
Abstract
In this study, we investigated differentially expressed genes between the original chrysanthemum cultivar ‘Argus’ with white flower color and its gamma-ray irradiated mutant ‘ARTI-purple’ with purple flower color. The expression levels of anthocyanin biosynthetic genes were not associated with anthocyanin accumulations of Argus and ARTI-purple. Expressed sequence tags (ESTs) analysis was performed to identify a novel cDNAs encoding enzymes of specific plant metabolic pathways and the biological effects of gamma-ray mutation through alterations in expression in each flower. A total of 796 unigenes were isolated from chrysanthemum ray florets. These unigenes were functionally classified using gene ontologies and tentative pathway associations were established to 99 sequences in the Kyoto encyclopedia of genes and genomes. The expressions of the isolated ESTs were screened by cDNA dot blot hybridization. Seven differentially expressed genes were identified as being involved in carbohydrate and lipid metabolic pathways and five as transcription factor or signal transduction genes. Of particular note, decreased expression of CmMYB1 was identified at the ‘ARTI-purple’. The CmMYB1 shared high similarity with AtMYB4 and AtMYBL2 which is a negative regulator of anthocyanin and flavonol accumulation. Furthermore, two genes involved in lipid metabolism, enoyl-ACP reductase and [acyl-carrier-protein] S-malonyltransferase, were decreased in the ‘ARTI-purple’ flower. Our results suggest that the purple pigmentation of the ‘ARTI-purple’ is not just dependent on the expression of anthocyanin synthesis genes, and that the pigmentation may also affect other metabolic processing and the plant cell environment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exposure to radiation is a hypothesized risk factor for mutation in all creatures, and the gamma ray is one of the most powerful forms of ionizing radiation, causing random damage to DNA and cytoplasmic organelles. The biological effects of ionizing radiation have been studied in plants, animals, cultured human cells and microorganisms through approaches such as SNP detection, measurement of nuclear DNA content, and microarrays [1–4]. Gamma radiation can result in chromosomal reduction in plants [5]. Gamma ray exposure can also increase the amount of superoxides and other reactive oxygen species (ROS) in cells [6]. Indirect effects of gamma rays in plants are damage to cellular macromolecular components including the cell walls, membranes and DNA, by water radiolysis [7]. Through these mechanisms, relevant genes in plants were observed to respond in ROS scavenging and signal transduction pathways [8], and gamma irradiation can also impact the capacity of a specific plant to produce antioxidants and phytochemicals such as flavonoids, phenolic compounds and anthocyanins at different levels [9, 10].
The chrysanthemum, a popular ornamental plant, comes in various colors, forms, sizes, growth habits and disease-resistant characters. In response to recent demands on the horticultural industry to produce more valuable and varied plants, the industry has begun to use gamma-ray mutagenesis as a way to produce new varieties. The understanding of its effects is an important factor in the use of gamma-ray mutagenesis. Petal colors are mainly determined by the content and type of a plant’s anthocyanins, which, in various chemical forms, are part of the plant’s secondary metabolites. Anthocyanins are synthesized by phenylpropanoid pathway enzymes such as chalcone synthesis (CHS), chalconeisomerase (CHI), flavonone 3-hydroxylase (F3H), dihydroflavonol 4-reductase (DFR), anthocyanidin synthase (ANS), glucosyltransferase (GT), acyltransferase (ACT) and methyltransferase (MAT) [11–13]. While some of the remaining structural enzymes of anthocyanins are well known, the specific interactions of anthocyanins in the living cell are uncharacterized. Anthocyanins are highly unstable and easily susceptible to degradation under extraction conditions. It has been reported that their stability can be altered by pH level, storage temperature, peroxidase, light, oxygen, the structure and concentration of the anthocyanin itself, or by the presence of other compounds such as other flavonoids, proteins and minerals [14]. Similarly, the stability of anthocyanins in vivo can be modified by intra-vacuolar conditions such as pH concentration, co-pigmentation with coexisting colorless flavonoids, the formation of complexes with metal ions, and plants hormones [15–18]. Moreover, Some MYB domain factors have been revealed as negative regulators of anthocyanins, such as AtMYBL2, AtMYB4 and FaMYB1 [19–21]. Such MYB domain factors bind to specific DNA sites, controlling the flow of metabolic process by promotion or repression.
Expressed sequence tags (ESTs) are an efficient gene discovery tool that provides an overview of transcriptional activities within specific tissues [22, 23]. Furthermore, identified EST clones can be used for gene expression analysis using cDNA dot blot hybridization. cDNA dot blot analysis is a technique for immobilizing specific oligonucleotide probes on a nylon membrane rather than in individual RNA samples. In this format, multiple pairs of mutant and normal specific oligonucleotide probes can be attached to strips of nylon membranes. For each diagnostic test, a spotted strip containing many normal and mutant oligonucleotides is hybridized with a specific DNA probe to screen for differences.
