
Abstract
Breast cancer is the most common cancer worldwide that costs lives of millions of people every year. Plant products with potential anticancer activities have become a vital source of novel agents in treating cancer. Adiantum capillus veneris (ACV) and Pteris Quadriureta (PQ) are such traditional herbs with potential pharmacological properties. In this study, both crude methanol extract and gold nanoparticles of ACV and PQ were tested for their anticancer activities in MCF7 and BT47 cell lines. By using GC–MS, we have identified 23 and 28 bioactive compounds in ACV and PQ respectively. We analysed the effects of ACV and PQ nanoparticles on various proteins involved in cell cycle and apoptosis using western blotting and PCR. With the help of flow cytometry, we measured number of cells undergoing apoptosis. We found that both the crude extract and nanoparticles have anti-proliferative and apoptosis inducing properties against MCF7 and BT47 cell lines. We also performed molecular docking to check whether there were any interactions between proteins involved in apoptosis and cell cycle and bioactive compounds present in the plant extracts. By using docking analysis, we also showed that phytol and eicosapentaenoic acid present in ACV and PQ interact with Bcl2 and cyclin D1. These findings demonstrate that ACV and PQ possess anticancer activities by modulating proteins involved in cell cycle and apoptosis. ACV and PQ that effectively modulate various oncogenic molecules can be used as promising agent for cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is characterized by key hallmarks such as cell proliferation, apoptosis evasion, invasion and angiogenesis [1]. Breast cancer is the most common cancer in women, accounting for 522,000 deaths worldwide [2, 3]. Around 1.67 million new cases of breast cancer were diagnosed in 2012 accounting for 25% of all cancers [4, 5]. Currently, research in breast cancer is mainly focused on chemoprevention and targeted therapies [6,7,8]. Past two decades, medicinal plants rich in antioxidant phytochemicals have received growing attention as potential preventive and therapeutic agents. Use of crude extract is a strategic approach to administer a cocktail of phytochemical entities that could modulate key mechanisms important for carcinogenesis and tumour promotion [9,10,11,12]. Furthermore, synergistic interactions among the phytochemicals can ensure higher potency in addition to overcoming problems of toxicity and resistance [13, 14].
In recent years, application of nanotechnology is widely implemented in diverse areas such as medicine, engineering, textile, life science, biomedical, tissue engineering, environmental industries, drug delivery and gene delivery [15, 16]. Metal nanoparticles have great potential towards biological sensing and cancer therapy. In particular, gold nanoparticles are used in diagnosis and treatment of various diseases including diabetes, cancer, Parkinson’s, Alzheimer’s, HIV/AIDS, tuberculosis and cardiovascular diseases due to their biocompatibility and non-cytotoxicity [17,18,19]. Of late, synthesis of nanoparticles using plant extract provides advancement over other methods as it is simple, single step, cost-effective, environment friendly and relatively reproducible, less toxic and more effective [20, 21].
In our previous study (paper in press), we showed that the crude extracts of Adiantum capillus veneris (ACV) and Pteris quadriureta (PQ) have antioxidant and antimicrobial activities. Phytochemical analysis of ACV and PQ showed presence of several secondary metabolites including flavonoid, phenol, tannin and terpenoids, which have been reported to possess anticancer activity. Therefore, this study was designed to synthesize and characterize stable gold nanoparticles of ACV and PQ and investigate their effects on MCF7 and BT47 cell lines. We have analysed the effects of ACV and PQ on cell proliferation and apoptosis in MCF7 and BT47 cells.
Materials and methods
Chemicals
Chemicals were obtained from Sigma-Aldrich Chemical Company (St. Louis, MO, USA) and HiMedia Labs (Mumbai, India). Primary and secondary antibodies for glyeraldehyde-3-phosphate dehydrogenase (GAPDH), Bax, Bcl2, proliferating cell nuclear antigen (PCNA), cyclin D1, cyclin-dependent kinase (CDK)-4, caspase-3, caspase-9, cytochrome c, p21, and histone were purchased from Santa-Cruz Biotechnology, USA. Power SYBR Green PCR master mix was obtained from Applied Biosystems, California, USA. Oligonucleotide primers were procured from Sigma Genosys, USA. All other reagents used were of analytical grade.
