole-transcriptome analysis and novel in-silico techniques for transcriptome-wide circRNA identification have shown that circRNAs are large, single-stranded RNA transcripts. Previously considered non-canonical by-products of back-splicing, they originate from pre-mRNAs and are composed of introns, exons, and intergenic regions, forming covalently closed continuous loops.
To date, multiple studies have reported an abundance of circRNA molecules capable of regulating gene expression [9]. These circRNAs have physiological significance as biological biomarkers and function as non-coding microRNA molecules [10]. Interactions between circRNAs and RNA-binding proteins (RBPs) can have bidirectional effects on gene regulation [1, 11, 12]. Dysregulated circRNAs play critical roles in the pathophysiological pathways of various diseases, affecting cellular developmental processes [13]. Recent evidence has revealed that circRNAs could serve as advanced biomarkers and therapeutic targets for cancer prognosis, as they are associated with several cancer hallmarks, such as inducing angiogenesis, sustaining proliferative signals, and obstructing apoptotic pathways [13,14,15].
Despite recent advancements in the field, the molecular intricacies of circRNAs remain largely uncharted, there is a pressing need for further research to provide a more comprehensive understanding of the multifaceted roles that circRNAs play in the prognosis of cancer.
Methodology
This comprehensive review aims to systematically review and analyze the current literature to elucidate the biological role and regulation of circular RNA (circRNA) in the context of its potential as a biomarker and therapeutic target for cancer. It has been conducted an extensive literature search using the following electronic databases: PubMed/MedLined, Google Scholar, Wiley, EMBASE, Scopus, and Web of Science. The following keywords: “circular RNA”, “circRNA”, “biomarker”, “therapeutic target”, cancer”, “oncology”, “tumor” and MeSH terms “Circular RNA”, “Biomarkers”, “Therapeutics”, “Neoplasms”, “Gene Regulation”, were used for the search strategy. Search queries were constructed by combining keywords and MeSH terms using the Boolean operators ‘AND’ and ‘OR’. An example search query in PubMed would be: (“Circular RNA”[MeSH] OR circRNA) AND (“Biomarkers”[MeSH] OR “Therapeutics”[MeSH]) AND “Neoplasms”[MeSH].
Inclusion criteria: original research articles focusing on the role and regulation of circRNA in cancer; studies exploring circRNA as a biomarker and/or therapeutic target in cancer; articles published in English; peer-reviewed studies.
Exclusion criteria: non-original articles (letter to editor, commentaries, editorials); studies not related to cancer; articles focused solely on other types of non-coding RNA; studies with insufficient data or methodological flaws; Articles not published in English.
From the selected articles, the following information was extracted: study design and methodology; type of cancer studied; role of circRNA in the specific type of cancer; therapeutic implications of circRNA; outcome measures and results. The most representative data has been summarized in Tables and Figures.
General characteristics of circRNA
A recently discovered class of intrinsic non-coding RNAs (ncRNA), known as circular RNAs (circRNAs), is primarily produced from 1 to 5 exons and is mainly found within cytoplasmic organelles. Along with intronic regions, circRNAs are also produced inside the nucleus, albeit in a negligible proportion [16, 17]. The average length of circRNAs is 547 nucleotides, ranging from just a few hundred to 1000 [16, 18]. Numerous studies have shown that circRNAs can originate from a wide range of genomic subsequences [19]. One significant characteristic of circRNAs is that they possess single-stranded, covalently closed-loop structures without free ends [8]. This characteristic distinguishes circRNAs from their linear counterparts and enhances their stability in body tissues and plasma, as well as their resistance to exonucleases [20]. CircRNAs are particularly abundant in neural tissues due to the high rate of circRNA molecule accumulation and formation in these tissues [21]. CircRNA expression is often low, leading to the suggestion that they may simply be 'splicing noise' with limited functional value [21]. CircRNAs may play roles by binding, engaging, and directing their content to specific intracellular compartments, as they can bind to RNA-binding proteins (RBPs) similarly to miRNAs; furthermore, circRNAs could compete for RBPs that are present in limited quantities in specific subcellular locations. A few circRNAs have been identified in vesicles, and since vesicles can transport to targeted body tissues, circRNAs might also serve as delivery mechanisms [22, 23]
Biogenesis of circRNA
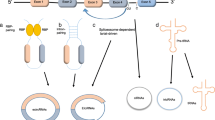
It is not yet fully understood how circRNAs are generated. They are commonly produced from pre-mRNAs using the conventional spliceosomal machinery, which is the most widely accepted method [24]. CircRNAs exist in three different forms and are considerably more stable than linear RNAs. Exosomal transport or other mechanisms could modify their function in cancer cells [25]. Exonic circRNAs are primarily located in the cytoplasm [26]. A distinct subgroup of circRNAs, called exon–intron circRNAs (EIciRNAs), contains both exons and introns [27]. Circular intronic RNAs (ciRNAs), along with intronic regions, are generally found within the nucleus and result from the failure of lariat introns to undergo debranching at the branch point, followed by the cleavage of the lariat end [28]. Back-splicing connects a 5′-splice region, the splice donor, to a 3′-splice end, the splice acceptor, forming a covalently closed loop. This is in contrast to linear RNAs, which are typically terminated with 3′ tails and 5′ caps [29]. Due to the absence of a polyadenylated tail and 5′-3′ polarity, circRNAs are more stable compared to linear RNAs and are not degraded by RNase R or exonucleases [5]. The circularization process may be induced by RNA-binding proteins (RBPs). Transacting factors such as Quaking, Muscleblind, and Fused-in Sarcoma can enhance circularization by binding to homologous intronic sequences [30]. The 30–50 ends of the circularized exons may come closer together, facilitating splicing, when aligned with upstream and downstream segments of the circularized exons and with RBP dimerization [31]. The presence of an equivalent inverted sequence can promote back-splicing [32]. As a result, the nucleophilic attack and cleavage are enhanced because the splice donor can now be near the splice acceptor [33]. It is generally accepted that most introns will be rapidly degraded through debranching, and only a few will form lariat structures during splicing. A small number of nucleotide sequences, such as a GU-rich motif of seven nucleotides near the 5′ splicing end and a C-rich motif of eleven nucleotides near the branch point, can inhibit debranching after splicing, leading to the formation of intronic circRNAs [34]. Variations in the 50 splice sites indicate the involvement of spliceosome-mediated exon circularization [24]. The spliceosomes that aggregate at the back-splicing site connect the 50 donor and 30 acceptor sites [35]. Another possibility is post-transcriptional and co-transcriptional back-splicing, which could involve a single exon or a set of exons along with their associated introns (Fig. 1) [31].

Schematical description of circRNA biogenesis and its mechanism. a Canonical splicing machinery produces linear mRNA through conventional splicing. b Exonic circRNA is produced via head-to-tail joining, which involves the backsplicing of a 5′-splice donor site to a 3′ splice acceptor region. c A close loop structure known as ciRNA can be formed by pairing reverse complementary lariat intron removed from pre-mRNA sequences. d The 5′ splice ends among exon 3 and the 3′ splicing regions of 2 exon are brought together by the removal of intron2 to create a circRNA that has multiple exons. e Intron 3 will also be intact, and Exons 3 and 4 will combine to produce an EIciRNA. f Stable ciRNA stimulates transcription by attaching to elongating RNA Pol II. g These circRNAs transport from the nucleus to the cytoplasm via nuclear pore. h Mechanism of circRNAs—Protein sponge, -miRNA sponge, -translation
Mechanism of action of circRNA
Due to its enormous potential, circRNA has recently gained popularity in the domain of molecular biology [36]. CircRNAs are known to perform a diverse array of functions, including acting as miRNA sponges, interacting with RNA-binding proteins (RBPs), regulating alternative splicing and transcription, facilitating translation, generating pseudogenes, transporting molecules, and mediating cell-to-cell communication. Their involvement in the translation process further enables them to modulate gene expression [31].
