
Abstract
Sika deer is one of the best-known and highly valued animals of China. Despite its economic, cultural, and biological importance, there has not been a large-scale sequencing project for Sika deer to date. With the ultimate goal of sequencing the complete genome of this organism, we first established a bone marrow cDNA library for Sika deer and generated a total of 2,025 reads. After processing the sequences, 2,017 high-quality expressed sequence tags (ESTs) were obtained. These ESTs were assembled into 1,157 unigenes, including 238 contigs and 919 singletons. Comparative analyses indicated that 888 (76.75%) of the unigenes had significant matches to sequences in the non-redundant protein database, In addition to highly expressed genes, such as stearoyl-CoA desaturase, cytochrome c oxidase, adipocyte-type fatty acid-binding protein, adiponectin and thymosin beta-4, we also obtained vascular endothelial growth factor-A and heparin-binding growth-associated molecule, both of which are of great importance for angiogenesis research. There were 244 (21.09%) unigenes with no significant match to any sequence in current protein or nucleotide databases, and these sequences may represent genes with unknown function in Sika deer. Open reading frame analysis of the sequences was performed using the getorf program. In addition, the sequences were functionally classified using the gene ontology hierarchy, clusters of orthologous groups of proteins and Kyoto encyclopedia of genes and genomes databases. Analysis of ESTs described in this paper provides an important resource for the transcriptome exploration of Sika deer, and will also facilitate further studies on functional genomics, gene discovery and genome annotation of Sika deer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sika deer (Cervus nippon) are historically widespread throughout northeastern Asia, from the Ussuri region to Vietnam, including the Korean Peninsula, mainland China and Taiwan, and the Japanese Archipelago, and up to 13 subspecies have been described [1]. It is one of the best-known and highly valued animals of China. In ancient times, it was regarded as an auspicious animal and referred to as “a divine creature”. The whole body of Sika deer is valuable, including its velvet antlers, pizzle, fetus, sinew, bone, blood, and skin, as well as venison, and these body parts are recorded as highly valued in traditional Chinese medicine and listed as tonics in Shennong’s Classic of Materia Medica, the Compendium of Materia Medica and other pharmaceutical monographs of ancient China. Sika deer products have strong tonic, anti-aging, accelerated ulcer healing and other pharmacological activities [2]. Shuangyang Sika deer from Jilin Province, which belong to Cervus nippon hortulorum, are a prized variety of these deer in the central part of northeastern China. In 1990, a Sika deer breeding project received the first prize of the State Science and Technology Awards.
Expressed sequence tag (EST) analysis is an effective method for discovering novel genes and investigating gene expression in different organs and tissues. This technology has become a commonly used approach to identify genes involved in specific biological functions, especially in organisms where genomic data are not available [3]. It can be performed more easily than whole-genome sequencing, enabling us to collect nucleotide sequence information of protein-coding regions rapidly and efficiently [4]. Thus, ESTs represent a cost-effective approach for gene discovery. In addition to humans [5–7], analyses of ESTs have also been carried out in various mammalian organisms, including cattle [8–11], sheep [12, 13], horse [14] and camel [15]. However, few genome resources have been developed from Sika deer, and there are only approximately 727 ESTs of Sika deer available in the NCBI database. Furthermore, reports of such studies are almost nonexistent in the literature. The lack of sequence information in this species has limited the progress of gene discovery and characterization. Such an approach can aid in understanding the biological functions of Sika deer at the transcriptome level, which is beneficial for finding functional genes and active proteins, as well as for studies on the genomics of Sika deer in the future.