Synthesis of gold nanoparticles
The leaves of ACV and PQ were washed thoroughly thrice with distilled water and were shade dried for 5 days. The fine powder was obtained from the dried leaves by using blender and sterilized at 121 °C for 15 min. 20 g of powder was mixed with 200 ml of distilled water and kept in a boiling water bath at 60 °C for 10 min. The extracts were filtered with Whatman filter paper No. 1 and stored at 4 °C for further studies. For the biosynthesis of gold nanoparticles, 1.5 ml of plant extract was mixed with 30 ml of auric chloride solution (1 mM/ml) and incubated at 28 °C for 24 h [22].
SEM and TEM Analysis of gold nanoparticles
To gain further insight into the features of the gold nanoparticles, the sample was analysed using a scanning electron microscope (JEOL 6380A; Tokyo, Japan) to record the micrographs of synthesized Au-NPs. The samples were also characterized morphologically by TEM. TEM images were formed using transmitted electrons (instead of the visible light) which can produce magnification details up to ×1,000,000 with resolution better than 10 Å. The images can be resolved over a fluorescent screen or a photographic film. Furthermore, the analysis of the X-ray produced by the interaction between the accelerated electrons with the sample allows determining the elemental composition of the sample with high spatial resolution. The sample can be loaded on carbon coated copper grid or can be made in disc form, 3 mm diameter, with a thinned (electron transparency) central area.
Cell culture
MCF7 and BT47 cell lines procured from Imgenex India were cultured in DMEM basal medium with 10% foetal bovine serum and antibiotics. Confluent cultures of MCF7 and BT47 cells were sub-cultured and maintained in a CO2 incubator at 37 °C.
MTT assay
Cells were plated at a density of 1 × 105 cells/ml into 96-well plates. After overnight growth, cells were incubated with various concentrations of ACV and PQ extracts and their nanoparticles for 24 h. Cells were then washed with PBS and 100 µl of 500 µg/ml MTT solution was added to each well. After incubation for 4 h at 37 °C, the medium was removed, and the insoluble formazan product was dissolved in DMSO. Absorbance was measured using an ELISA plate reader at 570 nm and cell viability was calculated as the percentage of viable cells that produce a dark blue formazan product in the treated cells versus untreated cells.
TUNEL assay
TUNEL assay was performed using APO-BrdU TUNEL assay kit. The cells were gently fixed with 70% ice cold ethanol at − 20 °C for 1 h, suspended in PBS containing 0.5 µg/ml RNase, and incubated at 37 °C for 30 min. Following this, cells suspended in 500 µl of PBS were added to 5 ml of 1% p-formaldehyde (w/v in PBS), placed on ice for 15 min, centrifuged at 300×g for 5 min, and washed with PBS. The pellet containing cells was re-suspended in 500 µl of PBS and incubated with 50 µl of the DNA-labeling solution (10 µl of reaction buffer, 0.75 µl of TdT enzyme, 8.0 µl of BrdUTP and 31.25 µl of dH2O) for 60 min at 37 °C in a shaker. At the end of the incubation time, 1 ml of rinse buffer was added to each tube and centrifuged at 300×g for 5 min. The cell pellet was resuspended in 100 µl of antibody solution, incubated at room temperature for 30 min and stained with 500 µl of the propidium iodide/RNase A staining buffer followed by incubation for 30 min at room temperature. Cells were analyzed by flow cytometry to determine the percentage of apoptotic cells.
