
Abstract
Background
Methicillin-resistant Staphylococcus aureus (MRSA), is considered a potential and aggressive nosocomial pathogen. It accounts for 50% of S. aureus isolates in tertiary hospitals in Iran, however, there is no sufficient evolutionary and epidemiological investigation about this medically important bacterium. We aimed to study the lineage and evolution of MRSA in Northwest Iran during 2021–2022 based on the obtained phenotypic and genotypic characteristics.
Materials and methods
Seventy-two non-duplicate MRSA isolates were collected from 3 referral hospitals in Tabriz, Ardebil, and Urmia cities. The antimicrobial susceptibility patterns were determined by disk diffusion test and micro broth dilution methods. Thereafter 4 virulence genes (eta, etb, pvl, tst) and 5 types of staphylococcal cassette chromosome mec (SCCmec) were detected by PCR. In the final step, representative isolates were selected to be studied by Multilocus sequence typing (MLST).
Results
The highest resistance was observed to erythromycin and clindamycin at a rate of 76.4%, followed by ciprofloxacin (61.1%), gentamicin (54.2%), rifampin (38.9%), and co-trimoxazole (27.8%). All isolates were susceptible to vancomycin. The virulence genes of etb, pvl, tst, and eta were detected in 50%, 29.2%, 21.8%, and 13.9% of isolates, respectively. SCCmec types III and I were the most prevalent types, followed by types IV, II, and V. MLST analysis revealed 6 sequence types: ST6854, ST5282, ST127, ST7804, ST1607, and ST7784. Two MLST-based clonal complexes (CC8, and CC97) were identified as well.
Conclusion
The ST numbers were non-repetitive. CC8 as a pandemic clone and an individual lineage and clinically significant clade was reported as the most prevalent clonal complex. It is essential periodic evaluations of antibiotic susceptibility patterns and study the evolutionary characteristics of medical-challenging microorganisms in particular MRSA to effectively treat and restrict the outbreaks.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Staphylococcus aureus is considered an important and accountable pathogen for mild to severe infections such as septicemia, endocarditis, and osteomyelitis in hospitalized patients and a crucial agent in high mortality rates in hemodialysis patients and intensive care unit (ICU) and surgery units in humans and animals, with the potential to cause coincidental nosocomial infections in part with Pseudomonas aeruginosa, Escherichia coli, and Acinetobacter baumannii [1, 2]. The strong pathogenicity and various virulence factors return to the acquisition of resistance agents to antibiotics [3]. In early 1940, penicillin was introduced for the treatment of infections, however, the emerging resistance was immediately reported as a result of penicillinase enzyme production encoded by plasmid genes [4]. In 1959, methicillin the first semisynthetic penicillin was developed and introduced to overcome the existing infections, unfortunately, the rapid resistance was repeated for this antibiotic as well. This event caused to increase in the numbers of Meticillin-Resistanant Staphylococcus aureus (MRSA), and therefore, the appropriate treatment option changed to vancomycin [5,6,7]. In 2005, MRSA infection deaths overtook AIDS mortalities in the USA [8], and in Iran, these infections have dramatically risen and this is considered an important medical challenge in hospitals [9]. Methicillin resistance is confirmed by the presence of mecA gene located on the Staphylococcal Cassette Chromosome mec (SCCmec) which includes the encoding ability of Penicillin Binding Protein 2a (PBP2a) and consequently prevention of β-lactams effects. SCCmec typing has been developed to determine the epidemiology, outbreaks, and lineage of MRSA isolates. Until now, 13 SCCmec types for MRSA strains have been described based on the combination of ccr and mec gene complexes, and only I–V types are distributed globally, whereas the others exist in local strains of the origin country [5,6,7]. Nowadays, the frequency of MRSA strains has risen to 50–60% in some countries, and Iran’s different provinces, therefore, microbiological research and special attention to treatment, and control policies seem more necessary [10, 11]. MRSA can be divided into healthcare-associated MRSA (HA-MRSA), and community-acquired MRSA (CA-MRSA), according to the molecular epidemiological features. CA-MRSA is a relatively new clone reported globally since the 1990s with the ability to cause infections in healthy and immunocompromised individuals. The high pathogenicity of CA-MRSA returns to the production of Panton–Valentine leucocidin (pvl) [12]. As mentioned earlier, S. aureus has extensive storage of virulence factors including adhesins, host-cell damaging agents, and immunomodulatory molecules that are different in specificity and presence between clones, and a major diversity between infections. The superantigen tst-1 is unique in its ability to cross mucosal surfaces and is the only pyrogenic toxin superantigen known to reactivate bacterial cell wall-induced arthritis and increase the lethal effects of endotoxin on renal tubular cells [13]. Exfoliative toxins (ETs) are defined as extracellular proteins that cause blisters in bullous impetigo and, in the disseminated form of staphylococcal scalded-skin syndrome (SSSS). Previous investigations demonstrate that eta gene is harbored on the temperate phage genome integrated into the S. aureus chromosome, whereas the etb gene is harbored on a large plasmid known as pETB. The eta-carrying S. aureus strains are frequently isolated from bullous impetigo- patients, whereas etb-carrying strains are isolated from SSSS patients [14].
