
Abstract
Chalkiness is one of the key determinants of rice quality and is a highly undesirable trait for breeding and marketing. In this study, qWCR7, a major quantitative trait locus (QTL) of white-core rate (WCR), was genetically validated using a BC3F2 segregation population and further fine mapped using a near isogenic line (NIL) population, of which both were derived from a cross between the donor parent DL208 and the recurrent parent ZS97. qWCR7 was finally narrowed to a genomic interval of ~ 68 kb, containing seven annotated genes. Among those, two genes displayed markedly different expression levels in endosperm of NILs. Transcriptome analysis showed that the synthesis and accumulation of metabolites played a key role in chalkiness formation. The contents of storage components and expression levels of related genes were detected, suggesting that starch and storage protein were closely related to white-core trait. Our findings have laid the foundation of map-based cloning of qWCR7, which may have potential value in quality improvement during rice breeding.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rice grain quality is a multi-faceted trait including appearance quality, cooking and eating quality (ECQ), processing quality, nutritional quality, and hygiene quality (Zhou et al. 2020). Among them, appearance quality is mainly determined by grain shape, translucency, and chalkiness, which are immediately noted to consumers and thus are the key factors affecting the commercial value (Fitzgerald et al. 2009). Grain chalkiness refers to the opaque region in endosperm and is classified into white-belly, white-core, white-back, and floury endosperm, according to its position (Lisle et al. 2000; Zhou et al. 2009b; Siebenmorgen et al. 2013). Except for its role in decreasing appearance quality, chalkiness also has a negative effect on ECQ and processing quality. Therefore, grain chalkiness is highly unpopular for marketing, and reducing chalkiness has become an important goal in improving grain quality (Cheng et al. 2005; Misra et al. 2019).
Chalky endosperm is often accompanied by changes in the morphology and arrangement of starch granules in rice and maize (Wang et al. 2008; Li et al. 2014; Zhang et al. 2018). Scanning electron microscope analysis has revealed that round and loosely packed starch granules are filled in chalky endosperm, which is completely different from polyhedral and densely packed starch granules in transparent endosperm; the loose arrangement of starch granules leads to the presence of air spaces and finally opaque phenotype (Kang et al. 2005; Fujita et al. 2007; Guo et al. 2011). Moreover, chalky grains are easier to break in the milling process due to reduced grain density, resulting in the decrease of head rice yield (Zhao and Fitzgerald 2013; Li et al. 2014). Hence, cultivating rice varieties with low chalkiness rate may be a potential strategy to enhance milling yield.
Previous reports have shown that rice chalkiness is a complex quantitative trait dominated by polygenes and environment (Siebenmorgen et al. 2013; Jagadish et al. 2015; Kaneko et al. 2016; Yun et al. 2016; Tang et al. 2019). In the past 20 years, numerous quantitative trait loci (QTL) for chalkiness have been mapped across all 12 pairs of rice chromosomes, but only a few QTL have been fine mapped or cloned (Zhou et al. 2009a; Li et al. 2014; Gao et al. 2016; Zhu et al. 2018). Zhou et al. (2009a) detected two QTL associated with the percentage of chalky grain using a chromosome segmental substitution line (CSSL) population of PA64S and 9311 and narrowed qPGWC-7 to a 44 kb DNA segment. Gao et al. (2016) detected a major QTL controlling area of chalky endosperm qACE9 from a recombinant inbred lines’ (RILs) population of PA64S and 9311 and fine mapped it into an interval of 22 kb region. Zhu et al. (2018) mapped a QTL for percentage of chalky grain qPCG1 in an interval of ~ 139 kb using residual heterozygous lines (RHLs) of Xieqingzao B and Zhonghui 9308. Yang et al. (2021) fine mapped two QTL for grain chalkiness on chromosomes 9 and 11, qPGC9 and qPGC11 respectively, using two single-segment substitution lines (SSSLs), 11–09 with substitution segment from Basmati 370 and HP97-11 with substitution segment from Oryza glaberrima. Chalk5 is the firstly cloned major QTL positively controlling white-belly and encodes a vacuolar H+-translocating pyrophosphatase (V-PPase) with inorganic pyrophosphate (PPi) hydrolysis and H+-translocation activity (Li et al. 2014). Yet, more investigations are needed to unearth the genetic basis of chalkiness.