Annexin V assay
Induction of apoptosis in breast cancer cells upon treatment with ACV and PQ was tested by flow cytometry analysis. Cells were seeded in a 6-well plate with 5 × 104 cells in each well. After 24 h of treatment with ACV and PQ, cells were harvested by trypsinization and washed twice with ice-cold PBS. Cells were then stained with annexin-FITC/PI using annexin staining kit as per the manufacturer’s instructions. At the end of the assay, cells were subjected to flow cytometry to determine percent of apoptotic cells. A minimum of 20,000 cells were collected for each measurement.
Determination of mitochondrial transmembrane potential
Changes in the mitochondrial transmembrane potential were determined using JC-1 assay. Briefly, cells were plated at a seeding density of 2 × 105 cells/well in a 6-well plate. After 24 h of treatment, cells were incubated with 5 µM JC-1 for 30 min at room temperature in the dark. The presence of JC-1 monomers/dimers was examined under a fluorescent microscope using filter pairs of 530/590 nm (dimers) and 485/538 nm (monomers).
RNA extraction and quantitative real-time RT-PCR
Total RNA from cell lines was extracted using Trizol reagent as described previously [23]. 5 µg of isolated total RNA was reverse-transcribed to cDNA in a reaction mixture containing 4 µl of 5× reaction buffer, 2 µl of dNTP mixture (10 mM), 20 units of RNase inhibitor, 200 units of avian-myeloblastosis virus (AMV) reverse transcriptase and 0.5 µg of oligo(dT) primer in a total volume of 20 µl. The reaction mixture was incubated at 42 °C for 60 min and the reaction terminated by heating at 70 °C for 10 min. The cDNA was stored at − 80 °C until further use. Quantitative RT-PCR was performed using Power SYBR Green master mix according to the manufacturer’s instructions using a StepOne Plus thermocycler (Applied Biosystems). To the 1 × PCR master mix, 2.5 µl of each cDNA was added in a 20 µl final volume. The PCR conditions were as follows: 95 °C for 5 min, 40 cycles of 30 s at 95 °C, 30 s at 52–60 °C (based on the target), and 60 s at 72 °C. All reactions were carried out in triplicate, and the relative quantitative fold change compared to control was calculated using the comparative Ct method.
Western blotting
Proteins were extracted from cell lines using lysis buffer. The stacking gel consisted of 4.5% acrylamide/0.1% SDS, while resolving gel consisted of 7.5% acrylamide/0.1% SDS. The gels were polymerized using TEMED and freshly prepared ammonium persulphate. The protein samples were prepared by heating them in a boiling water bath in SDS-gel loading buffer containing 62.5 mM Tris (pH 6.8), 2% SDS, 5% 2-mercaptoethanol, 10% glycerol and bromophenol blue. Equivalent protein extracts (50 µg) from each sample were electrophoresed on 12% SDS–PAGE gels. The resolved proteins were electrophoretically transferred to nitrocellulose membrane. The membranes were then incubated in blocking solution (5% non-fat dry milk in PBS or 3% BSA) for 1 h to block non-specific binding sites. The membranes were then probed with primary and secondary antibodies as per manufacturer’s instructions. After extensive washes with high and low salt buffers, the immunoreactive proteins were visualized using enhanced chemiluminescence (ECL) and the specificity was determined by the apparent molecular weight of the target protein. Densitometry was performed on IISP flat-bed scanner and quantitated with Total Lab 1.11 software.