This study describes gene expression alteration in a gamma-irradiated mutant ‘ARTI-purple’, which has acquired purple petals in contrast to the light pink of original cultivar. Total anthocyanin content and the expression levels of anthocyanin biosynthetic genes were examined in the petals. In addition, the differentially expressed genes were identified using cDNA dot blot hybridization and confirmed via gene ontologies (GO) descriptions and KEGG pathways. These approaches have been successfully developed to detect expression changes in the two transcripts, and the results may help understand the biologicals effect of gamma-ray irradiation and be useful in molecular manipulation to improve chrysanthemum traits in the future.
Materials and methods
Plant materials
The ‘Argus’ is a spray type of chrysanthemum (Chrysanthemum × morifolium) that is composed of two morphologically visible florets, with a central tubular floret packed into a ray floret (surrounded ray floret) The mutant line ‘ARTI-purple’, derived from ‘Argus’ by a gamma-ray mutagenesis program (40 Gy to calli), has higher anthocyanin pigmentation in the petals than ‘Argus’ (Fig. 1 and Supplementary Material 1). The mutant plants were propagated vegetatively and grown for three years (M3) under natural conditions in a plastic greenhouse at the Advanced Radiation Technology Institute, Korea Atomic Energy Research Institute. The mutated colors have been visually inherited for 3 years.

Photographs of the five samples representing the Chrysanthemum varieties. a, b ‘Argus’ forward and reverse. c, d ‘ARTI-purple’ forward and reverse. e ‘Ford’. f ‘Yeonja’. g ‘Orlando’. h Anthocyanin concentrations of ‘Argus’ and ‘ARTI-purple’ at three developmental stages
Identification of total anthocyanin contents
The total anthocyanin contents of the ray florets in ‘Argus’ and ‘ARTI-purple’ were measured using the pH differential method [24]. Extracts from the petals of each plant were prepared by adding 1 mL of 0.2 M potassium chloride solution (adjusted to pH 1.0 with 1.0 M HCl) and 1 M sodium acetate buffer (adjusted to pH 4.5 with 1.0 M HCl) to 3 g of the extracted samples. The mixtures were then incubated overnight at 4 °C. After incubation, the extracts were centrifuged at 15,000 rpm for 15 min, and the optical density of the supernatants was measured at 528 and 700 nm with a spectrophotometer (Bio-Tek, USA). The concentration (mg/L) of each sample was calculated according to the following formula: TAC (μg/g) = (A × MW × DF × 103)/e; where A = (A530 nm − A700 nm)pH1.0 − (A530 nm − A700 nm)pH4.5 (The difference in absorbency), e = the molar extinction coefficient for cyaniding-3-glucoside (26,900), MW = the molecular weight of cyaniding-3-glucoside (449.2 g/mol), and DF = the dilution factor.
RNA extraction and construction of cDNA libraries
Total RNA was isolated from sampled flower petals with the TRIzol Reagent (Invitrogen, USA) according to the protocol provided. The chrysanthemum petals were found to contain high levels of polysaccharides. Thus, the extracted total RNA in the DEPC was precipitated for 3 h using a constant concentration of 3 M LiCl2 at −20 °C, and Poly A + mRNA was isolated using a Poly Tract mRNA isolation kit (Promega, USA). The total RNA and mRNA were quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA) at wavelengths of 230, 260 and 280 nm, and the mRNA was adjusted to a final concentration of 1 μg/μL. The integrity of the total RNA and mRNA was verified by electrophoresis on 1.2 % agarose gels. The mRNA was isolated from the whole flower of ‘ARTI-purple’, and a cDNA library was constructed using the SMART cDNA Library Construction Kit (Clontech, USA). Synthesized cDNAs were sequenced and edited automatically using the cross-match procedure to remove any vector and ambiguous sequences. Low-quality sequences or sequence outputs <100 bases in length were excluded.
General molecular procedures and GO analysis
To assign putative functions to the proteins, we performed blastx searches of The Reference Sequence (refseq) database (http://www.ncbi.nlm.nih.gov/blast/blast.cgi) for matches using the Blast2GO tool. For the annotation configuration, the default settings (E-value filter of 1.0E−3 and annotation cut-off of 33) were used. To obtain more information, we searched the best-matched sequences from the Arabidopsis thaliana sequence information with At-numbers using the matched sequences from Blast2GO via a NCBI blastx web search (http://www.ncbi.nlm.nih.gov/blast). The ESTs were named according to homologous sequences in the database, and while sequences of less than 50 homologous bp were classified as ‘unknown sequence’, each sequence matched more than one GO term either in different GO categories (Biological Process, Molecular Function and Cellular Component) or in the same category (http://www.Arabidopsis.org/tools/bulk/go/index.jsp). The sequences were mapped according to their domain/motif similarity, and the GO results were merged with the remaining annotations. Furthermore, the peptides were assigned into metabolic pathways using the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/kegg2.html) with the At-numbers.