The high incidence of MDR-MRSA isolates suggested a revision of policies for infection control [15]. These strains acquired resistance features to methicillin as a result of SCCmec presence and other antibiotics. The existing reports declare that SCCmec types I, II, and III are the most prevalent types among HA-MRSA, and SCCmec types IV and V among the CA-MRSA [16]. The smallest structural of SCCmec types returns to SCCmec type IV as the mobile version. The SCCmec type IV associated with CA-MRSA can be also pertinent to some nosocomial clones, and studying this type is extremely crucial for molecular and epidemiological investigations [17]. All existing challenges make the treatment of MRSA infections difficult and costlier more than ever [6] and have caused to development of molecular characterization including Pulsed Field Gel Electrophoresis (PFGE), SCCmec typing, and Multilocus Sequence Typing (MLST) [18]. These methods have high speed and reproducibility that make them widely used in the classification of infectious agents, particularly in S. aureus. MLST is developed in Macro-epidemiological and evolutionary investigations, which is based on analysis of polymorphisms nucleotides in the sequence of 7 housekeeping genes, and obtained sequences from each gene locus in bacterial species known as an allele [19, 20].
The majority of the reports in Iran related to MRSA are data restricted to local regions, and there has not been a comprehensive review in this regard [10, 15, 21]. Therefore, we aim to investigate the evolutionary and epidemiological characterization of MRSA isolates by SCCmec typing and MLST methods for the first time in Northwest Iran according to genotyping characteristics and antibiotic resistance profile which will be completely explained in the related sections of the text.
Materials and methods
Study design and setting
This cross-sectional study was conducted at 3 participating referral hospitals in Tabriz (East Azarbaijan province), Ardebil (Ardebil province), and Urmia (West Azarbaijan province) cities (Northwest Iran) from November 2021 to December 2022. All patients who met fulfilled criteria for MRSA infections were included in the study. Patient demographic data including gender, age, and source of infection were obtained from medical unit records in each hospital. (Table 1). The Ethics Committee of Tabriz University of Medical Sciences approved this study (Number: IR.TBZMED.VCR.REC.1400.112).
Bacterial isolation and detection of MRSA
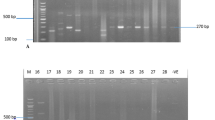
Seventy-two non-duplicate bacterial isolates were identified presumably as S. aureus (MRSA) biochemically and morphologically following positive DNase, catalase, mannitol fermentation, and coagulase tests from (Tabriz) (n = 26), (Ardebil) (n = 21), and (Urmia) (n = 25). After reliable confirmation, meticillin resistance was evaluated by detection of mecA gene through polymerase chain reaction (PCR), and disk diffusion test (DDT) using cefoxitin (30 µg) disk (Mast, UK), and interpreted according to the Clinical and Laboratory Standards Institute (CLSI) 2020 guidelines. Thereafter, the confirmed MRSA isolates were stored in a Tryptic Soy Broth (TSB) medium in part with 30% glycerol and preserved at − 70 °C for the next steps.
Antimicrobial susceptibility
DDT was performed to determine the antibacterial susceptibility patterns on Mueller–Hinton agar using gentamicin (10 µg), ciprofloxacin (15 µg ), co-trimoxazole (20 µg), erythromycin (15 µg), clindamycin (2 µg), and rifampin (20 µg) disks purchased from (Mast, UK) according to standard guidelines [22]. The D-test technique was conducted by clindamycin (2 µg) and erythromycin (15 µg) disks according to the CLSI recommendations. The minimum inhibitory concentration (MIC) was determined for studying the vancomycin susceptibility pattern using the broth dilution method as previously described [22]. S.aureus ATCC 25923 was considered a control strain in described methods. The obtained results were interpreted according to CLSI 2020 guidelines (https://www.clsi.org).
PCR
DNA extraction
DNA extraction was conducted as previously described by Sadeghi et al. [5] with some modifications. Briefly, a primary culture of colonies was prepared. After overnight incubation, 3–5 colonies were dissolved in 450 µL of TE buffer (10 mM Tris, 1 mM EDTA, pH 8). Cell lysis was achieved by treatment with 5 µL of proteinase K (20 mg/mL) for 20 min at 50 °C followed by the addition of 60 µL of 10% SDS for 10 min at 68 °C. Thereafter, 100 µL of 5 M NaCl and 80 µL of cetyltrimethylammonium bromide (CTAB)/NaCl were added and incubated at 65 °C for 10 min. In the next step, 700 µL of chloroform/isoamyl alcohol was added and centrifuged at 12,000×g for 8 min. The supernatant was transferred to a fresh tube and the DNA was precipitated with isopropanol, washed with 70% ethanol, dried, and dissolved in 100 µL of deionized water.
The presence of eta, etb, tst, and pvl genes was detected by the primers (Metabion-Germany) as previously described [23] (Table 2).
SCCmec typing: The optimization of multiplex PCR for SCCmec typing (types I, II, III, IV, and V) was carried out by using 5 previously designed primer pairs (Metabion-Germany) [24] (Table 2).