Gas chromatography–mass spectrometry (GC/MS) analysis
GC/MS analysis of the methanolic extract of ACV and PQ was carried out using a Perkin Elmer GC Claurus 500 system and Gas chromatograph coupled with mass spectrometer (GC/MS) along with silica capillary column (30 m × 0.25 mm × 1 µm) which made of 100% dimethyl polysiloxane). For GC/MS identification, an electron ionization system with ionization energy of 70 eV was used. Helium was used as carrier gas at a constant flow rate of 1 ml/min and 2 µl of sample was injected. Injector temperature was set as 260 °C and ion-source temperature was set as 250 °C. The oven temperature was set from 110 °C (isothermal for 2 min), with a raise of 10 °C/min, to 200 °C, then 5 °C/min to 280 °C. The relative percentage quantity of all components was estimated by comparing its average peak area to the total areas. Identification of components was done by National Institute of Standard Technology (NIST) database and WILEY library. The mass spectrum of the unknown compound was compared with spectrum of known compound stored in NIST and WILEY library. The name, molecular weight and structure of each component of plant extract were ascertained.
Docking study
The crystal structures of human protein cyclin D1 (PDB ID: 2W96, Resolution: 2.3 Å) and Bcl2 (PDB ID: 2XA0, Resolution: 2.7 Å) were retrieved from RCSB PDB (http://www.rcsb.org/) and refined using WHAT IF Web Interface. Structures of eicosapentaenoic acid (PubChem CID: 446284) and phytol (PubChem CID: 5280435) were retrieved from PubChem open chemistry database and refined using MarwinSketch software and saved in SDF format prior to docking.
Protein–ligand docking procedures were carried out using the AutoDock docking program. We have set up the docking to search entire surface of the protein using AutoGrid. AutoGrid created a very large 3D grid which can fit entire surface of protein. Docking was performed using genetic algorithm search parameter and output docking parameter was set up with Lamarckian Algorithm. The hydrophobic and hydrogen bond interactions between the receptor and ligands of docked complexes were analysed using the Ligplot program. Chimera visualization software was used to visualize the complexes in three-dimension. Among the analysed natural compounds, the top scored complex of these three proteins with compounds were further studies.
Statistical analysis
The data are expressed as mean ± standard deviation (SD). Statistical analysis was performed using one-way ANOVA followed by Tukey posthoc test for the in vitro experiments. A probability value of < 0.05 was considered significant.
Results
SEM and TEM analysis
SEM images of extracts of both ACV and PQ showed accumulation of AuNPs formed with diameter ranging from 55 to 85 nm. Morphologically nanoparticles were polydispersed, non-coagulated and anisotropy with different shapes including triangular, hexagonal, pentagonal, and truncated triangular shapes and some were spherical in shape. TEM analysis has also revealed the shape and size of the nanoparticles. ACV showed both anisotropy and spherical shaped nanoparticles. The spherical shaped nanoparticles of ACV ranged from 5 to 20 nm, whereas the size of anisotropic nanoparticles was in the range of 20–100 nm. In case of PQ, the size of the spherical nanoparticles was 5–50 nm and the size of the anisotropy nanoparticles was 20–100 nm like. The bright circular spots recorded in the selected area electron diffraction (SAED) patterns corresponding to the Bragg’s planes confirmed the crystalline nature of all types of GNPs (Figs. 1, 2).

Analysis of ACV gold nanoparticles. a Scanning electron micrograph of ACV synthesized gold nanoparticles. b–h Transmission electron micrograph of ACV synthesized gold nanoparticles

Analysis of PQ gold nanoparticles. a Scanning electron micrograph of PQ synthesized gold nanoparticles. b–h Transmission electron micrograph of PQ synthesized gold nanoparticles
Effect of ACV and PQ on Cell viability
We next analysed the effect of these gold nanoparticles on BT47 and MCF7 cells using MTT assay. We determined the concentration of ACV, PQ and their nanoparticles at which it inhibits the viability of BT47 and MCF-7 cells by 50% (IC50). Incubation of cells for 24 h with ACV, PQ and their nanoparticles significantly decreased the cell viability of BT47 and MCF-7 cells in a dose-dependent manner (Fig. 3). We used IC50 concentration for further experiments.