In a previous study, we repeatedly detected qWCR7 for white-core rate (WCR) on chromosome 7 using a RIL population derived from a cross between two rice cultivars (ZS97 and DL208) (Peng et al. 2014). In this study, we performed genetic validation with a BC3F2 segregating population and fine mapping with a near isogenic line (NIL) population, of which both were derived from the same cross between DL208 and the recurrent parent ZS97. Furthermore, we conducted transcriptome analysis in endosperm to analyze the causes of chalkiness formation. We also observed starch granule morphology of white-core grain by scanning electron microscopy. Finally, we analyzed the effect of qWCR7 on storage components by detecting the contents (oil, protein, and starch) and expression levels of related genes.
Materials and methods
Population and field experiment
A NIL population of qWCR7 was developed from a cross between indica cultivar DL208 (the donor parent) and ZS97 (the recurrent parent). To verify the genetic effect of qWCR7, a BC3F1 plant carrying the heterozygous region of qWCR7 was selected and self-pollinated to generate the BC3F2 population consisting of 180 plants. To conduct fine mapping of qWCR7, a total of 6300 NIL-F2 individuals were used to screen recombinants in the qWCR7 region, which were subjected to progeny test analysis.
The BC3F2 population and NIL-F2 population were planted under natural field conditions at the experimental station of Huazhong Agricultural University at Wuhan (N 30.49°, E 114.36°), Hubei province, in 2013 and 2014, respectively. The progenies of recombinants were grown at Lingshui (N 18.51°, E 110.04°), Hainan province, in 2015. Twelve 30-day-old seedlings of each line were transplanted with 16.5 cm spacing in single row plots in the field; rows were 26.4 cm apart. Field management followed local practices. Ten plants from the middle of each row were harvested individually for trait measurement.
Phenotyping
Harvested seeds from each plant were air dried and stored at room temperature for 3 months before further phenotyping. White-core rate was defined as percentage of white-core grains in random samples of more than 100 dehulled grains from each plant through visual assessment. Flour ground from milled grain was used for measuring total starch content, amylose content, and storage proteins. Total starch content was detected by anthrone colorimetry using a professional Kit (Grace Biotechnology, G0548W, Suzhou). Detection of amylose content was based on Iodine colorimetry (Bao et al. 2006). Measurement of glutelin, prolamin, globulin, and albumin contents in the flour was based on previously published methods (Kumamaru 1988). The brown rice was ground into flour to analyze total lipid content using gas chromatograph-mass spectrometry (GC–MS) (Wu et al. 2005).
Marker development and genotyping
All mapping primers were designed by referring to the genomes of ZS97 and MH63 (http://ricevarmap.ncpgr.cn/) (Zhao et al. 2015) and Rice EST Data Base (http://redb.ncpgr.cn/). Genomic DNA was extracted from young leaves, according to the cetyltrimethylammonium bromide (CTAB) procedure (Murray and Thompson 1980). PCR amplification reaction was performed at an initial 94 °C for 5 min, followed by 32 cycles of 30 s at 94 °C, 30 s/1 min at 55 °C, and 30 s at 72 °C, and then 72 °C for 5 min. The PCR products were identified by sequencing or using 4% non-denaturing polyacrylamide gel electrophoresis (PAGE) and visualized using silver staining. The polymorphic simple sequence repeat (SSR) or insertion/deletion (InDel) markers within the QTL interval were applied to identify the genotype of individuals (Supplementary Table 1).
The parental lines ZS97 and DL208 were sequenced by the illumine HiSeq2000 that generate 100 bp paired-end reads. The paired-end reads of the two parental lines were aligned against the rice reference genome (IRGSP 7.0) using BWA software (Li and Durbin 2009). SNPs were identified using SAMtools and BCFtools. Only alignments with mapping quality ≥ 40 were used, and bases with base quality ≥ 10 were used to identify SNPs. Only the reads uniquely mapped to the genome sequence were retained for further analysis. Genotypes of 4,726 varieties (http://ricevarmap.ncpgr.cn/) were used to impute missing genotypes of the parents using Beagle software (Browning et al. 2018).