Expression analysis
For expression analysis, RNA samples were treated with DNase I (amplification grade, Invitrogen) at 37 °C for 15 min prior to RT-PCR. First-strand cDNA synthesis was performed using a transcript or high-fidelity cDNA synthesis Version 6.0 kit (Roche, Germany), and anthocyanin biosynthetic genes were identified in the NCBI nucleotide databases (dihydroflavonol 4-reductase, Accession: GU324979.1 and anthocyanidin synthase, EU810810.2) and ESTs database. The primers were designed using in silico information on chrysanthemum from the conserved sequence regions and the expression level of each were estimated in a 1 % agarose gel by altering the 25 cycles of PCR amplification. The relative expression levels between ‘Argus’ and ‘ARTI-purple’ were obtained by quantitative RT-PCR using SYBR Green II Master Mix Kit (TAKARA, Japan). Primers for the PCR were designed using the Primer 3 web interface (http://frodo.wi.mit.edu/primer3/), and palindrome structures were avoided; the product size was approximately 80–160 bp. The total RNA (100 ng) was treated with DNase (Promega, USA), and reverse transcription was performed according to the PrimeScript RT Reagent Kit instructions. For the qPCR, 100 ng of total RNA was used for reactions in the Eco™ Real-Time PCR System (Illumine, USA), and the expression levels were analyzed with Eco Software v3.0.16.0 and normalized with the results of α-tubulin. The reaction was performed in three replicates for each set of conditions, and the data were presented as mean ± SD (n = 4).
To confirm the differential expression of the isolated cDNA clones, we used a reverse dot blot hybridization technique. The collected plasmid cDNA clones were amplified in a 25 μL reaction mix using a cloning vector primer (M13 forward and reverse). The conditions for PCR were 94 °C for 5 min, 40 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 1.5 min, followed by a 5 min final extension at 72 °C. The amplified products were denatured at 94 °C with 0.4 M NaOH and 10 mM EDTA for 10 min. After incubation, the cDNAs were dotted onto Hybond-N membranes (Amersham, Germany) using a vacuum blotter (Gibco BRL, USA). The blotted membranes were fixed by UV crosslinking using CL-1000 Ultraviolet Crosslinker (UVP Inc, England) and stored at 4 °C for future use. Two digoxigenin-labeled cDNAs were synthesized using the isolated total RNAs from ‘Argus’ and ‘ARTI-purple’ with the DIG DNA Labeling Kit (Roche, Switzerland) according to the manufacturer’s instructions. The membranes were pre-hybridized for 2 h at 62 °C and then hybridized for 16 h at 62 °C with 1 μg of the probes generated from the total RNAs. Membrane washing and detection were performed according to the protocols specified in the DIG Nucleic Acid Detection Kit (Roche, Switzerland). Both membranes were exposed to X-ray films for 1 h and scanned, and the results from the two hybridizations were compared for each clone.
Results and discussion
Total anthocyanin contents and expression of anthocyanin biosynthetic genes
The relationship between anthocyanin accumulation and gene expression was examined in ‘Argus’, ‘ARTI-purple’ and three other cultivars (Fig. 1e–g). The original cv. ‘Argus’ has light pink petals, while its derived mutant, ‘ARTI-purple’, has pure purple petals (Fig. 1a–d). We analyzed the total anthocyanin content by measuring cyanidin 3-glucoside in the petals. In ‘ARTI-purple’, the concentration of anthocyanin was increased approximately 3.5-fold over that of ‘Argus’.
Expression levels were determined for six anthocyanin biosynthetic genes including CHS, CHI, F3H, DFR and ANS. RT-PCR was performed using gene-specific primers (Supplementary Material 2). In the three Chrysanthemum spp. with white, pink and purple petals, the expression patterns of the anthocyanin biosynthetic genes correlated with the anthocyanin pigmentation levels of each flower (Fig. 2). For example, the white variety named ‘Ford’ (Fig. 1e) had low levels of anthocyanin biosynthetic gene expression (Fig. 2). The pink (Fig. 1f) and purple cultivars (Fig. 1g) showed expression levels of anthocyanin biosynthetic genes that corresponded with each flower’s pigmentation (Fig. 2). However, ‘Argus’ showed higher expression levels of the biosynthetic genes than ‘ARTI-purple’ despite the low anthocyanin concentration in the petals (Figs. 1, 2). And no significant differences according to the flower development were identified (Supplementary Material 3). To understand this difference, we conducted DMACA staining of petals from the two flowers, but we did not find any proanthocyanidins or condensed tannins (data not shown).