Effect of ACV and PQ on viability of BT47 and MCF7 cells. IC50 value for ACV and PQ and their nanoparticles on BT47 and MCF cells. All doses were tested in triplicates and the experiment was repeated at least three times
ACV and PQ inhibit cell proliferation
As cell proliferation and apoptosis evasion are the key cancer hallmarks, we next investigated the effect of ACV and PQ on cell proliferation. Treatment with ACV and PQ and their nanoparticles significantly reduced the mRNA and protein expression of cyclin D1, CDK4, and PCNA. Furthermore, ACV and PQ increased nuclear p21 expression relative to the cytosolic fraction. Interestingly, ACV and PQ nanoparticles were found to be more effective than crude extracts (Fig. 4).

ACV and PQ Inhibit Cell Proliferation. a Representative qRT-PCR analysis of p21 and cyclin D1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control (mean ± SD; n = 3). b Representative immunoblots of p21, cyclin D1, cyclin-dependent kinase (CDK)-4, and proliferating cell nuclear antigen (PCNA). GAPDH was used as a loading control. c Densitometric analysis. Protein expression from control was designated as 100% in the graph (n = 3). *Significantly different from control (p < 0.001), #Significantly different from control (p < 0.01),  Significantly different from control (p < 0.05). ACV Adiantum capillus veneris, PQ Pteris quadriureta, ACVN ACV nanoparticles, PQN PQ nanoparticles
Significantly different from control (p < 0.05). ACV Adiantum capillus veneris, PQ Pteris quadriureta, ACVN ACV nanoparticles, PQN PQ nanoparticles
ACV and PQ induce intrinsic apoptosis
To test whether the inhibitory effect of ACV and PQ on cell proliferation was also associated with apoptosis induction, we performed TUNEL assay which revealed TUNEL positive cells (upper right column) in the groups treated with ACV, PQ and their nanoparticles compared to untreated cells, confirming apoptosis induction (Fig. 5). Next, we performed annexin V assay which evaluates phospholipid turnover, an event typically associated with apoptosis. ACV and PQ treatment increased annexin positive cells compared to untreated cells, indicating apoptosis induction. Further to know whether apoptosis induced by ACV and PQ are mitochondria mediated, we performed JC-1 assay. Treatment with ACV and PQ induced depolarization of the mitochondrial membrane which resulted in change in fluorescence from red to green compared to untreated cells (Fig. 6).

ACV and PQ Induce Apoptosis. TUNEL assay by flow cytometry. Q1 and Q2 region shows TUNEL positive cells. ACV Adiantum capillus veneris PQ Pteris quadriureta, ACVN ACV nanoparticles, PQN PQ nanoparticles

ACV and PQ Induce phospholipid turn over and loss of mitochondrial membrane potential. a Plots showing annexin-FITC/PI staining of ACV and PQ treated breast cancer cells. The cells at early apoptosis are in the Q4 quadrant (Annexin V+/PI−), and the ones at late apoptosis are in the Q2 quadrant (Annexin V+/PI−). b Red fluorescence is visible in cell areas with high mitochondrial membrane potential, while yellowish green fluorescence of JC-1 monomer is seen in cell areas with low mitochondrial membrane potential. (Color figure online)
Next, we examined the effect of ACV and PQ on proteins involved in apoptosis using PCR and western blotting. Our results revealed that treatment with ACV, PQ and their nanoparticles has significantly increased the expression of Bax, caspase-9, and -3 and decreased the expression of Bcl-2 compared to untreated cells. Furthermore, ACV and PQ treatment induced the release of cytochrome C into the cytosol as evidenced by increased cytosolic levels of these proteins relative to the mitochondrial fraction (Fig. 7).