MLST
The protocol of MLST for MRSA was completely implemented as previously described [25] based on seven housekeeping genes (arc (carbamate kinase), aroE (shikimate dehydrogenase), glpF (glycerol kinase), gmK (guanylate kinase), pta (phosphate acetyltransferase), tpi (triosephosphate isomerase), yqiL (acetyl coenzyme A acetyltransferase). (Table 2). Allelic profiles and Sequence type designations were assigned by comparison with those previously characterized strains using the MLST database via the internet (http://www.mlst.net). Briefly, amplification of MLST genes, Purification of PCR products, reaction sequencing, electrophoresis, assessment of data quality, assigning allele numbers and STs, comparison of strains by the eBURST, and comparison of strains using concatenated allele sequences had been implemented.
Statistical analysis
Data were calculated by SPSSTM software Version 21.0 (IBM Corp., USA). Chi-square or Fisher’s exact tests were employed to determine the significance of the differences. A p-value less than 0.05 was considered statistically significant.
Results
In the present study, seventy-two nonduplicate MRSA isolates were collected from 58.4% male and 41.6% female patients from different clinical sources including wounds, blood, CSF, body fluids, and trachea. The mean age of the patients was 48 + 13 years. All isolates were resistant to cefoxitin disk (30 µg/mL) according to the CLSI breakpoints and harbored the mecA gene. According to the DDT, the highest resistance was observed to erythromycin and clindamycin at a rate of 76.4%, followed by ciprofloxacin (61.1%), gentamicin (54.2%), rifampin (38.9%), and co-trimoxazole (27.8%) in all isolates. Figure 1 depicts the antibiotic resistance patterns separately in each city, and Table 3 has more information on antibiotic combination resistance in 72 isolates. The multi-drug resistance (MDR) was obtained at a rate of 62.5%. We reported the combination resistance for studied antibiotics, in which this rate was determined at 65.27% for clindamycin-erythromycin. Two isolates had been influenced by the D-test phenomenon.
According to the MIC values, all isolates were susceptible to vancomycin. The vancomycin MIC range was recommended 0.25–16 µg/mL by the CLSI, and in the current research, MIC50, and MIC90 were 0.5 and 1 µg/mL, respectively in all isolates.

The antibiotic resistance patterns in Tabriz, Ardebil, and Urmia
The distribution rate of virulence genes in all isolates was 50% for etb, followed by 29.2%, 21.8%, and 13.9% for pvl, tst, and eta genes respectively. The SCCmec types prevalence rate was reported as SCCmec I 22.2%, SCCmec II 9.7%, SCCmec III 43.1%, SCCmec IV 12.5%, and SCCmec V 6.9%. Table 4 describes the gene statistics in each city in detail and Table 5 the genes positive cases in combination.
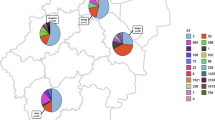
MLST results revealed diverse ST numbers and 2 CCs (CC8 as a pandemic MRSA clone and CC97) in 18 representative isolates (Table 6; Fig. 2).

Dendrogram using average linkage between groups (All isolates)
Discussion
The mortality rate of MRSA infections surpasses 20,000 cases annually in hospitalized patients in the United States; which is comparable with the death rates resulting from acquired immune deficiency syndrome (AIDS), in part due to viral hepatitis, and tuberculosis. In 2005, MRSA-associated deaths overtook AIDS mortalities in the USA [10]. In Iran, these infections have dramatically risen and this is considered an important medical challenge, particularly in hospitals [9]. The treatment procedures for MRSA infections rely on antibiotics, however, medicine nowadays is faced with a growing problem of antibiotic resistance [26,27,28]. The main goal of this study was to determine the lineage and evolution of 72 MRSA isolates in three provinces of Northwest Iran based on the obtained critical phenotypic and genotypic characteristics including the most prevalent SCCmec types and harboring toxin genes and critical resistance patterns. In this regard, we investigated the resistance rate for 7 antibiotics with DDT and micro broth dilution methods. The results elucidated the equal resistance rate to erythromycin and clindamycin in 72 isolates at a rate of 76.4%, and the lowest resistance rate for co-trimoxazole at a rate of 27.8%. Regardless of intermediate results, rifampin, co-trimoxazole, and gentamicin were reported most effective antibiotics against MRSA isolates. Also, the isolates from Tabriz had a higher resistance to all investigated antibiotics. The resistance in MRSA occurs through drug target alteration, inactivation of drug enzymes, altered drug accessibility, increased efflux of antimicrobial compounds, and a multitude of mobile genetic elements [29]. The MDR rate in the present study was reported at 62.5%, which following the recent research by Ahmadishoar et al. in 2021 in Tabriz that demonstrated a 68.2% MDR and also a whole-susceptible rate for vancomycin [15], and Dibah et al. in 2014 that reported co-trimoxazole as an effective antibiotic against the 41 MRSA isolates with 100% susceptibility for vancomycin in Ardebil [30]. We also reported the combination resistance, in which this rate was determined at 65.27% for clindamycin-erythromycin. This resistance is considered significant because two antibiotics had been influenced by the D-test phenomenon. D-test has been employed for screening inducible resistance that is affected by erm gene. Therapeutic failure for clindamycin is one of the consequences of inducible resistance [31]. A rather new study in the central provinces of Iran is under our research. In this way, Co-trimoxazole had a lower resistance (33.3%) in comparison with erythromycin, ciprofloxacin, gentamicin, and clindamycin [6]. The high resistance to beta-lactams has been reported in other Iranian studies as well [32, 33]. Due to the high prevalence of MRSA, precise and comprehensive policies that describe accurately epidemiological data must be available in each country to control the infections and subsequent medical challenges.