Bone marrow is a soft connective tissue within the medullary cavities of bones. Most cellular components of the immune system originate in the bone marrow, including B cells, natural killer (NK) cells, and macrophages [16]. It is also the only known organ in which two separate and distinct stem cell types not only coexist, but also functionally cooperate [17]. Bone marrow stem cells (BMSC) are classified into hematopoietic stem cells (HSC), which give rise to all cells of the hematopoietic system, and mesenchymal stem cells (MSC), which support hematopoiesis. In addition to their role in hematopoiesis, both HSC and MSC have the ability to differentiate into many other types of cells in vivo or in culture conditions [18]. As an example, stem cells isolated from bone marrow can differentiate not only into blood cells, but also into hepatocytes, skeletal muscle and cardiomyocytes, and recent studies have shown that stem cells may even give rise to neural cells in vivo [19]. The source of the cells that give rise to regenerating the antler has been a matter of controversy. Recently, it has been hypothesized that antler regeneration is a stem cell-based process, with stem cells located in the pedicle periosteum giving rise to progenitor cells of different lineages, such as chondro- and osteoprogenitors [20]. Sika deer also produce a series of functional polypeptides that have been reported to have effects on accelerating fracture healing by stimulating the proliferation of chondrocytes and osteoblast precursors, as well as maintaining the survival and promoting the growth of neural cells, particularly the regeneration of neural tissues [21–25]. However, little is known about the molecular biology of Sika deer. Identification of expressed genes and, in particular, their transcript profile, would be a key step in understanding their pharmacological activity and the regeneration mechanisms of these deer.
In this study, we generated and analyzed more than 2,000 ESTs from a bone marrow cDNA library of Chinese Sika deer, and most of these ESTs were reported in Sika deer for the first time. The availability of these EST sequences will allow the development of molecular markers for the establishment of a reference genetic map and for the design and construction of cDNA microarrays for global gene expression profiling. This new collection of sequences constitutes an important new resource for the genomics of Sika deer and related deer species.
Materials and methods
Materials
Bone marrow was collected from the hind femur of Shuangyang Sika deer, washed with RNase-free water several times, and then immediately placed in liquid nitrogen. The samples were kept at −80°C until use.
Construction of cDNA library
Total RNA was extracted from the marrow of Sika deer using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions, and poly (A) + mRNA was isolated from the total RNA using an mRNA purification kit (Takara, Japan). Then, the poly (A) + mRNA was reverse transcribed into cDNA using a cDNA Synthesis Kit (Stratagene, USA). After cDNA termini blunting, adapter ligation, and digestion, the cDNA was inserted into pBluescript_II_SK(+) (Stratagene, USA), digested with XhoI and EcoRI, and then introduced into the Escherichia coli strain DH10B (Takara, Japan).
EST sequencing
A total of 2,025 colonies were randomly picked from the cDNA library, transferred into 96-well plates containing LB medium and incubated for 18 h at 37°C for amplification. Bacterial plasmids were extracted using an AxyPrep™ Easy-96 Plasmid Purification Kit (Axygen) and were stored at −20°C until use. The cDNA inserts were single-pass sequenced from the 5′ end of the cDNA using BigDye® Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems) and a T3 primer (5′-AATTAACCCTCACTAAAG-3′) on a 3730XL DNA analyzer (Applied Biosystems) according to the manufacturer’s protocol.
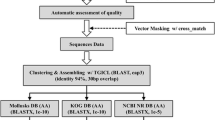
EST data analysis, processing and assembly
The raw sequences were processed using the Phred program [26, 27] to remove sub-standard reads, followed by Cross_match and RepeatMasker [28] to eliminate the vector and repeat sequences. High-quality ESTs (>100 bp) were then assembled and clustered into contiguous sequences (contigs) using Contig Assembly Program 3 (CAP3) [29]. Consed, an assembly viewer, was then used to assess contigs for misassemblies, in particular, mosaic clones resulting from polyA sequences [30].
EST annotation and analysis
We used the getorf program from the EMBOSS package [31] to find all open reading frames (ORFs) of at least 100 nucleotides, then selected the longest ORF for each sequence. Sequence similarity searches against different protein databases were conducted using an advanced BLAST program [32, 33]. The BLASTX program was used to compare the nucleotide sequences to the NCBI non-redundant (nr) protein database, and results with E-values equal to or less than 10−5 were treated as “significant matches”, whereas ESTs with no hits, or matches with E-values greater than 10−5 to proteins in NCBI were classified as “no significant matches”. The BLASTN program was used to compare the nucleotide sequences with the sequences in the Sika deer EST database at NCBI, and results with E-values equal to or less than 10−10 were treated as “significant matches”, whereas ESTs with no hits, or matches with E-values greater than 10−10 to nucleotides in NCBI were classified as “no significant matches”.
More detailed functional annotation was performed by mapping sequences to the Gene ontology (GO) database, Clusters of orthologous groups of proteins (COGs) database and Kyoto encyclopedia of genes and genomes (KEGG) database. GO classifications [34, 35] were acquired using Interproscan [36, 37], which was downloaded from the European Bioinformatics Institute (EBI). COGs classification [38, 39] was performed by BLASTX searching of the NCBI database. Furthermore, we also submitted the unigenes to the Kyoto Encyclopedia of Genes and Genomes (KEGG) [40, 41] to acquire concrete functional information.