ACV and PQ Induce Intrinsic Apoptosis. a Representative qRT-PCR analysis of Bax and Bcl2. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control (mean ± SD; n = 3). b Representative immunoblots of Bax, Bcl2, cytochrome (cyt) c, caspase-9, and caspase-3. GAPDH was used as a loading control. c Densitometric analysis. Protein expression from control was designated as 100% in the graph (n = 3). *Significantly different from control (p < 0.001). ACV Adiantum capillus veneris, PQ Pteris quadriureta, ACVN ACV nanoparticles, PQN PQ nanoparticles
GC–MS
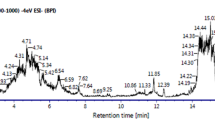
GC–MS analysis of the methanolic extracts of ACV and PQ was done to find out the bioactive compounds present in the plants. The active principles with their retention time (RT), molecular formula, and concentration (peak area %) are showed in Fig. 8 and Tables 1 and 2. GC–MS analysis showed presence of 23 and 28 bioactive phytochemical compounds in the methanolic extract of ACV and PQ respectively.

ACV and PQ GCMS chromatogram. a, b GCMS chromatogram of ACV and PQ
Docking
From GC–MS data, we selected eicosapentaenoic acid and phytol which were reported to possess anticancer activity, for docking analysis to investigate possible their interactions with Bcl2 and cyclin D1. Eicosapentaenoic acid was found to bind to Bcl2 with a docking score of − 5.6 Kcal/mol and formed hydrogen bonds with Arg107 and Arg110 in the BH3 domain of Bcl2. Phytol showed the docking score − 3.46 Kcal/mol with Bcl2 forming the hydrogen bond with Ser205 in the BH3 domain of Bcl2. Furthermore, eicosapentaenoic acid also showed a binding energy of 3.42 Kcal/mol while docking with cyclin D1 and formed hydrogen bonds with Glu75, Gln183 in the Cyclin box domain of cyclin D1. Phytol was found to bind with cyclin D1 with a docking score − 3.58 Kcal/mol by forming hydrogen bonds with Glu75, Glu69 in the Cyclin box domain of cyclin D1 (Fig. 9).

Molecular Docking. a Eicosapentaenoic acid forms hydrogen bonds with Glu75 and Gln183 of cyclin D1, while phytol forms hydrogen bonds with Glu75 and Glu69 of Cyclin D1. b Eicosapentaenoic acid forms hydrogen bonds with Arg107 and Arg110 of Bcl2, while phytol forms hydrogen bond with Ser205 of Bcl2
Discussion
Screening of herbal drugs against cancer has become more attractive and beneficial because of less side effects, cost effective and increased activity at the same time [11]. In the present study, we tested the anti-cancer effects of ACV and PQ methanolic extracts as well as their gold nanoparticles against BT47 and MCF7 breast cancer cell lines. Analysis of the gold nanoparticles of ACV and PQ using SEM and TEM showed that the nanoparticles are polydispersed in nature consisting of spherical, triangular, tetragonal and pentagonal with irregular contours.
MTT assay is useful in determining the viability of cells in vitro [24]. Our results from MTT assay revealed that the extracts and nanoparticles of ACV and PQ significantly inhibit the growth of BT47 and MCF-7 cells. The gold nanoparticles of ACV and PQ showed more significant effects against BT47 and MCF-7 cell lines compared to crude extracts. Similarly, several medicinal plants have been reported to inhibit growth of breast cancer cells [25, 26].
We report for the first time that ACV and PQ inhibit cell proliferation by downregulating the mRNA and protein expression of cyclin D1, CDK4, and PCNA while upregulating the expression of p21 in BT47 and MCF-7 cells. Cyclin D1-CDK4 complex is well-known to carry out G1-S phase transition. Hence, we examined effect of ACV and PQ on cyclin D1 and its inhibitor p21. Our results showed upregulation of p21 with consequent downregulation of cyclin D1 suggesting inhibition of cell cycle at G1/S phase. Similarly, Hedyotis diffusa Willd plant extract has been reported to inhibit cell proliferation by decreasing the expression of cyclin D1, CDK4, and PCNA [27, 28]. Furthermore, treatment with the methanol extracts and nanoparticles induced apoptosis which is evident from increased TUNEL and Annexin positive cells compared to untreated cells. Depolarization of mitochondrial membrane clearly indicates that apoptosis induced by ACV and PQ are mitochondria mediated. The increased expression of Bax, caspase-9, and -3 and decreased expression of Bcl-2 observed in the present study further confirms that ACV and PQ induces mitochondrial mediated apoptosis. Nepeta binaloudensis plant extract has been demonstrated to induce apoptosis by modulating the molecules involved in intrinsic apoptosis in breast cancer cells [26].