The presence of core genome recombination hotspots in the S. aureus genome has been revealed to be pertinent to the existence of mobile genetic elements [34]. Molecular detection of the mecA gene using PCR is commonly carried out to confirm the presence of MRSA isolates. This method is still the main recommendation even though it cannot be done routinely. However, the identification of MRSA with disk diffusion is still widely used because it can be done quickly and at a lower cost [35]. In the present study, both techniques were employed for accurate confirmation. 100% of our isolates were resistant to cefoxitin and harbored the mecA gene. On the other hand, MRSA isolates harbor various toxin-encoding genes. We screened 29.2% portion for pvl gene. It is produced by approximately 5% of S. aureus strains, and as the most widely investigated S. aureus virulence factor is expressed repeatedly in MRSA more than MSSAs [36]. This gene is commonly studied as a marker for community-acquired MRSA and epidemiologically associated with prevalent CA-MRSA strains carrying SCCmec type IV, V, VI, VII, and VIII, accountable for deep dermal and soft-tissue infections. This point should also be taken into account that the increasing prevalence of pvl-encoding HA-MRSA is a critical concern that can worsen infections [37]. Goudarzi et al. in Iran reported a 3.9% portion of isolates that harbor eta, etb, pvl, and tst genes together [33]. This is despite none of our isolates showing this condition. Another research in Iraq, the neighboring country of Iran, displayed a 3.95% portion for pvl, 80.26% tst, and 1.31 etb genes [38]. We screened tst at a rate of 21.8%. More results of these toxin-encoding genes have been implied in other studies from different countries [39, 40].
Direct analysis of the bacterial genome recently has been performed in epidemiological research [41]. The importance of molecular strain typing as an integral part of epidemiological studies is in the investigation of common sources and spread of infection from one patient to another, sporadic and nosocomial infections, tracking pathogenic strains, distinguishing endemic strains from epidemic strains, determining the antibiotics resistance and susceptibility patterns and proper strategy in the treatment and effectively managing and controlling of the related infections [24]. When comparing different molecular methods, it is important to know what region of the genome each method evaluates, for example, PFGE is considered a gold standard for epidemiological classifications in challenging nosocomial pathogens and assesses the entire chromosome based on the total size of the restriction fragments. Hence, minor genetic changes may go undetected. This is also useful to determine the genetic relatedness of MRSA strains isolated during a relatively short period (1–3 months), where presumably, the genetic variability is limited. On the other hand, PCR-based methods detect a specific region of the chromosome at a specific location [42, 43]. The genetic background based on the MLST and SCCmec typing is a better understanding of the epidemiology of long-term population relatedness of MRSA and defines the epidemic clones appropriately according to nucleotide variations [44].
The existing reports declare that SCCmec types (I, II, III) and (IV, V) are the most prevalent types among HA-MRSA and CA-MRSA respectively [16]. In the present study, we screened SCCmec III at 43.1%, followed by SCCmec I at 22.2%. Sedaghat et al. conducted a similar study and found the SCCmec III as the most prevalent type [45], which is under our results. Another Iranian research reported SCCmec type III as the most prominent type [46]. The high prevalence of SCCmec III probably highlights the nosocomial origin of MRSA isolates. The smallest structural of SCCmec types returns to SCCmec type IV as the mobile version. Besides being frequently related to CA-MRSA, SCCmec IV can be associated with some nosocomial clones, and studying type IV is extremely crucial for molecular epidemiological investigations of CA-MRSA strains [17]. However, this type is considered the third most common type in the present study. Indian research reported that all of the major SCCmec types, particularly SCCmec type III are distributed among (HA) MRSA isolates [47]. More results about SCCmec types are also available in other studies [48, 49].
In the current study, 18 representative isolates (details of isolates and selection criteria mentioned earlier) had been selected for the MLST analysis, and 6 ST numbers (6854, 5282, 127, 7804, 1607, 7784), and 2 colon complexes as CC8 (an individual lineage and clinically significant clade) and CC97 were identified. The CC8 consisted of SCCmec types I and III harboring isolates, however, CC97 included SCCmec type I carrying isolates. CC97 is one of the important S. aureus CCs in bovines, and recently, a livestock origin of the human pandemic CC97 MRSA strains has been displayed, resulting in two emergent human epidemics CC97 (CA-MRSA) clones [50]. Other studies approve the validity of this matter [50, 51].