Expression analysis of anthocyanin biosynthesis genes in the petals of ‘Argus’, ‘ARTI-purple’ and three control plants. The expression levels of five genes involved in general anthocyanin biosynthesis (CHS, CHI, F3H, DFR and ANS) were analyzed by RT-PCR. The ‘Argus’ and ‘ARTI-purple’ expression patterns were compared with three other Chrysanthemum varieties. Alpha tubulin was used as the quantitative control. Ag ‘Argus’, Ap ‘ARTI-purple’, W ‘Ford’, P ‘Yeonja’, Pp ‘Orlando’
Recently, Wei et al. [40] showed the relationships between anthocyanin contents and the expressions of anthocyanin biosynthetic genes with 12 litchi cultivars in which the expression levels of LcDFR and LcUFGT leads to enhanced anthocyanin accumulation in the pericarp. However, the anthocyanin content of two cultivars, ‘Sanyuehong’ and ‘Guinuo’, among the 12 litchi cultivars did not associated with the expression level of anthocyanin biosynthetic genes. Although ‘Guinuo’ has 4-fold higher anthocyanin concentration than ‘Sanyuehong’, the expression levels of LcDFR and LcUFGT was not notable at the ‘Guinuo’ [40]. These results imply that ‘Sanyuehong’ might have a different anthocyanin metabolism such as ‘Argus’ in our study.
EST analysis
A cDNA (complementary DNA) library was constructed from ‘ARTI-purple’ ray florets. A total of 1,137 clones were sequenced, and the sequences less than 100 bp in length were discarded as unreliable. The 1,137 clones contained 695 singletons and 289 redundant sequences clustered into 101 contigs (contiguous consensus sequences), corresponding to a redundancy level of 29.4 % (Table 1). Consequently, 769 unigenes were obtained with a mean length of 653 bp. The contig ESTs in abundance were assigned tentative functions following a blastx similarity search (Table 1).
The most highly clustered clones were sucrose: sucrose 1-fructosyltransferase (1.04 %), tubulin alpha chain, a putative protein (0.91 %), chalcone synthase (0.91 %), ribulose-1,5-bisphosphate carboxylase small subunit (0.78 %), lipid transfer protein precursor (0.78 %), C-4 methyl sterol oxidase, another putative protein (0.65 %) and a proline-rich protein (0.65 %) (Table 2). Among these, tubulin alpha chain, one of the putative proteins (0.91 %), ribulose-1,5-bisphosphate carboxylase small subunit (0.78 %) and the proline-rich protein (0.65 %) appeared in similar frequencies as in the chrysanthemum inflorescence library [23]. Lipid transfer proteins are highly expressed in carnation petals; a hypothetical protein (Accession number of best-matched sequence: XP_002285432.1) and a proline-rich protein (AAF78903.1) shared high similarity with lipid-transfer proteins in Arabidopsis (data not shown). In carnation petals, expression of many varied LTP genes was observed, suggesting their contribution to petal development is important, through the regulation of morphologically important cellular components such as the cell wall and cuticle [25]. In addition, we identified five clusters of chalcone synthase (0.91 %), which is a key enzyme involved in the biosynthesis of flavonoids and is required for the accumulation of purple anthocyanins in plant tissues [26, 27]. The abundant ESTs we confirmed in the ‘ARTI-purple’ petals can be used for chrysanthemum petal studies in genetics and molecular biology as ample expressed factors [22].
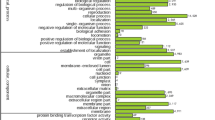
The three organizing principles of GO consist of the molecular function, biological process and cellular component categories. These three principles provide information that is necessary to describe the roles of genes and gene products [28]. A total of 769 unigenes were annotated with putative functions by Blastx matching, which reported best matches with Arabidopsis and other plant sequences. All of the unigenes were functionally classified into one or more GO annotations by referencing the Arabidopsis information resource (TAIR; www.arabidopsis.org) (Fig. 3). Functional categorizations for GO biological processes were grouped into 13 categories, and the majority of the sub-GO terms were cellular processes (27.9 %) and metabolic processes (24.2 %). In the biological process category, 725 genes were annotated using various terms. For molecular function, the majority groups appeared to be other binding (15.2 %) and other enzyme activity (19.4 %) based on 724 annotated genes. The majority of categorizations for cellular components included other intracellular components (21.4 %) and other cytoplasmic components (16.9 %) and these were listed for 653 genes. From the total unigenes, the percentage contributions of biological process, molecular function and cellular component were 92, 94 and 85 %, respectively (Fig. 3).