Results
Characterization of the cDNA library
The primary titer of the cDNA library constructed in this study consisted of 3.5 × 106 clones, with more than 90% recombinant clones, as revealed by PCR analysis (Fig. 1). The results indicated that the quality of the cDNA library should be sufficient to identify the expressed genes in the bone marrow of Sika deer.

PCR analysis of partial clones in the cDNA library. Lanes 1–16: PCR products from different clones. Lanes M: DNA ladder maker
EST sequencing and assembly
In total, 2,017 ESTs from single-pass 5′ sequencing of 2,025 cDNA clones passed the quality control analysis for high-confidence base calling, and 1,157 unigenes consisting of 238 contigs and 919 singletons were assembled. The GC content of the unigenes was approximately 51%, and 401 sequences included polyA(T) sequences (Table 1). The number of ESTs in the contigs ranged from 2 to 63, with more than 51.26% of contigs consisting of 2 ESTs, 18.91% of 3 ESTs, and 25.63% of 4-10 ESTs. Approximately 79.7% of the unigenes ranged from 500 to 1000 bp; the average length was 756 bp, and the longest unigene was 3127 bp.
ORF prediction
73.87% of contigs and 62.80% of singletons had an ORF longer than 300 bp. As expected, contigs yield longer ORFs (average ORF length: 501.69 bp) compared to those of singletons (average ORF length: 422.18 bp). Approximately 38.14% of the contigs had an ORF ranging from 500 to 1,000 bp; 33.33% were from 300 to 500 bp; and 3.43% were longer than 1,000 bp; the longest ORF was 1940 bp.
Functional annotation and classification
A total of 1,157 unigenes were compared to the NCBI nr database and the Sika deer EST database for homology identification. Among these sequences, 76.75% (total number: 888) had significant matches with sequences in the non-redundant protein database based on an E-value cut off that was equal to or less than 10−5; 23.25% (total number: 269) were homologous to genes with unknown functions. Comparing the 269 sequences against the Sika deer EST database using BLASTN demonstrated that the percentage of the sequences that had significant matches was only 9.29% based on an E-value cut off that was equal to or less than 10−10, and others had no matches to the sequences in the Sika deer EST database.
We further analyzed BLAST results separately for fifteen mammalian species. Nearly 92% of the EST hits came from mammalian species. Among these species, Chinese Sika deer genes had high similarity to cattle. In total, there were 496 ESTs that matched cattle (Bos taurus), corresponding to approximately 55.86% of all EST hits. Among other mammals, there were 55 ESTs (6.19%) that matched dog (Canis lupus familiaris); 51 (5.74%) matched laboratory mouse (Mus musculus); 45 (5.07%) matched horse (Equus caballus); 39 (4.39%) matched rhesus macaque (Macaca mulatta); 37 (4.17%) matched chimpanzee (Pan troglodytes), 25 (2.82%) matched sheep (Ovis aries); 23 (2.59%) matched rat (Rattus norvegicus); and 22 (2.48%) matched pig (Sus scrofa).
The highly expressed genes in the library that contained more than five ESTs in one contig are summarized in Table 2. The most frequently expressed gene was MSTP 008 (63 ESTs), followed by cytochrome c oxidase subunit I (60 ESTs), subunit III (55 ESTs) and stearoyl-CoA desaturase (SCD) (45 ESTs similar to Bubalus bubalis and 34 ESTs similar to Bos taurus).
Based on GO annotation, each sequence was assigned at least one GO term. Second-level GO terms were used to classify the sequences in terms of their involvement as having a cellular location, involved in molecular functions, and involved in biological process (Fig. 2). In total, 580 (50.1%) unigenes could be assigned to one or more ontologies. Of the sequences categorized as having cellular locations, 46.2% were categorized as “cell” and “cell part”, 23.79% as “organelles”, and 18.79% as “macromolecular complex”. Of the sequences categorized as having molecular functions, 45.69% were categorized as “binding”, followed by those involved in “catalytic activity (43.97%)”. The most represented biological processes were “cellular process” and “metabolic process”, which comprised 55.00% and 52.07% of these sequences, respectively.