Transcriptome assembly and functional annotation
Endosperm samples (two biological replicates) at the 14th day after flowering (DAF) were used for RNA sequencing by Hiseq-PE150 (Novogene Biotech. Company, Tianjin). Raw data were processed using NGS QC Toolkit (Patel and Jain 2012), and the reads containing ploy-N and the low-quality reads were removed to obtain the clean reads. The read counts of each gene were obtained by featureCounts and the fragments per kilobase of transcript per million fragments mapped (FPKM) value of each gene was calculated using R script. Differentially expression genes were identified using the DESeq2 (with replicates). P value ≤ 0.05 and |Log2FoldChange|≥ 1 determined as significantly differentially expression genes DEGs were selected and analyzed by GO (Gene Ontology) function and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment by clusterProfiler of R package.
Scanning electron microscopy (SEM)
Milled rice grains were transversely cut in the mid-section and coated with gold under vacuum conditions. Starch granule morphology at the central and peripheral parts of the endosperm was examined with a scanning electron microscope (JSM-6390LV, JEOL) at an accelerating voltage of 10 kV and spot size of 30 nm. SEM analysis involved at least three biological replications of mounted specimens. All procedures were carried out according to the manufacturer’s protocol.
RNA extraction, reverse transcription, and qRT-PCR
Total RNA was extracted from plant tissues using an RNA extraction kit (TRIzol, Invitrogen). First-strand cDNA was synthesized in 20 µl of reaction medium containing 2 µg of RNA and 200 U of M-MLV reverse transcriptase (Promega). Quantitative RT-PCR was performed on a QuantStudio6 Flex machine using SYBR Green PCR reagent according to the manufacturer’s instructions. All assays were performed with at least three biological and three technical replications. The rice actin1 gene served as the internal control to normalize gene expression. Primers of the genes involved in metabolism of storage components were used for qRT-PCR assays (Supplementary Table 2).
Statistical analysis
QTL analysis of the BC3F2 population was performed by WinQTLCart 2.5 and Mapmaker/Exp3.0 (Lander et al. 1987; Zeng 1994). The significance of differences for two sets of data was performed using two-tailed t test at a P < 0.05 level.
Results
Genetic validation of QTL effects of qWCR7
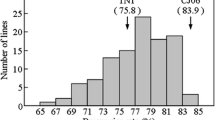
In a previous study, qWCR7 was located between markers RM445 and RM418 on chromosome 7 using a RIL population derived from a cross between DL208 and ZS97 (Peng et al. 2014). To validate the genetic effect of qWCR7, we developed a segregating population of 180 BC3F2 plants with ZS97 being the recurrent parent and DL208 the donor parent. The phenotypic variation of WCR in the population showed continuous bimodal distribution (Fig. 1). Meanwhile, we added an InDel marker WB1693 and analyzed the effect of the QTL. qWCR7 was located between markers WB1693 and RM445, explaining 81.40% of the phenotypic variance (Table 1). This result indicated that qWCR7 was a major genetic factor conferring WCR variation, and the ZS97-derived allele increased the WCR.

Frequency distribution of WCR in 180 BC3F2 population. Arrows indicate the average values of homozygous ZS97 (black) and homozygous DL208 (white), respectively
Fine mapping of qWCR7
To improve the mapping resolution of qWCR7, we developed a NIL-F2 population consisting of 6,300 individuals, from which a total of 45 recombinants were identified between WB1693 and RM445. Subsequently, 5 polymorphic indel markers were developed within the target region based on publicly available rice genome sequences (http://ricevarmap.ncpgr.cn/) and used in genotyping of recombinants (Fig. 2a). We undertook progeny tests to determine the qWCR7 genotype of each recombinant (Supplementary Table 3). qWCR7 showed incompletely dominant and was eventually narrowed to a genomic interval of ~ 68 kb flanked by markers M1711 and M1718 (Fig. 2a). To evaluate the effect of qWCR7 on chalkiness, we used a pair of NILs, NILqWCR7−ZS97 and NILqWCR7−DL208, which carried the qWCR7 locus from ZS97 and DL208, respectively, in the background of ZS97. Compared with NILqWCR7−DL208, NILqWCR7−ZS97 displayed ~ 24% higher WCR value (Fig. 2b).