Gene ontology (GO) assignment (2nd level GO terms) of 511 Chrysanthemum annotated unigenes. The three GO categories, biological process (a), molecular function (b) and cellular component (c), are presented
For the establishment of pathway associations, the KEGG within the GO terms were used. The representative pathways with enzyme numbers greater than five are presented in Table 3. Among the metabolic pathways identified, four metabolisms were included: carbohydrate metabolism, energy metabolism, amino acid metabolism and metabolism of other amino acids. The most abundant of these was carbohydrate metabolism, which comprised four KEGG pathways, namely, fructose and mannose metabolism, glycolysis/gluconeogenesis, amino sugar and nucleotide sugar metabolism and the pentose phosphate pathway (Table 3). The number of pathways identified involving biosynthesis of secondary metabolites (ko01110) was 48. Among them, the anthocyanin related-pathway (flavonoid: ko00941) included four enzymes, namely, chalconeisomerase (Enzyme Commission numbers: 5.5.1.6), dihydrokaempferol 4-reductase (ec: 1.1.1.219), flavanone 3-dioxygenase (ec: 1.14.11.9) and naringenin-chalcone synthase (ec: 2.3.1.74).
Further identification of anthocyanin biosynthetic enzymes and their expressions
Chrysanthemum secondary metabolism is important for understanding the regulation of pigment biosynthesis [29]. In Table 4, the putative genes involved in secondary metabolism are listed with putative annotations from the 511 unigenes. The phylogenetic relationships of isolated genes with homology to glucosyltransferase and malonyltransferase were analyzed to identify orthologs of the chrysanthemum flower pigment regulatory genes. Three clones (ED0113, EC0909 and EE0207) were clustered into the Arabidopsis UDP-glucose group, as well as anthocyanidin 3-glucosyltransferase (3GT), anthocyanidin 5′3-O-glycosyltransferase (5′3GT) and anthocyanidinmalonyltransferase (MAT), whose sequences were confirmed from several other plants (Fig. 4). The isolated 3GT (ED0113) and 5′3GT (EC0909) genes had 61 % maximal identity (126/205) and 100 % query coverage in the alignment results from the NCBI blastx search, while the amino acid sequence of the isolated MAT (EE0207) from our EST exhibited 54 % (156/290) sequence identity with both Dm3MaT1 and Dm3MaT2.

The phylogenetic relationships of glycosyltransferases. The amino acid sequences of Chrysanthemum 3GT (EST No.: EC0909), 5′3GT (ED0113) and MAT (EE0207) were aligned with Arabidopsis (AT numbers) and other plant glycosyltransferases using ClustalW2. The phylogenetic tree was constructed using the MEGA5.05 program with the minimum evolution test
The expression of anthocyanidin 3-glucosyltransferases is essential for anthocyanin biosynthesis in a variety of plant species [30]. In Arabidopsis, 3GT (NP_197207) is induced by PAP1 expression [31–33]. We compared the expression of this gene between ‘Argus’ and ‘ARTI-purple’, but no noticeable difference was detected (Fig. 5). In Rosa, Ogata et al. [34] suggested that the activation of RhGT1 (Rosa 5′3GT: Q4R1I9) could catalyze glycosylation at not just one, but two different sites on the anthocyanidin molecule; glycosylation occurs first at the 5-OH and then at the 3-OH group. Glycosylation also affects the stabilization of the anthocyanin structure. Chrysanthemum MAT function, structure and mutational establishment were studied previously [35, 36], and it was suggested that Dm3MaT1 (AAQ63615.1) and Dm3MaT2 (AAQ63616.1) function as both anthocyanidin 3-O-glucoside-6′-O-malonyltransferases and anthocyanidin 3-O-glucoside-3′6′-O-dimalonyltransferases [36]. We examined the expression levels of 5′3GT and MAT using the qPCR system (Fig. 5). However, we determined that ‘Argus’ had higher expression levels than ‘ARTI-purple’, although ‘ARTI-purple’ had higher anthocyanin content. This result suggested that 3GT, 5′3GT and MAT were not associated with the flower color change in ‘ARTI-purple’. Moreover, the expression of these three genes was decreased in the ‘ARTI-purple’ petal.