An Iranian investigation screened the ST239 as the prevalent ST number in the majority of isolates in two central provinces [52]. Phylogenetic evidence displayed a hospital transmission and intercontinental spread of CC8 (ST239) isolates through South America, North America, Europe, and Asia [53]. The spread of CC8 (ST239) was from South America to Europe and from Thailand to China in the 1990s [54]. The most frequently reported MRSA CCs worldwide between 1961 and 2008 were CC5, CC8, CC30 CC22, and CC45. In brief, CC5 and CC8 are the most prevalent global clonal complexes [55]. Recent shreds of evidence from Africa are limited, however, suggested a predominance of CC8, and infrequently CC5 and CC8 [56].
In comparison to the Japanese research that reported the prevalence of enterotoxin genes and drug-resistance profiles differ according to STs/CCs. Most CC5 clones represented enterotoxin gene cluster and tst-1, while pvl/ACME-positive ST8/SCCmec IVa-spa t008-agrI (USA300 clone) harbored sek, seq, sak, and speG genes [57], our findings reveal a separate ST number (ST7804 and ST7784 respectively) for tst-positive and pvl-positive isolates, and inclusion in the CC8 for the pvl- positive isolates and a SCCmec type III profile.
Harris et al. sequenced 63 isolates of ST239 as a globally disseminated HA clone defined by MLST. Their reports showed that this ST number is considered a global geographic sequence number and demonstrated the possibility of employing CGS analysis to track transmission within a single hospital [58]. Evaluation of genetic variation at single MLST loci displayed that yqiL as one of the housekeeping genes of S. aureus had various more polymorphic sites when compared with the other 6 gene fragments and there was suggestive evidence of a signature of recombination within the yqiL locus [59].
Overall, epidemiological studies are a broad discussion, and detected ST numbers in the current study were non-repetitive and had not been reported in any other Iranian investigations. We collected the isolates from 3 vast provinces in Northwest Iran, and their phenotypic and genotypic characteristics were studied. The profile of antibiotic resistance was still worrying and increasing. Fortunately, vancomycin has not shown any resistance. Considering the importance of the issue, it is suggested to evaluate the typing patterns of the isolates in each region alternately with applying different methods together, which can provide more useful and comprehensive information. With the availability of epidemiological and lineage data, the management and prevention of nosocomial infections that pose a serious burden on the health system and immunocompromised patients, have become easier. If these points are achieved, the mortality rate and the economic and psychological burden will have a significant reduction.
Conclusion
Our study presents an overview of antibiotic resistance profiles, and epidemiological information on MRSA isolates in Northwest Iran. MRSA rates continue to rise rapidly in many countries and have a globally dynamic spread. Its clones are defined by lineage and SCCmec variant types. They usually display successful and widely spread variants. Similar to the global evidence, CA-MRSA is reported as more invasive, transmissible, and increasingly difficult to differentiate from HA-MRSA. Many MRSA isolates originate from a restricted number of historically dominant clonal lineages, and some other clones are found worldwide, or restricted to certain geographic areas. They imply differences in transmission routes. Therefore, the Harmonization of surveillance will improve epidemiological studies in the future.
ST numbers were exclusive and CC8 as a pandemic, individual lineage, and clinically significant clade clone was reported as the most prevalent clonal complex. Generally, CC5 and CC8 are the most prevalent CCs across the world.
The flexibility of the S. aureus genome allows for the adaptive radiation of successful lineages. Mobile genetic elements that carry many of the virulence factors in S. aureus are often lineage-associated.
Continuous efforts to understand the variable epidemiology of S. aureus infection in humans and animals as a clinical pathogen are crucial steps for effective infection control, appropriate antimicrobial treatment, and monitoring the species’ evolution and organism’s phylogeny to the prevention of the spread of virulence lineages and for characterizing outbreaks, however, there have been few analyses employing whole genome sequencing, looking at the overall relationships among different clonal groups of this organisms. According to a remarkable raising in MRSA prevalence, a precise and updated report that describes accurately epidemiological data must be available in each country to control the infections and improve national policies.
Data availability
The data supporting this study’s findings are available and included in the article.