GO classification of unigenes. The bar chart shows the distribution of ESTs among three principal GO categories: cellular location, molecular function and biological process
We also analyzed the protein homology of 888 ESTs in the cluster of orthologous group (COGs) database. In this analysis, 412 ESTs were found to have significant protein homologs (E-value < 1e-05) and were sorted into 21 groups according to the functional categories of the database (Table 3). The largest group was assigned to the translation, ribosomal structure and biogenesis category (22.70%); the second largest group was genes involved in posttranslational modification, protein turnover, and chaperones (15.52%), followed by those with only a general function prediction (12.36%), energy production and conversion (11.21%) and lipid transport and metabolism (8.62%).
Using KEGG, 865 unigenes (74.76%) were assigned into specific pathways (Table 4). Thus, 37.69% of the unigenes were included in basic metabolism processes; most of these were involved in carbohydrate metabolism, energy metabolism, amino acid metabolism and lipid metabolism. The other unigenes were assigned into pathways involving human diseases (27.51%), cellular processes (20.11%), genetic information processing (12.14%) and environmental information processing (8.79%).
Discussion
EST analysis provides a powerful and rapid means of reconstructing the transcriptome of specific tissues for the identification of differentially expressed genes. In this study, 2,025 clones were isolated and sequenced, yielding 2,017 high-quality ESTs. In total, we obtained 1,157 unigenes, consisting of 238 contigs and 919 singletons. Approximately three-fourths of the ESTs were similar to sequences in nr protein databases, and the remaining sequences with no matches in the public databases may represent genes with unknown function. Sika deer were only represented by 727 sequences in the NCBI database prior to this study (http://www.ncbi.nlm.nih.gov/protein?term=Cervus%20nippon). Much of the previous EST sequencing in Sika deer has focused on cDNAs derived from antler (total number 701), liver (total number 3), spleen (total number 3), aorta (total number 3), and blood (total number 3), but little attention has been focused on bone marrow (total number 1). Bone marrow consists of different cell types and cells at different stages of differentiation. The major cellular groups in this tissue are hematopoietic cells and stromal cells [42], containing extracellular matrix components, cytokines, and growth factors [43]. In this study, we generated ESTs from the marrow of Sika deer that were not addressed by other research groups; most of the transcripts found in this study were the first representatives of these transcripts for Sika deer. Comparing the obtained sequences against the Sika deer EST database using BLASTN demonstrated that the percentage of the sequences that had significant matches was only 9.29% based on an E-value cut off that was equal to or less than 10−10. More than 90% of the ESTs had no matches to sequences in the Sika deer EST database, and these sequences may represent novel genes in Sika deer.
We listed 37 highly expressed genes observed in the EST collection, most of which were associated with fat-related genes and might originate in the yellow marrow consisting mainly of fat cells. The most highly represented transcript in the EST collection was MSTP008 (http://www.ncbi.nlm.nih.gov/protein/AAQ13510), but there exist almost no research reports on its functions. NCBI reports that it is also known as SCD stearoyl-CoA desaturase (SCD), which may suggest that these transcripts have similar functions. SCD (consisting of 79 ESTs) is a delta-9 fatty acid desaturase that converts saturated fatty acids into monounsaturated fatty acids (MUFAs). These MUFAs are key substrates for the formation of complex lipids, such as triglycerides, cholesterol esters, wax esters and diacylglycerols. Additionally, increased cellular SCD activity has been suggested to influence fatty acid partitioning by promoting fatty acid synthesis while decreasing oxidation [44]. The second most highly represented transcripts were cytochrome c oxidase subunit I (60 ESTs) and subunit III (55 ESTs). Cytochrome c oxidase is the terminal complex of the mitochondrial electron-transport chain. As the last key enzyme in the respiratory electron-transport chain of mitochondria, cytochrome c oxidase plays an important role in ATP generation by oxidative phosphorylation. In mammalian tissues, the holoenzyme is made up of 13 separate subunits, and the largest three of these (subunit I, II, III) are encoded by mitochondrial DNA, forming the functional catalytic core of the enzyme [45–47]. Both subunits I and III are more expressed in Sika deer bone marrow, suggesting their important roles in ATP generation during fat metabolism, as well as cell differentiation. Adipocyte-type fatty acid-binding protein (A-FABP) and adiponectin were also highly expressed. A-FABP belongs to the fatty acid-binding protein super-family in which members have relative molecular masses (Mr) of ~15,000, and it is highly expressed in adipose tissue. It is a predominantly cytosolic protein of mature adipocytes, accounting for ~6% of total cellular proteins. This protein may be an important regulator of systemic insulin sensitivity and lipid and glucose metabolism [48]. Adiponectin, an adipocyte-derived hormone, has been shown to have potential therapeutic applications in obesity, cardiovascular disease, and diabetes because of its influence on glucose and lipid metabolism. It is also found in bone marrow, with the potential to have an inhibitory effect on adipocyte differentiation, regulate some of the earliest events in blood cell formation and mediate functional responses involving hematopoietic cells [49]. In addition, thymosin beta 4 (Tβ4), which contains 15 ESTs, is a low molecular weight protein present in all cells except erythrocytes and has been demonstrated to promote corneal re-epithelialization, reduce inflammation and inhibit apoptosis. It may be used as a wound healing and anti-inflammatory agent for clinical application to promote corneal healing and properly modulate the inflammatory response in cases ranging from routine corneal injuries, such as abrasions and recurrent erosions, to more severe inflammatory-mediated pathologies [50].