Fine mapping of qWCR7 for grain chalkiness. a Substitution mapping of qWCR7. Fine mapping of the qWCR7 region using NIL-F2 population consisting of 6,300 individuals. The number of recombinants indicates the number of recombinants between the target site and flanking molecular markers. Genotypes and phenotypes of recombinants, each of which was confirmed by progeny test. Black, white, and gray blocks represent the genotypes of homozygous ZS97, homozygous DL208, and heterozygote, respectively. b The head rice appearance and WCR values of the NILqWCR7−ZS97 and NILqWCR7−DL208. Scale bar: 10 mm. Significant difference were based on two-tailed t test, **P ≤ 0.01. Error bars, s.e.m. c Scanning electron microscopic analysis of transverse sections of mature seeds from the NILs. Magenta and green rectangles represent the center and the edge positions of endosperm. Scale bars: 500 µm (entire grain), 10 µm (boxes)
According to the annotation information of Nipponbare (MSU Rice Genome Annotation Project), the target region of qWCR7 contains seven annotated genes (LOC_Os07g29220, LOC_Os07g29224, LOC_Os07g29230, LOC_Os07g29240, LOC_Os07g29280, LOC_Os07g29290, LOC_Os07g29300). Among these genes, LOC_Os07g29220, LOC_Os07g29230, and LOC_Os07g29290 encode a putative cyclopropane-fatty-acyl-phospholipid synthase, an LTP family protein precursor and an expansin precursor, respectively, whereas the remaining genes encode expressed proteins. We further compared the sequence variations of these candidate genes including the 2 kb promoter and the coding sequence between ZS97 and DL208 (Table 2). The result showed that the variations occurred in the promoter, intron, and 5′ and 3′ untranslated regions (UTR).
Many studies have shown that the formation of chalkiness is related to the morphology and arrangement of starch granules. Then, we examined the starch granule morphology of mature seeds using scanning electron microscopy. Results showed that starch granules of white-core grains from NILqWCR7−ZS97 were small, round, and loosely packed in the center of endosperm, which were notably different from polyhedral and densely packed starch granules from NILqWCR7−DL208; however, no difference was observed in the edge of endosperm of both the grains (Fig. 2c). This result indicated that altered morphology and spatial distribution of starch granules caused white-core endosperm.
Transcriptome analysis in endosperm
To detect putative differentially expressed genes (DEGs) affecting WCR, we performed transcriptome analysis in the 14th day after flowering (DAF) endosperm of NILqWCR7−ZS97 and NILqWCR7−DL208, which were denoised as 14EZ and 14ED, respectively. DEGs were identified using the following criteria: P value ≤ 0.05 and |Log2Fold Change|≥ 1. There were 1849 DEGs in 14ED vs 14EZ (1010 downregulated and 839 upregulated) (Fig. 3a and b). Gene Ontology (GO) enrichment analysis of DEGs showed that the most enriched genes participated in “biological process,” followed by “molecular function” and “cellular component.” The “biological process” category was dominated by photosynthesis (GO:0,015,979), epigenetic (GO:0,040,029), DNA metabolic process (GO:0,006,259), cell cycle (GO:0,007,049), flower development (GO:0,009,908), cell differentiation (GO:0,030,154), cell death (GO:0,008,219), cell growth (GO:0,016,049), secondary metabolic process (GO:0,019,748), and metabolites and energy (GO:0,006,091). The “molecular function” category was dominated by lipid binding (GO:0,008,289), motor activity (GO:0,003,774), and obsolete signal transducer activity (GO:0,004,871). In the “cellular component” category, thylakoid (GO:0,009,579), extracellular region (GO:0,005,576), and nuclear envelope (GO:0,005,635) were prominently represented (Fig. 3c). The above results showed that lipid binding, photosynthesis, and metabolic regulation may be tightly related to white-core.

DEGs and Gene Ontology (GO) enrichment analysis. a and b DEGs obtained by 14ED vs. 14EZ. DEGs were differentially expressed with statistical significance (P value ≤ 0.05 and |Log2FoldChange|≥ 1). c Comparison of GO classifications of DEGs in 14 DAF endosperm
To identify metabolic pathways in which DEGs were involved, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. DEGs mainly belonged to metabolic pathways, biosynthesis of secondary metabolites, starch and sucrose metabolism, photosynthesis, ABC transporters, phenylpropanoid biosynthesis, fatty acid elongation, plant–pathogen interaction, plant hormone signal transduction etc. (Fig. 4a). Further analysis exhibited that the total number of DEGs related to metabolic pathways, biosynthesis of secondary metabolites, and starch and sucrose metabolism accounted for a very large proportion, indicating that the accumulation of metabolites such as starch and protein may plays an important role in the process of chalkiness formation.