Circular RNA as a micro-RNA sponge and a competing endogenous RNA
The competitive endogenous RNA (ceRNA) theory states that transcripts with similar miRNA binding regions can inhibit miRNA function and increase the production of miRNA targets [37]. The majority of circRNAs have previously been referred to as “miRNA sponges” [38]. miRNAs are crucial in the progression of tumors [39]. It has been demonstrated that ceRNAs serve as miRNAs’ sponges [21]. Most circRNAs are found inside the cytoplasm and include several miRNA response elements (MREs), which suggests that they may compete with miRNAs for binding. [27] Therefore, by inhibiting the action of ceRNA, circRNAs could control miRNA functioning. MiR-7 is the most widely recognized miRNA in the domain of circRNA (ciRS-7; also known as CDR1as) [40] The first circRNA, CiRS-7, seems to have miR-7 binding regions above seventy and functions as a microRNA-7 sponge, minimizing the effect of microRNA-7 on target mRNA [41]. Even though the “miRNA sponge” is the standard model for how circular RNA functions. According to the latest report, the majority of circular RNAs cannot act as “bona fide” miRNA sponges [42].
Interaction of circRNA with RNA binding proteins (RBPS)
CircRNAs can bind to RBPs in a sequence-specific manner, the circular structure of circRNAs provides them with unique conformations that may be recognized by specific RBPs [43, 44] and have the next functional implications:
-
(i)
Sequestration of RBPs: by binding to RBPs, circRNAs can prevent these proteins from binding to their target linear RNAs. This sequestration can modulate the functions of RBPs and their downstream targets [43]
-
(ii)
Stabilization of circRNAs: some RBPs can enhance the stability of circRNAs, preventing their degradation and ensuring sustained function [44].
-
(iii)
Transport and localization: RBPs can guide circRNAs to specific cellular compartments, ensuring their localized functions [44, 45]. Regarding the clinical implications, the interaction of circRNAs with RBPs can influence disease pathways, especially in cancers [46]; disruptions in circRNA-RBP interactions might lead to aberrant post-transcriptional regulation, contributing to disease pathogenesis. Several experimental approaches, such as RNA immunoprecipitation (RIP) followed by sequencing, are used to identify and validate circRNA-RBP interactions. The interaction between circRNAs and RBPs is a complex and dynamic aspect of cellular regulation; circRNAs can associate with RBPs like circ-Foxo3, circ-MBL (muscleblind), and several circRNAs besides acting as miRNA sponges [22, 47]. Circ-Foxo3 has a wide range of protein-binding partners. Through associations with CDK2 and p21, it can inhibit the cell cycle and prevent the passage from G1 to the S phase [22]. Researchers also discovered that intron lariats can build up in the cytoplasm and attach to the protein TDP43, decreasing TDP43 toxicity in amyotrophic lateral sclerosis (ALS) [47]. Given the potential implications for health and disease, it remains an active area of research. However, for detailed data and the most recent findings, one should refer to specialized research papers or databases dedicated to circRNA research, as this is a rapidly advancing field.
Circular RNAs role in transcriptional regulation and alternative splicing
According to a few lines of evidence, circRNAs may act as helpful regulators of the transcription of RNA Pol II., which include studies showing that the downregulation of circular RNA acquired from the ANKRD52 intronic site (Ci-ankrd52) results in diminished appearance of their parent genetic makeup by combining RNA Pol II [34]. Additionally, thorough research revealed that muscleblind, a splicing factor that interferes with canonical pre-mRNA splicing, produces circMbl in its second exon [24]. The fact that circMbl's flanked introns and circMbl alone contain retained MBL binding regions suggests that generalized splicing factors like MBL possibly have an impact on alternate splicing which modifies the equilibrium amongst circular RNA synthesis and conventional splicing mechanism [24].
Translating proteins
Endogenous circRNAs have recently been shown to have the ability to translate into proteins. Legnini I. et al. discovered that circ-ZNF609 may read proteins in murine myoblasts when triggered by IRES (internal ribosome entry site) [48]. The possibility of using IRES-driven mechanisms to interpret certain circRNAs was demonstrated in three situations in 2017 with solid supporting evidence [49]. Thousands of such endogenous translatable circRNAs were discovered by Yang et al. and most of them had m6 A regions. This research suggested that circRNA translation may follow a common pattern [49]. Furthermore, it has been demonstrated that circMbl produced from the mbl domain could yield a quantifiable protein and that deprivation and FOXO can conceivably control the translation of a circMbl variant [50]. circ-ZNF609 was developed by Legnini et al. It had an accessible reading framework that started right at the start codon and could translate into a protein via cap-independent and a splicing-dependent pathway [48] The interpretation of literally hundreds of circRNAs had changed the coding architecture of the human transcriptome regardless of the disagreement and concern about the translation's ineffectiveness [51].
Table 1 summarizes the mechanism of action of circRNA.
Evidence linking circRNAs to cancer initiation and progression: mechanistic insights in different type of cancers
circRNAs, as a novel class of non-coding RNAs, have garnered significant attention for their roles in cancer [52]. Characterized by their unique covalently closed loop structures, circRNAs are intricately involved in the regulation of gene expression and are emerging as key players in the oncogenic process [53].
In colorectal cancer, specific circRNAs have been implicated in oncogenesis through their interaction with microRNAs and modulation of key signaling pathways. For instance, certain circRNAs are known to influence the Wnt/β-Catenin signaling, a critical pathway in colorectal cancer progression [54].
Breast cancer research has revealed a dual functionality of circRNAs; some circRNAs have been found to suppress tumor growth by interacting with miRNAs and inhibiting pivotal signaling pathways [55]. In contrast, others facilitate tumor progression, highlighting the diverse roles of circRNAs in breast cancer biology.
In the context of non-small cell lung cancer, studies have identified circRNAs that contribute to cancer proliferation and invasion; these circRNAs often function by sponging specific miRNAs, thereby impacting crucial oncogenic pathways [56].
The involvement of circRNAs in hepatocellular carcinoma is particularly notable; research has shown that certain circRNAs can suppress HCC progression and metastasis, offering insights into their potential therapeutic applications in liver cancer [57].
In melanoma, research has uncovered circRNAs that promote tumor progression by regulating key signaling pathways, such as the JAK2/STAT3 pathway, which is pivotal in melanoma pathogenesis [58].
The evidence accumulated to date highlights the complex and multifaceted role of circRNAs in cancer. Their regulatory functions across various signaling pathways, often through mechanisms like miRNA sponging, underscore their potential as biomarkers and therapeutic targets in the field of oncology.
Molecular and cellular mechanisms underlying circRNA manipulation in tumor phenotypes: in vitro and in vivo perspectives
The field of circular RNA (circRNA) research has unveiled their pivotal role in modulating cellular mechanisms and molecular pathways in cancer and functional studies, both in vitro and in vivo, have been instrumental in dissecting the mechanistic underpinnings of circRNA-mediated regulation of tumor phenotypes [59].
In cellular models, the manipulation of specific circRNAs has illustrated their role in key molecular pathways [52]. For example, overexpressing ciRS-7 in colorectal cancer cell lines revealed its regulatory function in the PI3K/Akt/mTOR pathway, a central axis in cell survival and proliferation [60]. ciRS-7 achieves this by sequestering miR-7, leading to the upregulation of PIK3CD and mTOR, and subsequently enhancing tumor cell proliferation and survival [61]. Similarly, silencing circRNA CDR1as in glioblastoma cell cultures resulted in marked alterations in the MAPK/ERK signaling cascade, reducing cell proliferation and increasing apoptosis [62].