Among the ESTs produced, we also obtained the vascular endothelial growth factor-A (VEGF-A) and heparin-binding growth-associated molecule (HB-GAM) genes, which are of great importance for angiogenesis research and have been proven to be regeneration factors involved in rapid angiogenesis during red deer antler growth [51]. VEGF-A, a member of the VEGF family, is an interesting inducer of angiogenesis and lymphangiogenesis because it is a highly specific mitogen for endothelial cells. Signal transduction involves binding to tyrosine kinase receptors and results in endothelial cell proliferation, migration, and new vessel formation [52]. It is expressed during articular cartilage growth, becomes quiescent at maturity, and is re-expressed in osteoarthritis [53]. HB-GAM, which is also known as PTN, OSF-1, HARP, and HBNF [54], is a heparin-binding, secretory protein that functions to induce mitogenesis, angiogenesis, differentiation, and transformation and has a vital role in neovascular formation after acute ischemic brain injury [55]. New research shows that pleiotrophin is a secreted growth factor for hematopoietic stem cells (HSCs) and efficiently promotes HSC expansion in vitro and HSC regeneration in vivo [56]. Furthermore, we found some genes that have previously been reported in human bone marrow, including collagen I (α1, α2), ferritin, decorin, annexin II, vimentin and tubulin [57, 58]. Their relative abundance suggests that they encode proteins with important roles in the biology of bone marrow.
In each of the three main categories (cellular location, molecular function and biological process) of the GO classification, “cell” and “cell part”, “binding” and “cellular process” were dominant, as expected. We also noted a high percentage of genes from the categories “organelle”, “catalytic activity”, and “metabolic process”, but only a few genes from the categories “extracellular matrix part”, “antioxidant activity”, “metallochaperone activity”, and “multi-organism process”. It was particularly interesting that the “catalytic activity” and “metabolic process” categories were overrepresented. This may be related to a large number of enzymes involved in fat metabolism and cell differentiation in the bone marrow. However, in the cellular location category, we did not find any genes in the clusters of “symplast”, “synapse”, “synapse part”, “virion” or “virion part”. In the molecular function category, there were no genes in the “auxiliary transport protein”, “chemoattractant”, “chemorepellent”, “nutrient reservoir”, “proteasome regulator”, or “protein tag” categories. In the biological processes category, “anatomical structure formation”, “cell killing”, “death”, “locomotion”, “reproduction”, “reproductive process”, “rhythmic process” and “viral reproduction” genes were not found. The GO analysis suggested that the ESTs we obtained exhibited a wide coverage of the cellular location, molecular function and biological process categories but were mainly related to genes involved in cellar structure and metabolism.
To further evaluate the completeness of our transcriptome library and the effectiveness of our annotation process, we searched the annotated sequences for genes involved in COGs classifications. In total, out of 888 nr hits, 412 sequences had a COG classification. Among the 21 COGs categories, the cluster for “translation, ribosomal structure and biogenesis” represented the largest group (79, 22.70%) followed by “posttranslational modification, protein turnover, chaperones” (54, 15.52%) and “general function prediction only (43, 12.36%)”. The following categories: chromatin structure and dynamics (2, 0.57%), RNA processing and modification (2, 0.57%) and cell motility (1, 0.29%) represented the smallest groups.