KEGG pathway assignments and expression analysis of the putative genes at qWCR7 locus. a KEGG pathway enrichment analysis of DEGs by 14ED vs. 14EZ. The major 20 categories are shown. b Expression patterns of the 7 candidate genes in 14 DAF endosperm of NILs, which are shown as fragments per kilobase of transcript per million fragments mapped (FPKM)
In order to analyze the transcription level of the 7 putative genes in the qWCR7 region, we extracted their transcriptome data of endosperm. As shown in Fig. 4b, LOC_Os07g29230, LOC_Os07g29280, and LOC_Os07g29300 were not expressed in 14 DAF endosperm, and the expression level of LOC_Os07g29290 was the highest. Importantly, both LOC_Os07g29224 and LOC_Os07g29240 had markedly different expression levels between NILqWCR7−ZS97 and NILqWCR7−DL208, suggesting that they might be the causal gene underlying qWCR7.
Effects of qWCR7 on rice grain storage components
Chalkiness formation is closely related to changes in the contents of storage components such as oil, protein, and starch (Kang et al. 2005; Li et al. 2014). To detect the effects of qWCR7 on storage components, we investigated the contents in the mature grains of NILqWCR7−ZS97 and NILqWCR7−DL208. Firstly, the oil composition consisting of various fatty acids was detected using GC–MS. Among identified fatty acids, myristic acid (C14:0), stearic acid (C18:0), and arachidic acid (C20:0) showed markedly higher values in NILqWCR7−ZS97 than in NILqWCR7−DL208. However, no difference was observed in other fatty acids accounting for more than 96% of the oil concentration between the two NILs, leading to similar content of the total oil (Fig. 5a). Subsequently, we detected storage protein contents including prolamin, glutelin, albumin, globulin, and total protein. Compared with NILqWCR7−DL208, NILqWCR7−ZS97 displayed significantly higher contents in glutelin but lower content in albumin. The content of total protein showed higher value in NILqWCR7−ZS97, which was related to the extremely high proportion of glutenin in storage protein (Fig. 5b). Then, we determined contents of total starch and amylose in milled rice. The amylose contents were similar between NILqWCR7−ZS97 and NILqWCR7−DL208, but the total starch content of NILqWCR7−ZS97 was significantly lower than that of NILqWCR7−DL208 (Fig. 5c).

The content of stored components in NILqWCR7−ZS97 and NILqWCR7−DL208. a Lipid content in brown rice. C14:0, myristic acid; C16:0, palmitic acid; C18:0, stearic acid; C18:1, oleic acid; C18:2, linoleic acid; C18:3, linolenic acid; C20:0, arachidic acid; C20:1, eicosenoic acid; oil, total lipid content. b Protein content in milled rice. c Starch content in milled rice. In a–c, the number of NILqWCR7−ZS97 and NILqWCR7−DL208 was 11 and 12 plants, respectively, and each sample consisted of at least 300 grains. Significant differences were based on two-tailed t tests, *P ≤ 0.05, **P ≤ 0.01. Error bars, s.e.m. d The effects of qWCR7 on transcription levels of genes related to storage components in 7 DAF endosperms. Endosperm samples were from different plants. Significant differences were based on two-tailed t tests (n = 4), *P ≤ 0.05, **P ≤ 0.01. Error bars, s.e.m
In addition, we examined the transcription level of genes related to storage components in 7 DAF endosperm. Relative expression levels of starch synthesis genes GBSSI, GBSSII, SSIVb, AGPL1, AGPL2, AGPS1, and AGPS2a displayed no difference, whereas that of SSI, SSIIc, SSIIIa, SBEI, SBEIIa, SBEIIb, AGPL3, AGPL4, and AGPS3b were generally higher in NILqWCR7−DL208 than in NILqWCR7−ZS97. The levels of glutelin synthesis genes GluA1, GluA2, GluA3, GluB1, and Glutelinλ were markedly lower in NILqWCR7−DL208 than in NILqWCR7−ZS97 (Fig. 5d). The above results suggested that qWCR7 had pleiotropic effects on storage components by affecting the expression levels of key genes related to starch and storage protein synthesis, which may influence the formation of white-core.
Discussion
Chalkiness is one of the key determinants of rice appearance quality. Chalky grains are also easier to crack during polishing due to the loose arrangement of starch granules of opaque endosperm, leading to a decreased head rice yield (Li et al. 2014). Moreover, the market value of rice largely depends on chalky grain rate and broken kernel rate, and chalkiness has a direct or indirect impact on both aspects (Fitzgerald et al. 2009). Therefore, cultivating varieties with low chalkiness not only helps to improve quality, but also contributes to marketable yield in rice.