Animal models have further substantiated the role of circRNAs in cancer progression. The knockout of circMTO1 in hepatocellular carcinoma (HCC) mouse models underscored its function in cell cycle regulation. The absence of circMTO1 led to the derepression of its target, miR-9, culminating in the aberrant activation of CDK6 and Cyclin D1, and thereby promoting cell cycle progression and tumor growth [7]. Additionally, the alteration of circRNAs involved in epithelial-to-mesenchymal transition (EMT) pathways demonstrated their capacity to modulate metastatic potential in vivo [63].
The impact of circRNAs extends to the tumor microenvironment, influencing angiogenesis, immune cell infiltration, and stromal interactions. For instance, the modulation of circRNAs involved in the hypoxia-inducible factor (HIF) pathway affects tumor angiogenesis, a crucial factor in tumor growth and metastasis [64]. circRNAs can also interact with components of the extracellular matrix, influencing the tumor-stroma crosstalk and thereby affecting tumor invasion and metastasis [65].
The elucidation of these molecular and cellular mechanisms offers promising avenues for circRNA-targeted therapies; the ability to modulate specific circRNAs and observe consequent effects on tumor-related pathways presents novel strategies for cancer treatment, with potential applications in targeted drug delivery and gene therapy [66].
The exploration of circRNAs in functional studies has shed light on their critical roles in cancer at the molecular and cellular levels; these findings not only enhance our understanding of cancer biology but also pave the way for innovative therapeutic approaches targeting circRNAs in the clinical oncology landscape.
Advances in circRNA-targeted therapies: current developments and effectiveness
The emerging field of circular RNA (circRNA) research has opened new avenues in cancer therapy; circRNAs, with their unique properties and diverse roles in gene regulation, present novel targets for therapeutic intervention [53, 67].
CircRNAs have been identified as crucial players in various oncogenic pathways, making them attractive targets for cancer therapy [67, 68]. Their stability, abundance, and cell-type specific expression patterns enhance their suitability as therapeutic targets; efforts are underway to develop strategies that either suppress oncogenic circRNAs or enhance the expression of tumor-suppressing circRNAs [53].
One of the most promising approaches involves the use of antisense oligonucleotides (ASOs). ASOs are short, synthetic strands of nucleic acids designed to specifically bind to circRNAs, thereby inhibiting their function [69]. For instance, ASOs targeting ciRS-7, a circRNA known to sponge tumor-suppressor miRNAs, have shown potential to reduce tumor growth in preclinical models [69, 70].
Researchers are also exploring the use of small molecule inhibitors to disrupt the biogenesis or function of oncogenic circRNAs; these molecules can interfere with the circularization process of circRNAs or inhibit their interaction with other molecules, like miRNAs or RNA-binding proteins, thus impeding their oncogenic activity [71].
RNA interference (RNAi) is another strategy being explored to silence specific circRNAs. By using siRNAs or shRNAs that target the back-splice junctions unique to circRNAs, researchers aim to selectively degrade these molecules without affecting the linear mRNA counterparts [72].
Despite the potential, circRNA-targeted therapies face challenges, including delivery mechanisms, off-target effects, and the complexity of circRNA-miRNA interactions [67]. Ongoing research is focused on optimizing these therapies for better specificity and efficacy, and understanding the broader impacts on cellular pathways.
Currently, several circRNA-targeted therapies are in the early stages of clinical trials [73]. Preliminary results are promising, indicating their potential effectiveness in reducing tumor progression and improving patient outcomes; these trials are pivotal in assessing the real-world efficacy and safety of circRNA-targeted interventions [73].
The development of circRNA-targeted therapies represents a significant stride in precision oncology. As our understanding of circRNA biology deepens, these therapies hold the promise of revolutionizing cancer treatment, offering more targeted, effective, and personalized options for patients.
CircRNA as a potential biomarker for cancer
Prognostic potential of circRNAs in various cancer types
Circular RNAs are considered as a potential biomarker in cancers, as the clinical use of biomarkers has become a significant approach in the diagnostic and prognostic procedures of cancer. Circular RNAs, linked by covalent bonding, are circular in structure. The covalently closed structure of CircRNA is highly stabilized which increases their efficacy of resistance toward exonuclease digestion (mainly preventing the degradation of RNase R) and aggregation of CircRNA in bodily fluids and tissues [21, 74, 75]. CirRNA can be detected in body fluids other than blood, including plasma, urine, cell-free saliva, tissue samples, and many other human components in a cell-specific manner. So it can be a novel biomarker for the detection of tumors along with surveillance [76]. Moreover, the efficiency of CircRNA to be considered a promising biomarker for cancer detection and prognosis can also be explained by the expression profiles and several other features such as increased stability, selectively abundant, highly conserved, and specified expressions. A study conducted by Memczak et al. [74] reveals that the level of CircRNA was higher in blood compared to the linear components present in low quantities in the blood. Therefore, CircRNA can be detected easily due to its elevated level in the blood. As a result, blood circRNAs may provide disease-related information whereas canonical RNA examination cannot [5, 9]. Another research was performed by Li et al. [77] which investigated that over 1000 circRNAs were detected in human serum exomes, suggesting that exomes contain a great number of circRNAs. Notably, circRNAs are at least two-fold overexpressed in exosomes as compared to producing cells (circRASSF2, circIARS, and circPTGR1) [78]. CircRNAs’ characteristics have prompted various investigations on their potential as cancer biomarkers. Several circRNAs that are involved in cancer are known to date [13]. The mode of action, target sites, and expression levels of circRNA in cancers are elaborated in Table 2 and Fig. 2.

Circular RNAs as a diagnostic and prognostic biomarker among several cancers. The diagram provides a comprehensive depiction of the diagnostic role of circular RNAs (circRNAs) across diverse cancer types. Specific circRNAs, associated with cancers such as esophageal squamous cell carcinoma, breast cancer, and more, could be crucial in the stages of early detection, therapeutic assessment, prognosis, and tailored medical interventions. These circRNAs underscore the potential for shaping individualized treatment strategies, reinforcing the emerging prominence of precision medicine in the realm of oncology. circRNAs circular RNAs, Circ-TTC17 Circ-DLGI, hsa_circRNA_002178, etc.: These are specific types of circRNAs associated with different cancers. Their names are typically derived from their circular RNA sequence identification. ↓ and ↑ represents the upregulation/downregulation of the respective circRNA about the particular cancer type
Many studies have been conducted on expression profiling and the functions of circRNAs to investigate their potential as a prognostic and diagnostic biomarker among tumors which opens up a new window to increase the efficacy of clinical diagnosis and therapeutics [13, 90].
circRNAs in personalized treatment and therapeutic monitoring
circRNAs have emerged not only as biomarkers for cancer diagnosis but also as pivotal tools in prognostication and guiding treatment strategies [53]. The prognostic significance of circRNAs in cancer is increasingly recognized; circRNAs are also instrumental in personalized medicine, particularly in guiding treatment decisions.
In lung cancer, the expression profile of circRNAs has been used to predict the responsiveness to EGFR tyrosine kinase inhibitors; patients exhibiting specific circRNA patterns showed differential responses, aiding in the selection of the most effective therapeutic regimen [91].
In the context of lung adenocarcinoma and resistance to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs), Dai et al. explored the circRNA-miRNA-mRNA network. Their study identified differentially expressed genes between sensitive and resistant cell lines to EGFR-TKIs. They constructed a network containing 18 circRNAs, 17 microRNAs, and 175 messenger RNAs, indicating the significance of circRNAs in the resistance mechanism. Notably, the circ-0007312- miR-764-MAPK1 axis was found to play a key role in EGFR-TKI resistance [92].
Beyond initial treatment decisions, circRNAs serve as dynamic biomarkers in monitoring treatment efficacy and the development of resistance. Recent studies emphasized that a large number of circRNAs are involved in carcinogenesis, metastasis, or chemoresistance of breast cancer through the transcriptional regulation of RNAs, including miRNA and mRNA [93,94,95]. In breast cancer, changes in circRNA levels have been observed in patients undergoing hormone therapy, providing insights into treatment responsiveness and the potential need for therapeutic adjustments [94, 95]. These circRNAs also show promise as stable biomarkers for monitoring breast cancer progression.