Relative expression analysis of Chrysanthemum 3GT, 5′3GT and MAT in ‘Argus’ and ‘ARTI-puprle’
Identification of differentially expressed genes
Total RNA was used to generate cDNA, which was labeled with digoxigenin according to the techniques recommended for cDNA dot blot hybridization. The amplicons of the ESTs, dotted on to the nylon membranes, were hybridized with labeled cDNA, as described in the “Materials and methods” section. cDNA dot blot hybridization has proven to be a good tool for detecting differentially expressed genes. In our results, 769 unigenes were screened in total, and their expression levels indicated that 19 clones were up-regulated and 53 clones were down-regulated. Most of the gene expression patterns in the gamma-ray mutant were at a level detectable by cDNA dot blot hybridization (Supplementary Material 4).
Both the up- and down-regulated genes were assigned to known pathways using the KEGG pathway databases. The identified clones showing expression changes were grouped into five KEGG pathways. The most abundant groups were carbohydrate and lipid metabolisms. Carbohydrate metabolism included four pathways, glycolysis/gluconeogenesis (ko00010), pentose phosphate metabolism (ko00030), ascorbate and aldarate metabolism (ko00053) and pyruvate metabolism (ko00620), with five enzymes (up-regulated in the mutant: L-lactate dehydrogenase, down-regulated: FBA1, LOS2, ribose 5-phosphate isomerase A, MIOX2) that showed changes in expression (Table 5). The expression of two clones, enoyl-ACP reductase (FabI) and [acyl-carrier-protein] S-malonyltransferase (FabD), involved in the fatty acid biosynthesis pathway (ko00071) was reduced in the mutant (Supplementary Material 5). These results suggest some interesting implications for the sharing of malonyl-CoA between fatty acid and anthocyanin biosynthesis. That is, malonyl-CoA may be used competitively in the metabolic processes of fatty acid and anthocyanin biosynthesis. Therefore, the reduction of these two enzymes, enoyl-ACP reductase (FabI) and [acyl-carrier-protein] S-malonyltransferase (FabD), may result in different malonyl-CoA utilization.
At the transcription factor and signal transduction level, five clones were differentially expressed, including a R2R3-MYB transcription factor, a CDM86 transcription factor, a MADS-box factor, a GRCD4 protein and a rapid alkalinization factor precursor (Table 6). These five clones were down-regulated from twofold to several-fold (Fig. 6). Among them, the R2R3-MYB transcription factor encoded by CmMYB1 (Accession No: AEO27497), appeared to be a full-length cDNA (858 bp) encoding a protein of 286 amino acids. CmMYB1 expression was reduced significantly in the ‘ARTI-purple’ petals (Fig. 6) and it clustered with Arabidopsis AtMYB4 and AtMYBL2, Malus MdMYB16, Populus MYB156, Vitis MYB4B and Glycine MYBZ2 in the amino acid alignment (Fig. 7).

Relative expression analysis of transcription factor and signal transduction mRNAs from ‘Argus’ and ‘ARTI-purple’ petals

Phylogenetic tree from CmMYB1 amino acid sequence alignment with best-matched Arabidopsis and other plant MYB-domain factors
Recent studies have demonstrated that some MYB-type transcription factors acts as a negative regulators of anthocyanin and flavonol accumulation [19, 20]. An Arabidopsis knockout mutant of AtMYB4 showed the down-regulation of cinnamate 4-hydroxylase (C4H) and 4-coumaroyl-CoA ligase (4CL) in the biosynthesis of anthocyanins [20]. The expression level of AtMYBL2 caused changes in plant color and its expression was induced by high light and sucrose concentrations [21, 37–39]. In the functional study of AtMYBL2, it showed as a transcriptional repressor and negatively regulates the biosynthesis of anthocyanin. Loss of function of AtMYBL2 (atmybl2) enhanced anthocyanin accumulation via induction of the expression of DFR and TT8. These results suggested that putative role of AtMYBL2 might be critical in determining whether a complex is in-active, repressive or active with respect to the expression of the target genes [21]. However, the expression pattern of AtMYBL2 appears to be the different with CmMYB1 in chrysanthemum petal. The anthocyanin biosynthetic genes such as DFR is not expressed correspond closely to anthocyanin accumulation between ‘ARTI-purple’ and ‘Argus’. If CmMYB1 function is repressive regulator of anthocyanin accumulation, its role is suggested as an inhibition of metabolic processes affecting the anthocyanin contents rather than the directly transcriptional inhibition of anthocyanin biosynthetic genes. However, the exact mechanism of action is not known.
Further studies are necessary to identify the mechanisms involved in a relationship between the anthocyanin accumulating facility and the candidate targets including transcription factor and the genes related to the two suggested metabolisms. In addition, to explain their complicated relationship, model plant-based transgenic approaches, as well as statistical bioinformatics research is required. Perhaps, in a metabolic process due to the limited materials, anthocyanin and lipid metabolism might be an inevitably competitive relationship using as a structural material of malonyl-CoA and glucose.