Until now, a lot of QTL for rice quality have been mapped, but only a few QTL/genes related to chalkiness have been fine mapped or cloned. The main reason is that chalkiness phenotype is greatly influenced by numerous genetic factors and external stress (Nevame et al. 2018; Deng et al. 2021). To obtain genetically stable QTL of chalkiness, researchers often used different populations under multiple environmental conditions (Zhou et al. 2009a; Gao et al. 2016; Yun et al. 2016; Zhu et al. 2018). The percentage of chalky grains was significantly different in Hainan and Zhejiang province using three populations, and the phenotypic variance of qPCG1 ranged from 6.8 to 21.9% (Zhu et al. 2018). Previously, qWCR7 was detected in RILs (ZS97/DL208) and F2 (ZS97/WG97) populations in multiple years, and the phenotypic variance ranged from 12.5 to 19.9% (Peng et al. 2014). To reduce the interference of genetic background, we constructed a NIL population of high generation in this study to fine map qWCR7 in two environments (Figs. 1 and 2). Compared with the RILs and F2 populations, the genetic effect of qWCR7 was more stable and greater in the NIL population (Table 1). Taken together, it is necessary to verify the reliability of chalkiness QTL in a variety of environmental and genetic backgrounds, which would facilitate the following utilization in rice breeding. At the same time, high-quality NIL population also plays an important role in fine mapping of minor QTL.
With the global warming, high temperature has become an important factor to promote the production of chalkiness in rice (Zhao and Fitzgerald 2013; Nevame et al. 2018). Lanning et al. (2011) reported that there is a significant positive correlation between night temperature and chalkiness rate during grain filling. Lyman et al. (2013) showed that high temperature stress increased percentages of chalky and broken kernels. Nevame et al. (2018) concluded that high temperature affected chalkiness formation by grain filling rate, carbohydrate biosynthesis, protein formation/degradation, redox homeostasis, and cell rescue/defense pathways. In our study, we found that the WCR of NILs in Wuhan was significantly higher than that in Lingshui (Figs. 1 and 2). Then we compared the average value of the maximum temperature of the two locations in grain filling stage and found that the temperature in Wuhan (30 °C) was significantly higher than that in Lingshui (20 °C). Thus, we speculated that chalkiness variation of the two locations is probably related to environmental conditions especially the temperature during grain filling stage.
Numerous studies have reported that chalkiness formation is influenced by the synthesis and accumulation of storage substances in endosperm (Fujita et al. 2007; Ryoo et al. 2007; Wang et al. 2008; Li et al. 2014). Flo5 encoded a soluble starch synthase SSIIIa involved in the biosynthesis of amylopectin, and its mutant displayed a white-core floury endosperm (Fujita et al. 2007; Ryoo et al. 2007). Wang et al. (2008) identified a cell-wall invertase gene GIF1 required for carbon partitioning, and its mutant gif1 showed markedly less sugar content and more grain chalkiness. In this study, the GO function and KEGG pathway analysis showed that a large number of DEGs participated in “secondary metabolic process” and “metabolites and energy” biological processes through “metabolic pathways,” “biosynthesis of secondary metabolites,” and “starch and sucrose metabolism” pathways, suggesting that the synthesis and accumulation of metabolites such as starch and protein may play a key role in chalkiness formation (Figs. 3c and 4a). We further detected the contents of starch and storage protein in NILs, and higher WCR was accompanied by lower starch content and higher protein content (Fig. 5b and c). Expression analysis showed that the difference of total starch was not induced by amylose but may be caused by amylopectin which is synthesized by soluble starch synthase (SS), starch branching enzyme (SBE), and ADP-glucose pyrophosphorylase (AGP) (Fig. 5d). In addition, we found that glutelin was the key storage protein causing the difference of total protein in NILs (Fig. 5b and d). Therefore, we concluded that the changes of these storage substances were closely related to the formation of chalkiness.
In summary, our results revealed that qWCR7 was a key genetic factor conferring WCR variation and was narrowed to a genomic interval of ~ 68 kb. Transcriptome analysis showed that the synthesis and accumulation of metabolites played a key role in chalkiness formation. The detection of storage components and expression levels of related genes exhibited that starch and storage protein were tightly related to white-core trait. Thus, our findings showed that qWCR7 may have potential value in the improvement of rice grain quality.