To identify the biological pathways that are active in the marrow of Sika deer, we mapped the 888 annotated sequences to the reference canonical pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG). In total, we assigned 865 sequences to 156 KEGG pathways. The pathways with the most representation among the unique sequences were the ribosome pathway (51 members), Parkinson’s disease pathway (47 members), oxidative phosphorylation pathway (46 members), Huntington’s disease pathway (44 members) and Alzheimer’s disease pathway (42 members). These results suggest that both the ribosome and oxidative phosphorylation pathways may be closely related to neurodegenerative diseases. Thus, they can be utilized not only for gene discovery, but also for studying neurodegenerative diseases. These annotations provide a valuable resource for investigating specific processes, functions and pathways in Sika deer research.
Despite the fact that a greater number of ESTs is required to have a good chance of finding any gene that is of interest and that it will likely take several years before this number can be achieved, we have decided to report the existing number of ESTs. This sequence information is important to the scientific community because the Sika deer genome has not been fully sequenced, and these data are not currently available in the public databases. Further study on this subject is required and will greatly expand our comprehensive understanding of the biological functions of the Sika deer. The results presented in this study could be considered a first step toward genomic and transcriptomic investigations of Sika deer.
Conclusion
This study is the first attempt to define and identify over 2,000 genes in Sika deer. A summary of the assignment of non-redundant ESTs to functional categories, as well as their relative abundances, are listed and discussed in this report. This collection of ESTs represents a starting point for studies in a number of different fundamental and applied areas. The EST sequences generated in this study will be a source of gene-targeted and tissue-specific markers. The dataset will also provide a useful resource for functional genetics studies and genetic divergence studies in Sika deer. This study demonstrates that the EST approach can be a very rapid and useful method for identifying genes that will help in understanding unique features of the biology of organisms of interest. From a wider perspective, only 727 Sika deer ESTs were available in the public databases at the time of submission of this paper. Therefore, the EST data described here represent an important contribution to the publicly accessible sequence data available for Sika deer.
Abbreviations
- ESTs:
-
Expressed sequence tags
- ORF:
-
Open reading frame
- GO:
-
Gene ontology
- COGs:
-
Clusters of orthologous groups of proteins
- KEGG:
-
Kyoto encyclopedia of genes and genomes
References
Lü X, Wei F, Li M, Yang G, Liu H (2006) Genetic diversity among Chinese Sika deer (Cervus nippon) populations and relationships between Chinese and Japanese Sika deer. Chin Sci Bull 51:433–440
Wei Z, Yang Y, Chen Y, Yu Y (2003) Partial cDNA sequence cloning of two housekeeping genes from Sika deer. J Jilin Univ (Medicine Edition) 29:713–718
Wang Y, Yang C, Liu G, Jiang J, Wu J (2006) Generation and analysis of expressed sequence tags from a cDNA library of Tamarix androssowii. Plant Sci 170:28–36
Akao T, Sano M, Yamada O, Akeno T, Fujii K, Goto K, Ohashi-Kunihiro S, Takase K, Yasukawa-Watanabe M, Yamaguchi K, Kurihara Y, Maruyama J, Juvvadi PR, Tanaka A, Hata Y, Koyama Y, Yamaguchi S, Kitamoto N, Gomi K, Abe K, Takeuchi M, Kobayashi T, Horiuchi H, Kitamoto K, Kashiwagi Y, Machida M, Akita O (2007) Analysis of expressed sequence tags from the fungus Aspergillus oryzae cultured under different conditions. DNA Res 14:47–57
Hwang DM, Fung YW, Wang RX, Laurenssen CM, Ng SH, Lam WY, Tsui KW, Fung KP, Waye M, Lee CY (1995) Analysis of expressed sequence tags from a fetal human heart cDNA library. Genomics 30:293–298