References
Goodhead DT (1994) Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol 65(1):7–17
H.J.M B (1961) Radiosensitivity of higher plants, and correlations with cell weight and DNA content. Radiat Bot 1:223–228
Uauy C, Paraiso F, Colasuonno P, Tran R, Tsai H, Berardi S, Comai L, Dubcovsky J (2009) A modified tilling approach to detect induced mutations in tetraploid and hexaploid wheat. BMC Plant Biol 9(1):115
Prithivirajsingh S, Story MD, Bergh SA, Geara FB, Ang KK, Ismail SM, Stevens CW, Buchholz TA, Brock WA (2004) Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett 571(1–3):227–232
Yamaguchi H, Shimizu A, Degi K, Morishita T (2008) Effects of dose and dose rate of gamma ray irradiation on mutation induction and nuclear DNA content in chrysanthemum. Breed Sci 58(3):331–335
Wi SG, Chung BY, Kim J-S, Kim J-H, Baek M-H, Lee J-W, Kim YS (2007) Effects of gamma irradiation on morphological changes and biological responses in plants. Micron 38(6):553–564
Nagata T, Todoriki S, Hayashi T, Shibata Y, Mori M, Kanegae H, Kikuchi S (1999) r-Radiation induces leaf trichome formation in Arabidopsis. Plant Physiol 120(1):113–120
Kim DS, Kim J-B, Goh EJ, Kim W-J, Kim SH, Seo YW, Jang CS, Kang S-Y (2011) Antioxidant response of Arabidopsis plants to gamma irradiation: genome-wide expression profiling of the ROS scavenging and signal transduction pathways. J Plant Physiol 168(16):1960–1971
Ayed N, Yu HL, Lacroix M (1999) Improvement of anthocyanin yield and shelf-life extension of grape pomace by gamma irradiation. Food Res Int 32(8):539–543
Alothman M, Bhat R, Karim AA (2009) Effects of radiation processing on phytochemicals and antioxidants in plant produce. Trends Food Sci Technol 20(5):201–212
Boss PK, Davies C, Robinson SP (1996) Analysis of the expression of anthocyanin pathway genes in developing Vitis vinifera L. cv shiraz grape berries and the implications for pathway regulation. Plant Physiol 111(4):1059–1066
Iida S, Hoshino A, Johzuka-Hisatomi Y, Habu Y, Inagaki Y (1999) Floricultural traits and transposable elements in the Japanese and common morning gloriesaa. Ann N Y Acad Sci 870(1):265–274
Yamaguchi TKusumi, Nishino T (2001) Malonyl-CoA: anthocyanin 5-O-glucoside-6′″-O-malonyltransferase from scarlet sage (Salvia splendens) flowers. J Biol Chem 276(52):49013–49019
Oren-Shamir M (2009) Does anthocyanin degradation play a significant role in determining pigment concentration in plants? Plant Sci 177(4):310–316
Asen S, Stewart RN, Norris KH (1975) Anthocyanin, flavonol copigments, and pH responsible for larkspur flower color. Phytochemistry 14(12):2677–2682
Shaked-Sachray L, Weiss D, Reuveni M, Nissim-Levi A, Oren-Shamir M (2002) Increased anthocyanin accumulation in aster flowers at elevated temperatures due to magnesium treatment. Physiol Plant 114(4):559–565
Loreti E, Povero G, Novi G, Solfanelli C, Alpi A, Perata P (2008) Gibberellins, jasmonate and abscisic acid modulate the sucrose-induced expression of anthocyanin biosynthetic genes in Arabidopsis. New Phytol 179(4):1004–1016
Jeong ST, Goto-Yamamoto N, Kobayashi S, Esaka M (2004) Effects of plant hormones and shading on the accumulation of anthocyanins and the expression of anthocyanin biosynthetic genes in grape berry skins. Plant Sci 167(2):247–252
Aharoni A, De Vos CHR, Wein M, Sun Z, Greco R, Kroon A, Mol JNM, O’Connell AP (2001) The strawberry FaMYB1 transcription factor suppresses anthocyanin and flavonol accumulation in transgenic tobacco. Plant J 28(3):319–332
Jin H, Cominelli E, Bailey P, Parr A, Mehrtens F, Jones J, Tonelli C, Weisshaar B, Martin C (2000) Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. EMBO J 19(22):6150–6161
Matsui K, Umemura Y, Ohme-Takagi M (2008) AtMYBL2, a protein with a single MYB domain, acts as a negative regulator of anthocyanin biosynthesis in Arabidopsis. Plant J 55(6):954–967
Ohlrogge J, Benning C (2000) Unraveling plant metabolism by EST analysis. Curr Opin Plant Biol 3(3):224–228