References
Hasani A, Asadi Faezi N, Ahangarzadeh Rezaee M, Sheykhsaran E, Darabi N, Ebrahimzadeh Leylabadlo H (2019) Determination of antimicrobial resistance patterns in bloodstream infections-isolated bacteria from a University Tertiary Hospital patients. Int J Enteric Pathog 7(2):49–54
Abad HEK, Sadeghi J, Aghazadeh M, Rezaee MA, Samadi H (2020) Frequency of fnbA, fnbB, hla and cna genes in Staphylococcus aureus isolates obtained from blood cultures and their antimicrobial susceptibility pattern in Tabriz, Iran. Arch Pharm Pract 1:137
Kumar S, Singh S, Kumar V, Datta S, Dhanjal DS, Sharma P et al (2020) Pathogenesis and antibiotic resistance of Staphylococcus aureus. Model organisms for microbial pathogenesis, biofilm formation and antimicrobial drug discovery. Springer, Singapore, pp 99–115
Peterson E, Kaur P (2018) Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front Microbiol 9:2928
Sadeghi J, Mansouri S (2014) Molecular characterization and antibiotic resistance of clinical isolates of methicillin-resistant Staphylococcus aureus obtained from Southeast of Iran (Kerman). APMIS 122(5):405–411
Firoozeh F, Omidi M, Saffari M, Sedaghat H, Zibaei M (2020) Molecular analysis of methicillin-resistant Staphylococcus aureus isolates from four teaching hospitals in Iran: the emergence of novel MRSA clones. Antimicrob Resist Infect Control 9(1):1–8
Sheykhsaran E, Abbasi A, Baghi HB, Ghotaslou R, Sharifi Y, Sefidan FY et al (2022) Staphylococcus aureus: a bacterial candidate for multiple sclerosis incidence and progression. Rev Res Med Microbiol 33(4):212–220
Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S et al (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298(15):1763–1771
Zeinalpour Ahrabi S, Rahbarnia L, Dehnad A, Naghili B, Ghaffari Agdam MH, Nazari A (2019) Incidence of oxacillin-susceptible meca-positive Staphylococcus aureus (OS-MRSA) isolates and TSST-1 virulence factor among high school students in Tabriz, Northwest of Iran. Arch Clin Infect Dis 14(4):1
Dadashi M, Nasiri MJ, Fallah F, Owlia P, Hajikhani B, Emaneini M et al (2018) Methicillin-resistant Staphylococcus aureus (MRSA) in Iran: a systematic review and meta-analysis. J Global Antimicrob Resist 12:96–103
Hosseini M, Shapouri Moghaddam A, Derakhshan S, Hashemipour SMA, Hadadi-Fishani M, Pirouzi A et al (2020) Correlation between biofilm formation and antibiotic resistance in MRSA and MSSA isolated from clinical samples in Iran: a systematic review and meta-analysis. Microb Drug Resist 26(9):1071–1080
Takadama S, Nakaminami H, Kaneko H, Noguchi N (2020) A novel community-acquired MRSA clone, USA300-LV/J, uniquely evolved in Japan. J Antimicrob Chemother 75(11):3131–3134
Etter D, Schelin J, Schuppler M, Johler S (2020) Staphylococcal enterotoxin C—an update on SEC variants, their structure and properties, and their role in foodborne intoxications. Toxins 12(9):584
Elbargisy RM (2022) Distribution of leukocidins, exfoliative toxins, and selected resistance genes among methicillin-resistant and methicillin-sensitive clinical strains in Egypt. Open Microbiol J 16:1
Ahmadishoar S, Pour NK, Sadeghi J, Nahaei MR, Kheirkhah B (2021) Molecular epidemiology of clinical isolates of methicillin-resistant Staphylococcus aureus by multilocus sequence typing in northwestern (Tabriz) and southern (Kerman) of Iran: the emergence of MRSA ST4848-SCCmec III. Gene Rep 24:101212
Monecke S, Coombs G, Shore AC, Coleman DC, Akpaka P, Borg M et al (2011) A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS ONE 6(4):e17936
Jin Y, Zhou W, Zhan Q, Zheng B, Chen Y, Luo Q et al (2021) Genomic epidemiology and characterization of methicillin-resistant Staphylococcus aureus from bloodstream infections in China. MSystems 6(6):e00837
Chen Y, Sun L, Wu D, Wang H, Ji S, Yu Y (2018) Using core-genome multilocus sequence typing to monitor the changing epidemiology of methicillin-resistant Staphylococcus aureus in a teaching hospital. Clin Infect Dis 67(suppl2):S241–S248
Cvetnić L, Samardžija M, Duvnjak S, Habrun B, Cvetnić M, Jaki Tkalec V et al (2021) Multi locus sequence typing and spa typing of Staphylococcus aureus isolated from the milk of cows with subclinical mastitis in Croatia. Microorganisms 9(4):725
Liu Y, Ji Y (2020) Multilocus sequence typing of Staphylococcus aureus. Methicillin-resistant Staphylococcus aureus (MRSA). Springer, London, pp 95–102
Tahbaz SV, Azimi L, Nowroozi J, Armin S, Fallah F (2019) Multilocus sequence typing and antibiotic resistant patterns of the meticillin-resistant Staphylococcus aureus isolates from different clinical specimens. Rev Med Microbiol 30(2):77–82
Schwalbe R, Steele-Moore L, Goodwin AC (2007) Antimicrobial susceptibility testing protocols. Crc Press
Japoni-Nejad A, Rezazadeh M, Kazemian H, Fardmousavi N, van Belkum A, Ghaznavi-Rad E (2013) Molecular characterization of the first community-acquired methicillin-resistant Staphylococcus aureus strains from Central Iran. Int J Infect Dis 17(11):e949–e954