Nelson PS, Ng WL, Schummer M, True LD, Liu AY, Bumgarner RE, Ferguson C, Dimak A, Hood L (1998) An expressed-sequence-tag database of the human prostate: sequence analysis of 1168 cDNA clones. Genomics 47:12–25
Ewing B, Green P (2000) Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet 25:232–234
da Mota AF, Sonstegard TS, Van Tassell CP, Shade LL, Matukumalli LK, Wood DL, Capuco AV, Brito MA, Connor EE, Martinez ML, Coutinho LL (2004) Characterization of open reading frame-expressed sequence tags generated from Bos indicus and B. taurus mammary gland cDNA libraries. Anim Genet 35:213–219
Lee SH, Park EW, Cho YM, Lee JW, Kim HY, Lee JH, Oh SJ, Cheong IC, Yoon DH (2006) Confirming single nucleotide polymorphisms from expressed sequence tag datasets derived from three cattle cDNA libraries. J Biochem Mol Biol 39:183–188
Lim D, Lee SH, Cho YM, Yoon D, Shin Y, Kim KW, Park HS, Kim H (2010) Transcript profiling of expressed sequence tags from intramuscular fat, longissimus dorsi muscle and liver in Korean cattle (Hanwoo). BMB Rep 43:115–121
Oishi M, Gohma H, Lejukole HY, Taniguchi Y, Yamada T, Suzuki K, Shinkai H, Uenishi H, Yasue H, Sasaki Y (2004) Generation of a total of 6483 expressed sequence tags from 60 day-old bovine whole fetus and fetal placenta. Anim Biotechnol 15:1–8
Li J, Zhang W (2009) Expression sequence tag and QTL/MAS of goat/sheep in China. Recent Pat DNA Gene Seq 3:213–218
Sheng X, Song X, Yu Y, Niu L, Li S, Li H, Wei C, Liu T, Zhang L, Du L (2010) Characterization of microRNAs from sheep (Ovis aries) using computational and experimental analyses. Mol Biol Rep 38(5):3161–3171. doi:10.1007/s11033-010-9987-3
Lieto LD, Cothran EG (2001) Characterization of expressed sequence tags generated from skin cDNA clones of Equus caballus by single pass sequencing. Anim Biotechnol 12:87–97
Al-Swailem AM, Shehata MM, Abu-Duhier FM, Al-Yamani EJ, Al-Busadah KA, Al-Arawi MS, Al-Khider AY, Al-Muhaimeed AN, Al-Qahtani FH, Manee MM, Al-Shomrani BM, Al-Qhtani SM, Al-Harthi AS, Akdemir KC, Inan MS, Out HH (2010) Sequencing, analysis, and annotation of expressed sequence tags for Camelus dromedarius. PLoS ONE 5(5):e10720. doi:10.1371/journal.pone.0010720
Krammer PH (2000) CD95’s deadly mission in the immune system. Nature 407:789–795
Bonnet D (2003) Biology of human bone marrow stem cells. Clin Exp Med 3:140–149
Lin F (2008) Renal repair: role of bone marrow stem cells. Pediatr Nephrol 23:851–861
Scintu F, Reali C, Pillai R, Badiali M, Sanna MA, Argiolu F, Ristaldi MS, Sogos V (2006) Differentiation of human bone marrow stem cells into cells with a neural phenotype: diverse effects of two specific treatments. BMC Neurosci 7:1186–1198
Kierdorf U, Kierdorf H, Szuwart T (2007) Deer antler regeneration: cells, concepts, and controversies. J Morphol 268:726–738
Zhou Q, Guo Y, Wang L, Wang Y, Liu Y, Wang Y, Wang B (1999) Velvet antler polypeptides promoted proliferation of chondrocytes and osteoblast precursors and fracture healing. Acta Pharmacol Sin 20:279–282
Guan S, Duan L, Li Y, Wang B, Zhou Q (2006) A novel polypeptide from Cervus nippon Temminck proliferation of epidermal cells and NIH3T3 cell line. Acta Biochim Pol 53:395–397
Zhang X, Chang Y, Chen Y, Yu Y (2002) Study on the growth factor-like action of the Sika deer immunocytes. J Jilin Univ (Medicine Edition) 28:603–605
Shao M, Wan M, Wang L, Yu Y (2005) Cloning and analysis of fibroblast growth factor 10 of Cervus nippon Temminck. J Jilin Univ (Medicine Edition) 28:215–218
Lu L, Chen L, Meng X, Yang F, Zhang Z, Chen D (2005) Biological effect of velvet antler polypeptides on neural stem cells from embryonic rat brain. J Chin Med Assoc 118:38–42
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics (Chapter 4: Unit 4.10)
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence. Genome Res 8:195–202
Olson SA (2002) EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite. Brief Bioinform 3:87–91