Clinical case studies further highlight the role of circRNAs in cancer management. For example, in hepatocellular carcinoma, the expression of circRNA_103809 has been linked to tumor size and recurrence, informing post-surgical monitoring and adjuvant therapy decisions [96]. Research by Li et al. focused on circRNA hsa_circ_103809 in HCC; the study revealed that hsa_circ_103809 expression was significantly down-regulated in HCC tissues and cell lines. The circRNA was found to inhibit HCC cell proliferation, migration, and invasion by sponging miR-620, suggesting its potential as a biomarker and therapeutic target for HCC [96].
These studies underscore the growing importance of circRNAs in cancer research and treatment. They highlight the potential of circRNAs as biomarkers for cancer prognosis, indicators of treatment responsiveness, and targets for therapy in various cancer types.
Figure 2 represents the expression profiles of some circRNAs that can be used as biomarkers in the therapeutic diagnosis and prognosis of different tumors.
Techniques to evaluate circular RNA
Employing techniques and methodologies that appropriately explore circRNA sequence, subcellular localization, length, physiological activities, engaging molecules, disease consequences, and therapeutic efficacy has become crucial due to the growing urge to evaluate circRNAs in detail and with clarity. The methods and tools currently available to investigate circRNAs are outlined in this section, along with their benefits and drawbacks. Numerous circRNAs have been detected due to advances in high-throughput analysis of circRNA sequencing. Though circRNAs were first identified decades ago, routine poly(A)-enrichment of RNA was unable to detect them [97]. To enrich extremely pure circRNAs, the novel technique known as RNase R protocol accompanied by poly (A) + RNA depletion and polyadenylation has been detected. In this technique, linear RNAs are first degraded from total RNAs by treating through RNase R. The residual RNAs having 3′-OH terminals are polyadenylated, thus eliminated through oligo beads and poly (A) enriched RNA depletion. The dependability of the results was significantly increased by the RPAD approach. However, the evaluation of circRNAs in association with other proteins, such as miRNAs and mRNAs, cannot be performed via RPAD. For the particular experimental objective, multiple approaches should be adopted by researchers for the optimization of RNA sequencing libraries. Up to now, numerous algorithms have been devised to locate circRNAs. Since canonical splicing is not the primary method utilized to make circRNAs, the mapping techniques employed in preliminary transcriptome studies cannot precisely correlate with the fragments to the genomic sequence. Consequently, additional genome sequencing and modification are required for sequencing read length that crosses back-splicing regions [98, 99].
Since circRNA annotations and evaluation algorithms are constantly being developed, various bioinformatics software and tools have been designed for circular RNA analysis and quantitation [98, 100]. CircRNADb can be used to investigate circRNAs that could potentially be translated into the proteins. Numerous databases, such as Circ2Traits, TSCD, CSCD and CircR2Disease, have been designed to study circRNAs in multiple disorders because of their potential association with certain diseases. Approximately 11 different tools are currently present for the analysis of circRNAs from RNA sequencing data and are broadly categorized into two classes [101,102,103]. In particular, to discover circRNAs, NCLScan, KNIFE and PTESFinder all need the potential circular RNA sequences to be generated using gene annotation data be available. This approach was referred to as “candidate-based” or “pseudo-reference-based” strategy. The second method, known as the “fragmented-based” in or “segmented read approach”, was employed by other algorithms to locate backsplicing connections using the mapping data provided by a various-split read alignments to the genomic sequence [101, 102]. Find circ and UROBORUS can be categorised together since they both collect the unmapped reads, retrieve the initial and final 20 base pair anchor points, and then deduce the backsplicing activities from the mapping data, whereas circRNA finder, DCC, CIRCexplorer, Segemehl and MapSplice are assigned to a subcategory as they developed spliced alignment to identify and analyses the backsplicing mechanisms. CIRI, being the most unique uses BWA-MEM to identify the signals of paired chiastic clipping and associates them with systematic screening procedures to eliminate any possible false positive results [104]. CircRNA finder depends upon the RNA sequence alignment programme known as STAR and involves sequencing information that is paired end [26, 105]. Following read alignment, a set of presumptive circularization connections is formed by filtering and collapsing the resultant putative chimeric connection reads. A Python-based tool called CIRCexplorer provides circular RNA identification results that are simple to understand. It first aligns reads to the genomic sequence by using TopHat, then removes the unmapped reads and uses TopHat-Fusion to determine backsplicing activities. In order to ensure that the acceptor and donor splice regions identified are compatible with canonical splice regions from specific gene annotation, the mapping sites of these reads are modified and realigned as required [32, 106, 107]. DCC is another software programme based on the application of STAR alignment program that is evaluated in this research. The DCC algorithm instructs programmers to execute an alignment of every section from read pairs sequentially in addition to the standard alignment of read pairing from paired end series as a whole to increase the identification of smaller circular RNAs. The computational cost for the alignment stage is practically doubled as a response [108]. Find circ, executes read alignment using Bowtie2. It mainly assembles the unmapped reads produced in the initial genome alignment round. The second alignment is carried out by removing the initial and final 20 base pair anchors in each unmapped sequence. Furthermore, it widens the alignment of the anchors, gathers and produces the detected splice junctions, and retains those reads that span those junctions. The filtering process is then applied to evaluate and verify credible circular RNA candidates [9, 109]. Another detection method designed on the Bowtie RNA alignment technique is UROBORUS [110]. It starts by performing splice alignment using TopHat. Next, it gathers both balanced and unbalanced mapped junctions by employing TopHat to realign the first and final 20 base pair of an unmapped read. Finally, the putative backspliced reads are inferred by handling separately the two types of junction-spanning anchors. Bowtie2 as well as Bowtie both are used in PTES Finder to achieve read alignment [111]. Only backsplicing junctions originating from specified splice sites are detected. It is intriguing that, although being available, it does not employ the paired-end data. Based on Bowtie2, KNIFE maps reads to the genomic sequences, transcriptome, rRNA sequences, and specifically designed backspliced and linear junction datasets independently [112]. When potential backspliced reads also map well to the other above mentioned databases, they are discarded. When paired-end information is available, the resulting backspliced junction-reads are further divided into circRNA as well as decoy reads depending upon the mapping details of the mate. Ultimately, it rectifies with a de novo investigation module to determine circular RNAs originating by unannotated splice regions for reads mapped with none of the data libraries described previously. The BWA-MEM alignment tool can automatically identify the break sites of query sequences inferred from circular RNAs, in contrast to the circRNA detection methods referenced above that rely on Bowtie and involve retrieving a definite size of anchors among the reads that are unmapped to spot possible backspliced junctions [113]. CIRI analyzes the alignment findings twice after BWA-MEM aligning. One of the software tools that can detect various sorts of splice junction occurrences is Map Splice [114]. De novo splice mapping technique is specifically used to locate noncanonical and canonical junctions from RNA-Sequencing datasets by segmenting reads into different anchors. A multi-split mapping software called Segemehl is also capable of detecting circulare RNA, Trans and canonical splicing, and gene fusion processes [115]. It is stated that it is more sensitive at identifying these activities than its counterparts. RNA-Seq evaluation method, NCLscan, professes to be efficient in spotting non collinear transcripts including circular RNAs through transcriptome data [116]. Microarrays are effective techniques for frequent assessments of the expression profiles of distinct circular RNAs and high-throughput evaluation of those expression levels. The combined pools of linear and circular RNA are treated with microarray probes that are particularly devised to identify the juncture regions of circular RNAs. Similar to Circ-sequence, the RNA samples for microarray investigation are often treated with RNase R to lower the proportion of linear RNAs and promote circRNA identification and quantification. The analysis of various circRNAs, including circPABPN1, circMTO1 and circEPSTI1, was successful using this [7, 117, 118]. Shorter probes that traverse the splice junction can be used to examine circular RNAs through Northern blot assay, however, extended probes corresponding to the whole circRNA can also be employed as demonstrated for circHIPK3 [119]. To assess high-throughput outcomes, it is essential to carry out reverse transcription accompanied by quantitative polymerase chain reaction. Circular RNA sequencing method provides significant data about circular RNA annotations and junctions [120]. Moreover, it provides a quantitative assessment of circular RNA concentration in various sub cellular compartments as well as its abundance in multiple circumstancess, including circRNA suppression, stress, and infection [14, 121]. Circular RNA copy number can be detected by a relatively recent technique known as digital droplet polymerase chain reaction. The target gene for amplification is comprised of nanoliter-sized nucleic acid particles. To assess the concentration of RNA, the percentage of positive droplets to negative droplets is evaluated [122, 123].