References
Bao J, Shen S, Sun M, Corke H (2006) Analysis of genotypic diversity in the starch physicochemical properties of nonwaxy rice: apparent amylose content, pasting viscosity and gel texture. Starch-Stärke 58:259–267
Browning BL, Zhou Y, Browning SR (2018) A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103:338–348
Cheng F, Zhong L, Wang F, Zhang G (2005) Differences in cooking and eating properties between chalky and translucent parts in rice grains. Food Chem 90:39–46
Deng F, Li Q, Chen H, Zeng Y, Li B, Zhong X, Wang L, Ren W (2021) Relationship between chalkiness and the structural and thermal properties of rice starch after shading during grain-filling stage. Carbohydr Polym 252:117212
Fitzgerald MA, McCouch SR, Hall RD (2009) Not just a grain of rice: the quest for quality. Trends Plant Sci 14:133–139
Fujita N, Yoshida M, Kondo T, Saito K, Utsumi Y, Tokunaga T, Nishi A, Satoh H, Park JH, Jane JL (2007) Characterization of SSIIIa-deficient mutants of rice: the function of SSIIIa and pleiotropic effects by SSIIIa deficiency in the rice endosperm. Plant Physiol 144:2009–2023
Gao Y, Liu C, Li Y, Zhang A, Dong G, Xie L, Zhang B, Ruan B, Hong K, Xue D (2016) QTL analysis for chalkiness of rice and fine mapping of a candidate gene for qACE9. Rice 9:41
Guo T, Liu X, Wan X, Weng J, Liu S, Liu X, Chen M, Li J, Su N, Wu F (2011) Identification of a stable quantitative trait locus for percentage grains with white chalkiness in rice (Oryza sativa). J Integr Plant Biol 53:598–607
Jagadish S, Murty M, Quick W (2015) Rice responses to rising temperatures–challenges, perspectives and future directions. Plant, Cell Environ 38:1686–1698
Kaneko K, Sasaki M, Kuribayashi N, Suzuki H, Sasuga Y, Shiraya T, Inomata T, Itoh K, Baslam M, Mitsui T (2016) Proteomic and glycomic characterization of rice chalky grains produced under moderate and high-temperature conditions in field system. Rice 9:26
Kang HG, Park S, Matsuoka M, An G (2005) White-core endosperm floury endosperm-4 in rice is generated by knockout mutations in the C4-type pyruvate orthophosphate dikinase gene (OsPPDKB). Plant J 42:901–911
Kumamaru T (1988) Mutants for rice storage proteins 1. Screening of mutants semidwarfism-related proteins and glutelin seed protein in rice (Oryza sativa L.). Theor Appl Genet 83:153–158
Lander E, Green P, Abrahamson J, Barlow A, Daly M, Lincoln S, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lanning SB, Siebenmorgen TJ, Counce PA, Ambardekar AA, Mauromoustakos A (2011) Extreme nighttime air temperatures in 2010 impact rice chalkiness and milling quality. Field Crops Res 124:132–136
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li Y, Fan C, Xing Y, Yun P, Luo L, Yan B, Peng B, Xie W, Wang G, Li X (2014) Chalk5 encodes a vacuolar H+-translocating pyrophosphatase influencing grain chalkiness in rice. Nat Genet 46:398
Lisle A, Martin M, Fitzgerald M (2000) Chalky and translucent rice grains differ in starch composition and structure and cooking properties. Cereal Chem 77:627–632
Lyman NB, Jagadish KS, Nalley LL, Dixon BL, Siebenmorgen T (2013) Neglecting rice milling yield and quality underestimates economic losses from high-temperature stress. PLoS One 8:e72157
Misra G, Anacleto R, Badoni S, Butardo V Jr, Molina L, Graner A, Demont M, Morell MK, Sreenivasulu N (2019) Dissecting the genome-wide genetic variants of milling and appearance quality traits in rice. J Exp Bot 70:5115–5130
Murray M, Thompson W (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4326
Nevame AYM, Emon RM, Malek MA, Hasan MM, Alam MA, Muharam FM, Aslani F, Rafii MY, Ismail MR (2018) Relationship between high temperature and formation of chalkiness and their effects on quality of rice. Biomed Res Int 2018:1653721
Patel RK, Jain M (2012) NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619