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
The Gene Ontology Consortium (2010) The gene ontology in 2010: extensions and refinements. Nucleic Acids Res 38:D331–D335
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120
Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38:W695–W699
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29:22–28
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27:29–34
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Orr SL, Hughes TP, Sawyers CL, Kato RM, Quan SG, Williams SP, Witte ON, Hood L (1994) Isolation of unknown genes from human bone marrow by differential screening and single-pass cDNA sequence determination. Proc Natl Acad Sci USA 91:11869–11873
Gregory CA, Prockop DJ, Spees JL (2005) Non-hematopoietic bone marrow stem cells: molecular control of expansion and differentiation. Exp Cell Res 306:330–335
Flowers MT, Ntambi JM (2010) Stearoyl-CoA desaturase and its relation to high-carbohydrate diets and obesity. Biochim Biophys Acta 1791:85–91
Hood DA (1990) Co-ordinate expression of cytochrome c oxidase subunit III and VIc mRNAs in rat tissues. Biochem J 269:503–506
Pridgeon JW, Becnel JJ, Clark GG, Linthicum KJ (2009) Permethrin induces overexpression of cytochrome c oxidase subunit 3 in Aedes aegypti. J Med Entomol 46:810–819
Kang H, Chung J, Lee S (1997) Involvement of cytochrome c oxidasesubunit I gene during neuronal differentiation of PC12 cells. J Biochem Mol Biol 30:285–291
Xu A, Wang Y, Xu JY, Stejskal D, Tam S, Zhang J, Wat NM, Wong WK, Lam KS (2006) Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin Chem 52:405–413
Yokota T, Meka CS, Kouro T, Medina KL, Igarashi H, Takahashi M, Oritani K, Funahashi T, Tomiyama Y, Matsuzawa Y, Kincade PW (2003) Adiponectin, a fat cell product, influences the earliest lymphocyte precursors in bone marrow cultures by activation of the cyclooxygenase–prostaglandin pathway in stromal cells. J Immunol 171:5091–5099
Sosne G, Qiu P, Kurpakus-Wheater M (2007) Thymosin beta 4: a novel corneal wound healing and anti-inflammatory agent. Clin Ophthalmol 1:201–207
Clark DE, Lord EA, Suttie JM (2006) Expression of VEGF and pleiotrophin in deer antler. Anat Rec A 288:1281–1293
Hoeben AN, Landuyt B, Highley MS, Wildiers H, Oosterom AT, De Bruijn EA (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56:549–580
Lingaraj K, Poh CK, Wang W (2010) Vascular endothelial growth factor (VEGF) is expressed during articular cartilage growth and re-expressed in osteoarthritis. Ann Acad Med Singa 39:399–403
Lu KV, Jong KA, Kim GY, Singh J, Dia EQ, Yoshimoto K, Wang MY, Cloughesy TF, Nelson SF, Mischel PS (2005) Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem 280:26953–26964
Yeh HJ, He YY, Xu J, Hsu CY, Deuel TF (1998) Upregulation of pleiotrophin gene expression in developing microvasculature, macrophages, and astrocytes after acute ischemic brain injury. J Neurosci 18:3699–3707
Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, Chi J, Salter AB, Lento WE, Reya T, Chao NJ, Chute JP (2010) Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med 16:475–482
Jia L, Young MF, Powell J, Yang L, Ho NC, Hotchkiss R, Robey PG, Francomano CA (2002) Gene expression profile of human bone marrow stromal cells: high-throughput expressed sequence tag sequencing analysis. Genomics 79:7–17
Silva WA Jr, Covas DT, Panepucci RA, Proto-Siqueira R, Siufi JL, Zanette DL, Santos AR, Zago MA (2003) The profile of gene expression of human marrow mesenchymal stem cells. Stem Cells 21:661–669
Acknowledgment
This work was supported by the National Key Technology R&D Program of China (No. 2007BAI38B06).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Yao, B., Zhao, Y., Zhang, M. et al. Generation and analysis of expressed sequence tags from the bone marrow of Chinese Sika deer. Mol Biol Rep 39, 2981–2990 (2012). https://doi.org/10.1007/s11033-011-1060-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-1060-3