Table 3 offers a succinct yet comprehensive overview of various techniques and algorithms utilized in the identification and analysis of circRNAs, their strengths and limitations, and their specific use cases.
Applications of circulating RNA in clinical practice
Circulating RNA, particularly circular RNA (cirRNA), has demonstrated significant potential in becoming an integral part of routine clinical practice due to its stability, abundance and unique expression profiles across various types of cancer [72].
-
(i)
Early cancer diagnosis and screening
One of the most immediate applications of cirRNA in routine clinical practice is in the early diagnosis of cancer [124]. cirRNA biomarkers found in liquid biopsies such as blood or urine samples can be indicative of malignancies at their earliest stages [125]. For instance, cirRNA hsa_circ_0001649 has been identified as a potential biomarker for gastric cancer and is detectable in both tissue and plasma samples [126]
-
(ii)
Prognostic monitoring
Monitoring the levels of specific cirRNAs can also serve as a prognostic indicator for patient outcomes [125]. For example, increased levels of cirRNA CDR1as have been correlated with poor prognosis in colorectal cancer patients [127].
-
(iii)
Treatment decision-making
In breast cancer, cirRNA profiles have been used to predict the likely success of chemotherapy regimens, thereby guiding clinicians in personalized treatment planning [128].
-
(iv)
Minimal residual disease monitoring
After initial treatment, tracking cirRNA levels can help identify the presence of minimal residual disease, guiding decisions on adjuvant therapies and monitoring for relapse [129, 130].
-
(v)
Targeted therapy
Emerging research indicates that cirRNA may even be a target for molecular therapies [68, 131]. Antisense oligonucleotides (ASOs) targeting cirRNA have shown promise in preclinical models for the treatment of hepatocellular carcinoma [132].
Limitations and challenges
The topic of circular RNAs (circRNAs) as potential biomarkers and therapeutic targets in cancer is a developing field and there are some possible limitations, challenges, and clinical pitfalls that might be associated. The diversity of cancer types means that the roles and regulation of circRNAs might differ across various tumors, making it challenging to establish a unified framework and the biological roles and mechanisms by which circRNAs influence cancer biology are not fully elucidated, limiting the potential therapeutic implications [133]. Also, the precise and reliable detection of circRNAs remains a technical challenge, with potential issues related to false positives or false negatives; the current knowledge is based on cross-sectional studies, while longitudinal studies which are necessary to validate circRNAs as potential predictive or prognostic markers are limited [134]. Efficiently isolating, quantifying, and characterizing circRNAs from biological samples is technically demanding and circRNAs might have multiple roles in the cell, from acting as miRNA sponges to influencing transcription and protein function. Moving from basic research to clinical application requires exhaustive validation, including ensuring the reproducibility of circRNA detection in clinical settings. Targeting circRNAs in a therapeutic context necessitates effective delivery mechanisms, which are still under development for many RNA-based therapies. An important clinical gap is represented by the expression levels of circRNAs that can vary between patients, stages of cancer, and even between samples from the same tumor, complicating their use as consistent biomarkers. Targeting circRNAs might inadvertently influence other cellular processes, given their multifunctional nature. Another cinical pitfall is represented by resistance Mechanisms because the tumors might develop resistance mechanisms against circRNA-targeted treatments. Also, introducing or blocking circRNAs could have unforeseen consequences on non-cancerous cells, potentially leading to adverse side effects. Implementing circRNA-based diagnostics or therapies might be expensive, potentially limiting their accessibility to a broader patient population.
When working with circulating RNA (cirRNA) in a clinical setting, several critical measures must be taken to ensure data accuracy and reliability. First, the isolation of cirRNA from biological samples should be performed using standardized protocols to minimize degradation and contamination. It is also vital to use high-quality reagents and to store cirRNA samples at appropriate temperatures to preserve their integrity; given the sensitive nature of cirRNA, even small variations in methodology can lead to significant differences in results [135]. Next, using controls and calibrators is essential for the normalization of cirRNA levels, as this allows for accurate quantification and comparison across different samples or time points [136]. Additionally, the source of the cirRNA, whether it is derived from plasma, serum, or other body fluids, should be consistently documented, as different sources may yield different profiles; careful attention should also be paid to potential confounding factors like patient age, medication, or disease stage, which could influence cirRNA levels [137]. Analytical validation of cirRNA detection methods, such as qRT-PCR or sequencing, is crucial for ensuring the robustness and reproducibility of the data [135, 138]. Lastly, bioinformatic analysis should be performed using validated algorithms to interpret cirRNA signatures in the context of specific clinical questions [139].
Conclusion
Recent discoveries in circRNA research have revealed that circRNAs play a wide range of roles in both biological and pathological processes, particularly in cancer. With the rapid advancements in technology, the study of circRNAs has emerged as a new research area. Although the biological functions of most circRNAs are still largely unknown, existing evidence suggests that they play important roles in various biological mechanisms, including transcriptional regulation, alternative splicing, protein translation, and interactions with RBPs and miRNAs. The advent of bioinformatics and RNA sequencing techniques has enabled the identification of numerous circRNAs. These circRNAs have been shown to perform diverse functions in gene regulation, affecting both basic biological processes and the onset and progression of diseases. The field has gained particular interest in cancer research, providing a new avenue for understanding tumor occurrence and growth. This has led to preliminary evidence suggesting that circRNAs may serve as potential biomarkers or therapeutic targets for cancer diagnosis and prognosis.