The compounds present in the methanolic crude leaf extracts of ACV and PQ were next analysed by GC–MS to find potential anti-cancer compounds. GC–MS results revealed presence of several bioactive compounds. In particular, phytol, a potent anti-cancer agent [29], is present in methanol extracts of both the plants but in different quantities. Eicosapentaenoic acid that possess antiproliferative activity, was also found in the methanol extract of PQ.
We performed docking of eicosapentaenoic acid and phytol with Bcl2 and Cyclin D1, respectively to find the possible interactions. We observed significant interactions with broad range of binding energies. These two phytochemicals interacted with the cyclin box domain of cyclin D1, which is required for binding of cyclin dependent kinase. The antiproliferative effect observed in the present study may be due to inhibition of cyclin D1 by eicosapentaenoic acid and phytol present in the extract. Furthermore, eicosapentaenoic acid and phytol also interacted with BH3 domain of Bcl-2, an anti-apoptotic protein, suggesting that ACV and PQ induces apoptosis by inhibiting Bcl-2. Taken together, our results demonstrate that ACV and PQ inhibit cell proliferation and induce apoptosis by interacting with cyclin D1 and Bcl-2 respectively.
Conclusion
ACV and PQ that effectively modulates various oncogenic molecules can be used as promising agent for cancer therapy. However, detailed mechanism of action of eicosapentaenoic acid and phytol against cancer cell lines not only BT47 and MCF7 cells but also various other cell lines and animal models is yet to be studied. Further, identification of more potential anti-cancer agents from ACV and PQ plants is warranted.
Abbreviations
- ACV:
-
Adiantum capillus veneris
- PQ:
-
Pteris quadriureta
- GC–MS:
-
Gas chromatography–mass spectrometry
- DMEM:
-
Dulbecco’s modified eagle medium
- PCR:
-
Polymerase chain reaction
- SEM:
-
Scanning electron microscope
- TEM:
-
Transmission electron microscope
- SDS:
-
Sodium dodecyl sulphate
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Fouad YA, Aanei C (2017) Revisiting the hallmarks of cancer. Am J Cancer Res 7(5):1016–1036
Carels N, Spinassé LB, Tilli TM, Tuszynski JA (2016) Toward precision medicine of breast cancer. Theor Biol Med Model 13:7
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN2012. Int J Cancer 136(5):E359–E386
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68(1):7–30
Hollander PD, Savage MI, Brown PH (2013) Targeted therapy for breast cancer prevention. Front Oncol 3:250
Ke X, Shen L (2017) Molecular targeted therapy of cancer: the progress and future prospect. Front Lab Med 1(2):69–75
Cazzaniga M, Bonanni B (2012) Breast cancer chemoprevention: old and new approaches. J Biomed Biotechnol. https://doi.org/10.1155/2012/985620
Wang H, Khor TO, Shu L, Su Z, Fuentes F, Lee JH et al (2012) Plants against cancer: a review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med Chem 12(10):1281–1305
Saxena M, Saxena J, Nema R, Singh D, Gupta A (2013) Phytochemistry of medicinal plants. J Pharmacogn Phytochem 1(6):168–182
Pan SY, Zhou SF, Gao SH, Yu ZL, Zhang SF, Tang MK et al (2013) New perspectives on how to discover drugs from herbal medicines: CAM’s outstanding contribution to modern therapeutics. Evid Based Complement Alterant Med 2013:627375