Japoni A, Jamalidoust M, Farshad S, Ziyaeyan M, Alborzi A, Japoni S et al (2011) Characterization of SCCmec types and antibacterial susceptibility patterns of methicillin-resistant Staphylococcus aureus in Southern Iran. Jpn J Infect Dis 64(1):28–33
Saunders NA, Holmes A (2014) Multilocus sequence typing (MLST) of Staphylococcus aureus. Methicillin-resistant Staphylococcus aureus (MRSA) protocols. Springer, London, p 113
Sheykhsaran E, Bannazadeh Baghi H, Soroush Barhaghi MH, Alizadeh N, Memar MY, Etemadi S et al (2018) The rate of resistance to tetracyclines and distribution of tetA, tetB, tetC, tetD, tetE, tetG, tetJ and tetY genes in Enterobacteriaceae isolated from Azerbaijan, Iran during 2017. Physiol Pharmacol 22(3):205–212
Sheykhsaran E, Baghi HB, Soroush MH, Ghotaslou R (2019) An overview of tetracyclines and related resistance mechanisms. Rev Res Med Microbiol 30(1):69–75
Lalehzadeh A, Soroush MH, Sadeghi J, Ahangarzadeh Rezaee M, Pirzadeh T, Yeganeh Sefidan F (2019) Determination of fosfomycin resistant Enterobacteriaceae in isolates and frequency of fos genes in Tabriz hospitals during 2018. J Biochem Technol 10(2):143–148
Vestergaard M, Frees D, Ingmer H (2019) Antibiotic resistance and the MRSA problem. Microbiol Spectr 7(2):7
Dibah S, Arzanlou M, Jannati E, Shapouri R (2014) Prevalence and antimicrobial resistance pattern of methicillin resistant Staphylococcus aureus (MRSA) strains isolated from clinical specimens in Ardabil, Iran. Iran J Microbiol 6(3):163
Khan AA, Farooq J, Abid M, Zahra R (2020) Assessment of inducible clindamycin resistance and hyper variable region (HVR) of mecA gene in clinical staphylococci. Pak J Med Sci 36(2):136
Shekarabi M, Hajikhani B, Salimi Chirani A, Fazeli M, Goudarzi M (2017) Molecular characterization of vancomycin-resistant Staphylococcus aureus strains isolated from clinical samples: a three year study in Tehran, Iran. PLoS ONE 12(8):e0183607
Goudarzi M, Seyedjavadi SS, Nasiri MJ, Goudarzi H, Nia RS, Dabiri H (2017) Molecular characteristics of methicillin-resistant Staphylococcus aureus (MRSA) strains isolated from patients with bacteremia based on MLST, SCCmec, spa, and agr locus types analysis. Microb Pathog 104:328–335
Everitt RG, Didelot X, Batty EM, Miller RR, Knox K, Young BC et al (2014) Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus. Nat Commun 5(1):3956
Khairullah AR, Rehman S, Sudjarwo SA, Effendi MH, Ramandinianto SC, Gololodo MA et al (2022) Detection of mecA gene and methicillin-resistant Staphylococcus aureus (MRSA) isolated from milk and risk factors from farms in probolinggo, Indonesia F1000Research:11
Labandeira-Rey M, Couzon F, Boisset S, Brown EL, Bes M, Benito Y et al (2007) Staphylococcus aureus Panton-valentine Leukocidin causes necrotizing pneumonia. Science 315(5815):1130–1133
Bhatta DR, Cavaco LM, Nath G, Kumar K, Gaur A, Gokhale S et al (2016) Association of panton valentine leukocidin (PVL) genes with methicillin resistant Staphylococcus aureus (MRSA) in Western Nepal: a matter of concern for community infections (a hospital based prospective study). BMC Infect Dis 16:1–6
Rasheed NA, Hussein NR (2020) Characterization of different virulent factors in methicillin-resistant Staphylococcus aureus isolates recovered from Iraqis and Syrian refugees in Duhok city, Iraq. PLoS ONE 15(8):e0237714
Tegegne HA, Florianová M, Gelbíčová T, Karpíšková R, Koláčková I (2019) Detection and molecular characterization of methicillin-resistant Staphylococcus aureus isolated from bulk tank milk of cows, sheep, and goats. Foodborne Pathog Dis 16(1):68–73
Doudoulakakis A, Spiliopoulou I, Giormezis N, Syridou G, Nika A, Bozavoutoglou E et al (2022) Methicillin-resistant Staphylococcus aureus transmission and hospital-acquired bacteremia in a neonatal intensive care unit in Greece. J Infect Chemother 28(2):176–180
Suzuki Y, Omoe K, Hu DL, Satoo Y, Ono HK, Monma C et al (2014) Molecular epidemiological characterization of Staphylococcus aureus isolates originating from food poisoning outbreaks that occurred in Tokyo, Japan. Microbiol Immunol 58(10):570–580
Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH et al (1995) Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33(9):2233–2239
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG (2000) Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38(3):1008–1015
Lima DF, Brazão NBV, Folescu TW, Neves FP, Ferreira AG, Santos EA et al (2014) Panton-valentine Leukocidin (PVL) gene carriage among Staphylococcus aureus strains with SCCmec types I, III, IV, and V recovered from cystic fibrosis pediatric patients in Brazil. Diagn Microbiol Infect Dis 78(1):59–62
Sedaghat H, Esfahani BN, Halaji M, Jazi AS, Mobasherizadeh S, Havaei SR et al (2018) Genetic diversity of Staphylococcus aureus strains from a teaching hospital in Isfahan, Iran: the emergence of MRSA ST639-SCCmec III and ST343-SCCmec III. Iran J Microbiol 10(2):82