Data availability
Not applicable.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ANKRD52:
-
Ankyrin repeat domain 52
- BWA-MEM:
-
Burrow-wheeler aligner’s maximal exact match
- CDR1as:
-
Cerebellar degeneration-related protein 1 antisense
- ceRNA:
-
Competitive endogenous RNA
- CDK2:
-
Cyclin-dependent kinase 2
- circRNA:
-
Circular RNA
- circ-Foxo3:
-
Circular RNA forkhead box O3
- circHIPK3:
-
Circular RNA homeodomain-interacting protein kinase 3
- circ-MBL:
-
Circular muscleblind
- circMTO1:
-
Circular RNA mitochondrial tRNA translation optimization 1
- circ-ZNF609:
-
Circular RNA zinc finger protein 609
- CIRI:
-
Circular RNA identifier
- CSCD:
-
Cancer-specific circRNA database
- EIciRNAs:
-
Exon–intron circRNAs
- IRES:
-
Internal ribosome entry site
- KNIFE:
-
Known and novel isoform explorer
- MREs:
-
MiRNA response elements
- miRNA:
-
MicroRNA
- ncRNAs:
-
Non-coding RNA
- NCLScan:
-
Non-co-linear transcripts scan
- PTESFinder:
-
Post-transcriptional exon shuffling finder
- RBPs:
-
RNA binding proteins
- RPAD:
-
RNase R protocol accompanied by poly (A) + RNA depletion and polyadenylation
- TDP-43:
-
TAR DNA-binding protein 43
- TSCD:
-
Tissue-specific CircRNA database
References
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495(7441):384–388
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK (1976) Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci 73(11):3852–3856
Wu W, Ji P, Zhao F (2020) CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol 21(1):1–14
Kristensen L, Andersen M, Stagsted L, Ebbesen K, Hansen T, Kjems J (2019) The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20(11):675–691
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 7(2):e30733
Barrett SP, Salzman J (2016) Circular RNAs: analysis, expression and potential functions. Development 143(11):1838
Han D, Li J, Wang H, Su X, Hou J, Gu Y et al (2017) Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology 66(4):1151–1164
Chen LL, Yang L (2015) Regulation of circRNA biogenesis. RNA Biol 12(4):381–388
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338
Xiao J (2018) Circular RNAs: biogenesis and functions. Springer, Singapore
Wilusz J, Sharp PJS (2013) A circuitous route to noncoding RNA NIH public access. Science 340:440–441
Huang A, Zheng H, Wu Z, Chen M, Huang Y (2020) Circular RNA-protein interactions: functions, mechanisms, and identification. Theranostics 10(8):3503
Liu J, Zhang X, Yan M, Li H (2020) Emerging role of circular RNAs in cancer. Front Oncol 10:663
Huang R, Zhang Y, Han B, Bai Y, Zhou R, Gan G et al (2017) Circular RNA HIPK2 regulates astrocyte activation via cooperation of autophagy and ER stress by targeting MIR124–2HG. Autophagy 13(10):1722–1741
Bach DH, Lee SK, Sood AK (2019) Circular RNAs in cancer. Mol Therap Nucleic Acids 16:118–129
Guo JU, Agarwal V, Guo H, Bartel DP (2014) Expanded identification and characterization of mammalian circular RNAs. Genom Biol 15(7):1–14
Zhu J, Yu Y, Meng X, Fan Y, Zhang Y, Zhou C et al (2013) De novo-generated small palindromes are characteristic of amplicon boundary junction of double minutes. Int J Cancer 133(4):797–806
Lasda E, Parker R (2014) Circular RNAs: diversity of form and function. RNA 20(12):1829–1842
Hayes M, Li J (2015) An integrative framework for the identification of double minute chromosomes using next generation sequencing data. BMC Genet 16(2):1–7
Tan S, Gou Q, Pu W, Guo C, Yang Y, Wu K et al (2018) Circular RNA F-circEA produced from EML4-ALK fusion gene as a novel liquid biopsy biomarker for non-small cell lung cancer. Cell Res 28(6):693–695
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO (2013) Cell-type specific features of circular RNA expression. PLoS Genet 9(9):e1003777
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB (2016) Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44(6):2846–2858
Preußer C, Hung LH, Schneider T, Schreiner S, Hardt M, Moebus A et al (2018) Selective release of circRNAs in platelet-derived extracellular vesicles. J Extracell Vesicles 7(1):1424473
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M et al (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56(1):55–66
Zhu LP, He YJ, Hou JC, Chen X, Zhou SY, Yang SJ et al (2017) The role of circRNAs in cancers. Biosci Rep. https://doi.org/10.1042/BSR20170750
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P et al (2014) Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 9(5):1966–1980
Chen LL (2016) The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol 17(4):205–211
Shen T, Han M, Wei G, Ni T (2015) An intriguing RNA species—perspectives of circularized RNA. Protein Cell 6(12):871–880
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J et al (2016) Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 26(9):1277–1287
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA et al (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 160(6):1125–1134
Tang X, Ren H, Guo M, Qian J, Yang Y, Gu C et al (2021) Review on circular RNAs and new insights into their roles in cancer. Cell 19:910–928
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L (2014) Complementary sequence-mediated exon circularization. Cell 159(1):134–147
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR et al (2015) Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10(2):170–177
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH et al (2013) Circular intronic long noncoding RNAs. Mol Cell 51(6):792–806
Kelly S, Greenman C, Cook PR, Papantonis A (2015) Exon skipping is correlated with exon circularization. J Mol Biol 427(15):2414–2417
Ebert MS, Sharp PA (2010) MicroRNA sponges: progress and possibilities. RNA 16(11):2043–2050
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146(3):353–358
Piwecka M, Glažar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf A et al (2017) Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357(6357):8526
Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME (2015) MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 34(48):5857–5868
Zhao J, He S, Minassian A, Li J, Feng P (2015) Recent advances on viral manipulation of NF-κB signaling pathway. Curr Opin Virol 15:103–111
Hansen TB, Kjems J, Damgaard CK (2013) Circular RNA and miR-7 in CancerCircular RNA and miR-7 in Cancer. Cancer Res 73(18):5609–5612
Militello G, Weirick T, John D, Döring C, Dimmeler S, Uchida S (2017) Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinfo 18(5):780–788
Ikeda Y, Morikawa S, Nakashima M, Yoshikawa S, Taniguchi K, Sawamura H et al (2023) CircRNAs and RNA-binding proteins involved in the pathogenesis of cancers or central nervous system disorders. Noncoding RNA. https://doi.org/10.3390/ncrna9020023
Zhao X, Zhong Y, Wang X, Shen J, An W (2022) Advances in circular RNA and its applications. Int J Med Sci 19(6):975–985. https://doi.org/10.7150/ijms.71840
Zang J, Lu D, Xu A (2020) The interaction of circRNAs and RNA binding proteins: an important part of circRNA maintenance and function. J Neurosci Res 98(1):87–97. https://doi.org/10.1002/jnr.24356
Wawrzyniak O, Zarębska Ż, Kuczyński K, Gotz-Więckowska A, Rolle K (2020) Protein-related circular RNAs in human pathologies. Cells. https://doi.org/10.3390/cells9081841
Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z et al (2012) Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44(12):1302–1309
Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O et al (2017) Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 66(1):22–37
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y et al (2017) Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res 27(5):626–641
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L et al (2017) Translation of circRNAs. Mol Cell 66(1):9–21
Chen L, Shan G (2021) CircRNA in cancer: Fundamental mechanism and clinical potential. Cancer Lett 505:49–57
Xiao W, Li J, Hu J, Wang L, Huang JR, Sethi G et al (2021) Circular RNAs in cell cycle regulation: Mechanisms to clinical significance. Cell Prolif 54(12):e13143. https://doi.org/10.1111/cpr.13143