Shaikh R, Pund M, Dawane A, Iliyas S (2014) Evaluation of anticancer, antioxidant, and possible ant-inflammatory properties of selected medicinal plants used in Indian traditional medication. J Tradit Complement Med 4(4):253–257
Shakya AK (2016) Medicinal plants: future source of new drugs. IJHM 4(4):59–64
Zhang YJ, Gan RY, Li S, Zhou Y, Li AN, Xu DP et al (2015) Antioxidant phytochemicals for the prevention and treatment of chronic diseases. Molecules 20(12):21138–21156
Shi J, Votruba AR, Farokhzad OC, Langer R (2010) Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett 10(9):3223–3230
Shrivastava S, Dash D (2009) Applying nanotechnology to human health: revolution in biomedical sciences. J Nanotech. https://doi.org/10.1155/2009/184702
Kumar A, Boruah BM, Liang XJ (2011) Gold nanoparticles: promising nanomaterials for the diagnosis of cancer and HIV/AIDS. J Nanomater 2011:17
Nikalje AP (2015) Nanotechnology and its applications in medicine. Med Chem 5:81–89
Bwatanglang IB, Mohammad F, Yusof NA (2014) Role of multifunctional nanomaterials in disease diagnosis and therapy. J Chem 6(11):821–844
Rai M, Ingle AP, Gaikwad S, Padovani FH, Alves M (2016) The role of nanotechnology in control of human diseases: perspective in ocular surface diseases. Crit Rev Biotechnol 36(5):777–787
Sett A, Gadewar M, Sharma P, Deka M, Bora U (2016) Green synthesis of gold nanoparticles using aqueous extract of Dillenia indica. Adv Nat Sci: Nanosci Nanotechnol 7(025005):8
Ahmed S, Ahmad M, Swami BL, Ikram S (2016) A review on plant extract mediated synthesis of silver nanoparticles for antimicrobial applications; a green expertise. J Adv Res 7(1):17–28
Kowshik J, Mishra R, Sophia J, Rautray S, Anbarasu K, Reddy GD, Dixit M, Mahalingam S, Nagini S (2017) Nimbolide upregulates RECK by targeting miR-21 and HIF-1α in cell lines and in a hamster oral carcinogenesis model. Sci Rep 7(1):2045
Sylvester PW (2011) Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods Mol Biol 716:156–168
Levitsky DO, Dembitsky VM (2015) Anti-breast cancer agents derived from plants. Nat Prod Bioprospect 5(1):1–16
Afshar AS, Nematpour FS, Meshkani M, Khaf A (2017) Growth inhibition of human breast cancer cells and down-regulation of ODC1 and ADA genes by Nepeta binaloudensis. Revista Brasileira de Farmacognosia 27:84–90
Lin M, Lin J, Wei L, Xu W, Hong Z, Cai Q et al (2012) Hedyotis diffusa Willd extract inhibits HT-29 cell proliferation via cell cycle arrest. Exp Ther Med 4(2):307–310
Cai Q, Lin J, Wei L, Zhang L, Wang L, Zhan Y et al (2012) Hedyotis diffusa Willd inhibits colorectal cancer growth in Vivo via inhibition of STAT3 signaling pathway. Int J Mol Sci 13(5):6117–6128
Casuga FP, Castillo AL, Corpuz MJAT (2016) Bioactive compounds and cytotoxicity of ethyl acetate extract from Broussonetia luzonica (Moraceae) blanco leaves against hepatocellular carcinoma (Hepg2) cell lines. PHCOG J 8(5):497–501
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors state that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Rautray, S., Panikar, S., Amutha, T. et al. Anticancer activity of Adiantum capillus veneris and Pteris quadriureta L. in human breast cancer cell lines. Mol Biol Rep 45, 1897–1911 (2018). https://doi.org/10.1007/s11033-018-4337-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4337-y