Chen S, Miao H, Chen F, Jiang B, Lu J, Fang W (2009) Analysis of expressed sequence tags (ESTs) collected from the inflorescence of chrysanthemum. Plant Mol Biol Rep 27(4):503–510
Jurd L, Asen S (1966) The formation of metal and “co-pigment” complexes of cyanidin 3-glucoside. Phytochemistry 5(6):1263–1271
Harada T, Torii Y, Morita S, Masumura T, Satoh S (2010) Differential expression of genes identified by suppression subtractive hybridization in petals of opening carnation flowers. J Exp Bot 61(9):2345–2354
Durbin ML, McCaig B, Clegg MT (2000) Molecular evolution of the chalcone synthase multigene family in the morning glory genome. Plant Mol Biol 42(1):79–92
Harker CL, Ellis T, Coen ES (1990) Identification and genetic regulation of the chalcone synthase multigene family in pea. Plant Cell 2(3):185–194
Berardini T, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller L, Yoon J, Doyle A, Lander G, Moseyko N, Yoo D, Xu I, Zoeckler B, Montoya M, Miller N, Weems D, Rhee S (2004) Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol 135:745–755
Courtney-Gutterson N, Firoozabady E, Lemieux C, Nicholas J, Morgan A, Robinson K, Otten A, Akerboom M (1993) Production of genetically engineered color-modified chrysanthemum plants carrying a homologous chalcone synthase gene and their field performance. Acta Hortic 336:57–62
Solfanelli C, Poggi A, Loreti E, Alpi A, Perata P (2006) Sucrose-specific induction of the anthocyanin biosynthetic pathway in Arabidopsis. Plant Physiol 140(2):637–646
Kobayashi S, Ishimaru M, Ding CK, Yakushiji H, Goto N (2001) Comparison of udp-glucose:flavonoid 3-O-glucosyltransferase (UFGT) gene sequences between white grapes (Vitis vinifera) and their sports with red skin. Plant Sci 160(3):543–550
Kovinich N, Saleem A, Arnason JT, Miki B (2010) Functional characterization of a UDP-glucose:flavonoid 3-O-glucosyltransferase from the seed coat of black soybean (Glycine max (L.) merr.). Phytochemistry 71(11–12):1253–1263
Suzuki H, Nakayama T, Yonekura-Sakakibara K, Fukui Y, Nakamura N, Nakao M, Tanaka Y, Tohge M-A, Nishiyama TY, Hirai MY, Yano M, Nakajima J-I, Awazuhara M, Inoue E, Takahashi H, Goodenowe DB, Kitayama M, Noji M, Yamazaki M, Saito K (2005) Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J 42(2):218–235
Ogata J, Kanno Y, Itoh Y, Tsugawa H, Suzuki M (2005) Plant biochemistry: anthocyanin biosynthesis in roses. Nature 435(7043):757–758
Unno H, Ichimaida F, Suzuki H, Takahashi S, Tanaka Y, Saito A, Nishino T, Kusunoki M, Nakayama T (2007) Structural and mutational studies of anthocyanin malonyltransferases establish the features of BAHD enzyme catalysis. J Biol Chem 282(21):15812–15822
Suzuki H, Nakayama T, Yamaguchi M-A, Nishino T (2004) cDNA cloning and characterization of two Dendranthema × morifolium anthocyanin malonyltransferases with different functional activities. Plant Sci 166(1):89–96
Dubos C, Le Gourrierec J, Baudry A, Huep G, Lanet E, Debeaujon I, Routaboul J-M, Alboresi A, Weisshaar B, Lepiniec L (2008) MYBL2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J 55(6):940–953
Gonzalez A (2009) Pigment loss in response to the environment: a new role for the WD/bHLH/MYB anthocyanin regulatory complex. New Phytol 182(1):1–3
Winkel-Shirley B (2002) Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Biol 5(3):218–223
Wei Y-Z, Hu F-C, Hu G-B, Li X-J, Huang X-M, Wang H-C (2011) Differential expression of anthocyanin biosynthetic genes in relation to anthocyanin accumulation in the pericarp of Litchi chinensis Sonn. PLoS One 6(4):e19455
Acknowledgments
This work was supported by a grant from the Korea Atomic Energy Research Institute (KAERI) and the Ministry of Education, Science, and Technology (MEST), Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sung, S.Y., Kim, S.H., Velusamy, V. et al. Comparative gene expression analysis in a highly anthocyanin pigmented mutant of colorless chrysanthemum. Mol Biol Rep 40, 5177–5189 (2013). https://doi.org/10.1007/s11033-013-2620-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-013-2620-5