Huang Y, Zhang C, Xiong J, Ren H (2021) Emerging important roles of circRNAs in human cancer and other diseases. Genes Dis 8(4):412–423. https://doi.org/10.1016/j.gendis.2020.07.012
Zhang Y, Luo J, Yang W, Ye WC (2023) CircRNAs in colorectal cancer: potential biomarkers and therapeutic targets. Cell Death Dis 14(6):353. https://doi.org/10.1038/s41419-023-05881-2
Zhang F, Li L, Fan Z (2022) circRNAs and their relationship with breast cancer: a review. World J Surg Oncol 20(1):373. https://doi.org/10.1186/s12957-022-02842-5
Zhang N, Nan A, Chen L, Li X, Jia Y, Qiu M et al (2020) Circular RNA circSATB2 promotes progression of non-small cell lung cancer cells. Mol Cancer 19(1):101. https://doi.org/10.1186/s12943-020-01221-6
Zhou Y, Mao X, Peng R, Bai D (2022) CircRNAs in hepatocellular carcinoma: characteristic, functions and clinical significance. Int J Med Sci 19(14):2033–2043. https://doi.org/10.7150/ijms.74713
Tang K, Zhang H, Li Y, Sun Q, Jin H (2021) Circular RNA as a potential biomarker for melanoma: a systematic review. Front Cell Dev Biol 9:638548. https://doi.org/10.3389/fcell.2021.638548
Ng WL, Mohd Mohidin TB, Shukla K (2018) Functional role of circular RNAs in cancer development and progression. RNA Biol 15(8):995–1005. https://doi.org/10.1080/15476286.2018.1486659
Riquelme I, Tapia O, Espinoza JA, Leal P, Buchegger K, Sandoval A et al (2016) The gene expression status of the PI3K/AKT/mTOR pathway in gastric cancer tissues and cell lines. Pathol Oncol Res 22(4):797–805. https://doi.org/10.1007/s12253-016-0066-5
Dou Z, Gao L, Ren W, Zhang H, Wang X, Li S et al (2020) CiRS-7 functions as a ceRNA of RAF-1/PIK3CD to promote metastatic progression of oral squamous cell carcinoma via MAPK/AKT signaling pathways. Exp Cell Res 396(2):112290. https://doi.org/10.1016/j.yexcr.2020.112290
Yang X, Li S, Wu Y, Ge F, Chen Y, Xiong Q (2020) The circular RNA CDR1as regulate cell proliferation via TMED2 and TMED10. BMC Cancer 20(1):312. https://doi.org/10.1186/s12885-020-06794-5
Jiang M, Fang S, Zhao X, Zhou C, Gong Z (2021) Epithelial-mesenchymal transition-related circular RNAs in lung carcinoma. Cancer Biol Med 18(2):411–420. https://doi.org/10.20892/j.issn.2095-3941.2020.0238
Wu Z, Yu X, Zhang S, He Y, Guo W (2022) Mechanism underlying circRNA dysregulation in the TME of digestive system cancer. Front Immunol. https://doi.org/10.3389/fimmu.2022.951561
Shao Y, Lu B (2020) The crosstalk between circular RNAs and the tumor microenvironment in cancer metastasis. Cancer Cell Int 20:448. https://doi.org/10.1186/s12935-020-01532-0
Begliarzade S, Sufianov A, Ilyasova T, Shumadalova A, Sufianov R, Beylerli O et al (2024) Circular RNA in cervical cancer: fundamental mechanism and clinical potential. Noncoding RNA Res 9(1):116–124. https://doi.org/10.1016/j.ncrna.2023.11.009
He AT, Liu J, Li F, Yang BB (2021) Targeting circular RNAs as a therapeutic approach: current strategies and challenges. Signal Transduct Target Ther 6(1):185. https://doi.org/10.1038/s41392-021-00569-5
Zhang F, Jiang J, Qian H, Yan Y, Xu W (2023) Exosomal circRNA: emerging insights into cancer progression and clinical application potential. J Hematol Oncol 16(1):67. https://doi.org/10.1186/s13045-023-01452-2
Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A et al (2020) Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. https://doi.org/10.3390/jcm9062004
Zheng XB, Zhang M, Xu MQ (2017) Detection and characterization of ciRS-7: a potential promoter of the development of cancer. Neoplasma 64(3):321–328. https://doi.org/10.4149/neo_2017_301
Sharma AR, Bhattacharya M, Bhakta S, Saha A, Lee SS, Chakraborty C (2021) Recent research progress on circular RNAs: biogenesis, properties, functions, and therapeutic potential. Mol Ther Nucleic Acids 25:355–371. https://doi.org/10.1016/j.omtn.2021.05.022
Pisignano G, Michael DC, Visal TH, Pirlog R, Ladomery M, Calin GA (2023) Going circular: history, present, and future of circRNAs in cancer. Oncogene 42(38):2783–2800. https://doi.org/10.1038/s41388-023-02780-w
Jagtap U, Anderson ES, Slack FJ (2023) The emerging value of circular noncoding RNA research in cancer diagnosis and treatment. Cancer Res 83(6):809–813. https://doi.org/10.1158/0008-5472.Can-22-3014
Memczak S, Papavasileiou P, Peters O, Rajewsky N (2015) Identification and characterization of circular RNAs as a new class of putative biomarkers in human blood. PLoS ONE 10(10):e0141214
Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A (2006) Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34(8):e63
Qu S, Liu Z, Yang X, Zhou J, Yu H, Zhang R et al (2018) The emerging functions and roles of circular RNAs in cancer. Cancer Lett 414:301–309
Li J, Li Z, Jiang P, Peng M, Zhang X, Chen K et al (2018) Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J Exp Clin Cancer Res 37(1):1–16
Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y et al (2015) The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem 61(1):221–230
Wang R, Zhang S, Chen X, Li N, Li J, Jia R et al (2018) CircNT5E acts as a sponge of miR-422a to promote glioblastoma tumorigenesis. Cancer Res 78(17):4812–4825. https://doi.org/10.1158/0008-5472.Can-18-0532
Wang R, Zhang S, Chen X, Li N, Li J, Jia R et al (2018) EIF4A3-induced circular RNA MMP9 (circMMP9) acts as a sponge of miR-124 and promotes glioblastoma multiforme cell tumorigenesis. Mol Cancer 17(1):166. https://doi.org/10.1186/s12943-018-0911-0
Lei Y, Chen L, Zhang G, Shan A, Ye C, Liang B et al (2020) MicroRNAs target the Wnt/β-catenin signaling pathway to regulate epithelial-mesenchymal transition in cancer (Review). Oncol Rep 44(4):1299–1313. https://doi.org/10.3892/or.2020.7703
Liu W, Ma W, Yuan Y, Zhang Y, Sun S (2018) Circular RNA hsa_circRNA_103809 promotes lung cancer progression via facilitating ZNF121-dependent MYC expression by sequestering miR-4302. Biochem Biophys Res Commun 500(4):846–851. https://doi.org/10.1016/j.bbrc.2018.04.172
Chen L, Nan A, Zhang N, Jia Y, Li X, Ling Y et al (2019) Circular RNA 100146 functions as an oncogene through direct binding to miR-361-3p and miR-615-5p in non-small cell lung cancer. Mol Cancer 18(1):13. https://doi.org/10.1186/s12943-019-0943-0
Tang H, Huang X, Wang J, Yang L, Kong Y, Gao G et al (2019) circKIF4A acts as a prognostic factor and mediator to regulate the progression of triple-negative breast cancer. Mol Cancer 18(1):23. https://doi.org/10.1186/s12943-019-0946-x
Zhang X, Wang S, Wang H, Cao J, Huang X, Chen Z et al (2019) Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol Cancer 18(1):20. https://doi.org/10.1186/s12943-018-0935-5
Li RC, Ke S, Meng FK, Lu J, Zou XJ, He ZG et al (2018) CiRS-7 promotes growth and metastasis of esophageal squamous cell carcinoma via regulation of miR-7/HOXB13. Cell Death Dis 9(8):838. https://doi.org/10.1038/s41419-018-0852-y
Xie F, Li Y, Wang M, Huang C, Tao D, Zheng F et al (2018) Circular RNA BCRC-3 suppresses bladder cancer proliferation through miR-182-5p/p27 axis. Mol Cancer 17(1):144. https://doi.org/10.1186/s12943-018-0892-z
Rao D, Yu C, Sheng J, Lv E, Huang W (2021) The emerging roles of circFOXO3 in cancer. Front Cell Dev Biol 9:659417. https://doi.org/10.3389/fcell.2021.659417
Zhang L, Zhou Q, Qiu Q, Hou L, Wu M, Li J et al (2019) CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol Cancer 18(1):144. https://doi.org/10.1186/s12943-019-1080-5
Tang Q, Hann SS (2020) Biological roles and mechanisms of circular RNA in human cancers. Onco Targets Therap 13:2067
Ren W, Yuan Y, Peng J, Mutti L, Jiang X (2022) The function and clinical implication of circular RNAs in lung cancer. Front Oncol 12:862602. https://doi.org/10.3389/fonc.2022.862602
Dai C, Liu B, Li S, Hong Y, Si J, Xiong Y et al (2021) Construction of a circRNA-miRNA-mRNA regulated pathway involved in EGFR-TKI lung adenocarcinoma resistance. Technol Cancer Res Treat 20:15330338211056808. https://doi.org/10.1177/15330338211056809