Peng B, Wang L, Fan C, Jiang G, Luo L, Li Y, He Y (2014) Comparative mapping of chalkiness components in rice grain using five populations across two environments. BMC Genet 15:49
Ryoo N, Yu C, Park C, Baik M, Park IM, Cho M, Bhoo SH, An G, Hahn T, Jeon JS (2007) Knockout of a starch synthase gene OsSSIIIa/Flo5 causes white-core floury endosperm in rice (Oryza sativa L.). Plant Cell Rep 26:1083–1095
Siebenmorgen TJ, Grigg BC, Lanning SB (2013) Impacts of preharvest factors during kernel development on rice quality and functionality. Annu Rev Food Sci Technol 4:101–115
Tang S, Zhang H, Liu W, Dou Z, Zhou Q, Chen W, Wang S, Ding Y (2019) Nitrogen fertilizer at heading stage effectively compensates for the deterioration of rice quality by affecting the starch-related properties under elevated temperatures. Food Chem 277:455–462
Wang E, Wang J, Zhu X, Hao W, Wang L, Li Q, Zhang L, He W, Lu B, Lin H (2008) Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat Genet 40:1370–1374
Wu G, Truksa M, Datla N, Vrinten P, Bauer J, Zank T, Cirpus P, Heinz E, Qiu X (2005) Stepwise engineering to produce high yields of very long-chain polyunsaturated fatty acids in plants. Nat Biotechnol 23:1013–1017
Yang W, Liang J, Hao Q, Luan X, Tan Q, Lin S, Zhu H, Liu G, Liu Z, Bu S et al (2021) Fine mapping of two grain chalkiness QTLs sensitive to high temperature in rice. Rice (n y) 14:33
Yun P, Zhu Y, Wu B, Gao G, Sun P, Zhang Q, He Y (2016) Genetic mapping and confirmation of quantitative trait loci for grain chalkiness in rice. Mol Breed 36:162
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang S, Zhan J, Yadegari R (2018) Maize opaque mutants are no longer so opaque. Plant Reproduction 31:319–326
Zhao H, Yao W, Ouyang Y, Yang W, Wang G, Lian X, Xing Y, Chen L, Xie W (2015) RiceVarMap: a comprehensive database of rice genomic variations. Nucleic Acids Res 43:D1018-1022
Zhao X, Fitzgerald M (2013) Climate change: implications for the yield of edible rice. PLoS One 8:e66218
Zhou H, Xia D, He Y (2020) Rice grain quality—traditional traits for high quality rice and health-plus substances. Mol Breeding 40:1
Zhou L, Chen L, Jiang L, Zhang W, Liu L, Liu X, Zhao Z, Liu S, Zhang L, Wang J (2009a) Fine mapping of the grain chalkiness QTL qPGWC-7 in rice (Oryza sativa L.). Theor Appl Genet 118:581–590
Zhou LJ, Jiang L, Zhai HQ, Wan JM (2009b) Current status and strategies for improvement of rice grain chalkiness. Hereditas 31:563–572
Zhu A, Zhang Y, Zhang Z, Wang B, Xue P, Cao Y, Chen Y, Li Z, Liu Q, Cheng S (2018) Genetic dissection of qPCG1 for a quantitative trait locus for percentage of chalky grain in rice (Oryza sativa L.). Front Plant Sci 9:1173
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology (Grant 2016YFD0100501), the National Program on R&D of Transgenic Plants (2016ZX08009004), and the Natural Science Foundation of China (91935303) and earmarked fund for the China Agriculture Research System (CARS-01-03).
Author information
Authors and Affiliations
Contributions
Bian Wu conducted most of the experiments, including genetic effect validation, fine mapping, expression analysis, electron microscopy, transcriptome analysis, and detection of storage substances. Duo Xia, Hao Zhou, and Shiyuan Cheng contributed to some statistical analysis. Yipei Wang and Minqi Li contributed to detection of storage substances. Guanjun Gao, Qinglu Zhang, and Xianghua Li participated in field management and logistics. Yuqing He designed and supervised the study. Yuqing and Bian Wu analyzed the data and wrote the paper.
Corresponding author
Ethics declarations
Ethics approval
The experiments comply with the ethical standards in the country in which they were performed.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, B., Xia, D., Zhou, H. et al. Fine mapping of qWCR7, a grain chalkiness QTL in rice. Mol Breeding 41, 68 (2021). https://doi.org/10.1007/s11032-021-01260-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-021-01260-x