Darban-Sarokhalil D, Khoramrooz SS, Marashifard M, Hosseini SAAM, Parhizgari N, Yazdanpanah M et al (2016) Molecular characterization of Staphylococcus aureus isolates from southwest of Iran using spa and SCCmec typing methods. Microb Pathog 98:88–92
Sunagar R, Hegde NR, Archana GJ, Sinha AY, Nagamani K, Isloor S (2016) Prevalence and genotype distribution of methicillin-resistant Staphylococcus aureus (MRSA) in India. J Global Antimicrob Resist 7:46–52
Monecke S, Slickers P, Gawlik D, Müller E, Reissig A, Ruppelt-Lorz A et al (2018) Variability of SCCmec elements in livestock-associated CC398 MRSA. Vet Microbiol 217:36–46
Youssef CRB, Kadry AA, El-Ganiny AM (2022) The alarming coincidence of toxin genes with staphylococcal cassette chromosome mec (SCCmec) in clinical MRSA isolates. Saudi J Biol Sci. https://doi.org/10.1016/j.sjbs.2022.02.026
Spoor LE, McAdam PR, Weinert LA, Rambaut A, Hasman H, Aarestrup FM et al (2013) Livestock origin for a human pandemic clone of community-associated methicillin-resistant Staphylococcus aureus. MBio 4(4):e00356
Luini M, Cremonesi P, Magro G, Bianchini V, Minozzi G, Castiglioni B et al (2015) Methicillin-resistant Staphylococcus aureus (MRSA) is associated with low within-herd prevalence of intra-mammary Infections in dairy cows: genotyping of isolates. Vet Microbiol 178(3–4):270–274
Havaei SA, Halaji M, Vidovic S, Dillon J-AR, Karbalaei M, Ghanbari F et al (2017) Prevalence and genotyping of methicillin-resistant and-susceptible Staphylococcus aureus strains isolated from patients in a university hospital, Isfahan, Iran. Jundishapur. J Microbiol 10:5
Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N et al (2010) Evolution of MRSA during hospital transmission and intercontinental spread. Science 327(5964):469–474
Gray RR, Tatem AJ, Johnson JA, Alekseyenko AV, Pybus OG, Suchard MA et al (2011) Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a bayesian framework. Mol Biol Evol 28(5):1593–1603
Stefani S, Chung DR, Lindsay JA, Friedrich AW, Kearns AM, Westh H et al (2012) Meticillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int J Antimicrob Agents 39(4):273–282
Moodley A, Oosthuysen W, Duse A, Marais E (2010) Molecular characterization of clinical methicillin-resistant Staphylococcus aureus isolates in South Africa. J Clin Microbiol 48(12):4608–4611
Aung MS, Urushibara N, Kawaguchiya M, Hirose M, Ito M, Habadera S et al (2021) Clonal diversity of methicillin-resistant Staphylococcus aureus (MRSA) from bloodstream infections in northern Japan: identification of spermidine N-acetyltransferase gene (speG) in staphylococcal cassette chromosomes (SCCs) associated with type II and IV SCCmec. J Global Antimicrob Resist 24:207–214
Driebe EM, Sahl JW, Roe C, Bowers JR, Schupp JM, Gillece JD et al (2015) Using whole genome analysis to examine recombination across diverse sequence types of Staphylococcus aureus. PLoS ONE 10(7):e0130955
Budd KE, McCoy F, Monecke S, Cormican P, Mitchell J, Keane OM (2015) Extensive genomic diversity among bovine-adapted Staphylococcus aureus: evidence for a genomic rearrangement within CC97. PLoS ONE 10(8):e0134592
Acknowledgements
We would like to all hospitals for their assistance and all institutes for providing expertise that greatly assisted.
Funding
This research was financially supported by the Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran, and was written based on a dataset of a Ph.D. thesis registered at Tabriz University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
ES: Conceptualization, Data curation, Writing—original draft, Writing—review & editing. AA: Formal analysis, Writing—original draft, Writing—review & editing. MYM: Writing—review & editing, Writing—original draft, Data curation. HBB: Writing—original draft, Data curation. RG: Writing—review & editing, Writing—original draft, Data curation, Conceptualization. DL: Writing—review & editing, Writing—original draft. JS: Project administration, Conceptualization, Data curation, Writing —original draft, Writing—review & editing. FYS: Data analysis, review. YS: Review, Data Curation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The Ethics Committee of Tabriz University of Medical Sciences approved this study (Number: IR.TBZMED.VCR.REC.1400.112). All ethical considerations have been observed during this research. The collection of isolates was conducted with the full consent of the patients and parents were legally authorized representatives of the minor subjects.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sheykhsaran, E., Sadeghi, J., Memar, M.Y. et al. Epidemiological characterization of clinical isolates of meticillin resistant Staphylococcus aureus through multilocus sequence typing and staphylococcal cassette chromosome mec typing in Northwest Iran. Mol Biol Rep 51, 58 (2024). https://doi.org/10.1007/s11033-023-08951-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-023-08951-y