Zepeda-Enríquez P, Silva-Cázares MB, López-Camarillo C (2023) Novel insights into circular RNAs in metastasis in breast cancer: an update. Noncoding RNA. https://doi.org/10.3390/ncrna9050055
Li Z, Chen Z, Hu G, Jiang Y (2019) Roles of circular RNA in breast cancer: present and future. Am J Transl Res 11(7):3945–3954
Gopikrishnan M, Hephzibah Cathryn R, Gnanasambandan R, Ashour HM, Pintus G, Hammad M et al (2023) Therapeutic and diagnostic applications of exosomal circRNAs in breast cancer. Funct Integr Genom 23(2):184. https://doi.org/10.1007/s10142-023-01083-3
Li X, Shen M (2019) Circular RNA hsa_circ_103809 suppresses hepatocellular carcinoma proliferation and invasion by sponging miR-620. Eur Rev Med Pharmacol Sci 23(2):555–566. https://doi.org/10.26355/eurrev_201902_16868
Panda AC, De S, Grammatikakis I, Munk R, Yang X, Piao Y et al (2017) High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res 45(12):e116
Zeng X, Lin W, Guo M, Zou Q (2017) A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 13(6):e1005420
Jakobi T, Dieterich C (2019) Computational approaches for circular RNA analysis. Wiley Interdiscip Rev 10(3):e1528
Hansen TB (2018) Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol 6:20
Chen I, Chen CY, Chuang TJ (2015) Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip Rev 6(5):563–579
Meng X, Li X, Zhang P, Wang J, Zhou Y, Chen M (2017) Circular RNA: an emerging key player in RNA world. Brief Bioinfo 18(4):547–557
Jeck WR, Sharpless NE (2014) Detecting and characterizing circular RNAs. Nat Biotechnol 32(453):461
Li HJ (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics. https://doi.org/10.48550/arXiv.1303.3997
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21
Kim D, Salzberg S (2011) TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 12:72
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9):1105–1111
Cheng J, Metge F, Dieterich C (2016) Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 32(7):1094–1096
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359
Song X, Zhang N, Han P, Moon BS, Lai RK, Wang K et al (2016) Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res 44(9):e87
Izuogu OG, Alhasan AA, Alafghani HM, Santibanez-Koref M, Elliott DJ, Jackson MS (2016) PTESFinder: a computational method to identify post-transcriptional exon shuffling (PTES) events. Nucleic Acids Res 17(1):1–11
Szabo L, Morey R, Palpant N, Wang P, Afari N, Jiang C, Parast MM, Murry CE, Laurent LC, Salzman J (2015) Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol 16:126
Gao Y, Wang J, Zhao F (2015) CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16(1):1–16
Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL et al (2010) MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38(18):e178
Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S et al (2014) A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol 15(2):1–11
Chuang TJ, Wu CS, Chen CY, Hung LY, Chiang TW, Yang MY (2016) NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res 44(3):e29
Chen B, Wei W, Huang X, Xie X, Kong Y, Dai D et al (2018) circEPSTI1 as a prognostic marker and mediator of triple-negative breast cancer progression. Theranostics 8(14):4003
Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S et al (2017) Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 14(3):361–369
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B et al (2016) Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7:11215
Dudekula DB, Panda AC, Grammatikakis I, Supriya D, Abdelmohsen K, Gorospe M (2016) CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol 13(1):34–42
Panda AC, Abdelmohsen K, Gorospe M (2017) RT-qPCR detection of senescence-associated circular RNAs. In: Nikiforov MA (ed) Oncogene-induced senescence. Springer, New York, pp 79–87
Quan PL, Sauzade M, Brouzes EJS (2018) dPCR: a technology review. Sensors 18(4):1271
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ et al (2011) High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83(22):8604–8610
Zhang Y, Wang Y, Su X, Wang P, Lin W (2021) The value of circulating circular RNA in cancer diagnosis, monitoring, prognosis, and guiding treatment. Front Oncol 11:736546. https://doi.org/10.3389/fonc.2021.736546
Wang S, Zhang K, Tan S, Xin J, Yuan Q, Xu H et al (2021) Circular RNAs in body fluids as cancer biomarkers: the new frontier of liquid biopsies. Mol Cancer 20(1):13. https://doi.org/10.1186/s12943-020-01298-z
Xu CY, Zeng XX, Xu LF, Liu M, Zhang F (2022) Circular RNAs as diagnostic biomarkers for gastric cancer: a comprehensive update from emerging functions to clinical significances. Front Genet. https://doi.org/10.3389/fgene.2022.1037120
Artemaki PI, Scorilas A, Kontos CK (2020) Circular RNAs: a new piece in the colorectal cancer puzzle. Cancers (Basel). https://doi.org/10.3390/cancers12092464
De Palma FDE, Salvatore F, Pol JG, Kroemer G, Maiuri MC (2022) Circular RNAs as potential biomarkers in breast cancer. Biomedicines. https://doi.org/10.3390/biomedicines10030725
Santini D, Botticelli A, Galvano A, Iuliani M, Incorvaia L, Gristina V et al (2023) Network approach in liquidomics landscape. J Exp Clin Cancer Res 42(1):193. https://doi.org/10.1186/s13046-023-02743-9
Souza VGP, Forder A, Brockley LJ, Pewarchuk ME, Telkar N, de Araújo RP et al (2023) Liquid biopsy in lung cancer: biomarkers for the management of recurrence and metastasis. Int J Mol Sci 24(10):8894
Szczepaniak A, Bronisz A, Godlewski J (2023) Circular RNAs-new kids on the block in cancer pathophysiology and management. Cells. https://doi.org/10.3390/cells12040552
Lu K, Fan Q, Zou X (2022) Antisense oligonucleotide is a promising intervention for liver diseases. Front Pharmacol 13:1061842. https://doi.org/10.3389/fphar.2022.1061842
Li F, Yang Q, He AT, Yang BB (2021) Circular RNAs in cancer: limitations in functional studies and diagnostic potential. Semin Cancer Biol 75:49–61. https://doi.org/10.1016/j.semcancer.2020.10.002
Kristensen LS, Hansen TB, Venø MT, Kjems J (2018) Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 37(5):555–565. https://doi.org/10.1038/onc.2017.361
Drula R, Braicu C, Chira S, Berindan-Neagoe I (2023) Investigating circular RNAs using qRT-PCR; roundup of optimization and processing steps. Int J Mol Sci 24(6):5721
Gharib E, Nasrabadi PN, Robichaud GA (2023) Circular RNA expression signatures provide promising diagnostic and therapeutic biomarkers for chronic lymphocytic leukemia. Cancers (Basel). https://doi.org/10.3390/cancers15051554
Tang X, Ren H, Guo M, Qian J, Yang Y, Gu C (2021) Review on circular RNAs and new insights into their roles in cancer. Comput Struct Biotechnol J 19:910–928. https://doi.org/10.1016/j.csbj.2021.01.018
Vromman M, Anckaert J, Bortoluzzi S, Buratin A, Chen CY, Chu Q et al (2023) Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision. Nat Methods 20(8):1159–1169. https://doi.org/10.1038/s41592-023-01944-6
Cochran KR, Gorospe M, De S (2022) Bioinformatic Analysis of CircRNA from RNA-seq Datasets. Methods Mol Biol 2399:9–19. https://doi.org/10.1007/978-1-0716-1831-8_2
Acknowledgements
The authors would like to express their gratitude to Dr. Irina Zamfir, MD, RCP London, Basildon University Hospital UK, for providing professional English editing of this manuscript and for editorial support.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas that is, revising or critically reviewing the article; giving final approval of the version to be published; agreeing on the journal to which the article has been submitted; and confirming to be accountable for all aspects of the work. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Saleem, A., Khan, M.U., Zahid, T. et al. Biological role and regulation of circular RNA as an emerging biomarker and potential therapeutic target for cancer. Mol Biol Rep 51, 296 (2024). https://doi.org/10.1007/s11033-024-09211-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09211-3