
Abstract
In pepper, the TMV resistance locus L is syntenic to the tomato I2 and the potato R3 loci on chromosome 11. In this report, we identified pepper bacterial artificial chromosome (BAC) clones corresponding to the I2 and R3 loci and developed L-linked markers using the BAC sequence information. A BAC library was screened using the tomato I2C-1 gene as a probe. The resulting clones were sorted further by PCR screening, sequencing, and genetic mapping. A linkage analysis revealed that BAC clone 082F03 could be anchored to the target region near TG36 on chromosome 11. Using the 082F03 sequence, more BAC clones were identified and a BAC contig spanning 224 kb was constructed. Gene prediction analysis showed that there were at least three I2/R3 R gene analogs (RGAs) in the BAC contig. Three DNA markers closely linked (about 1.2 cM) to the L 4 gene were developed by using the BAC contig sequence. The single nucleotide polymorphism marker 087H3T7 developed in this study was subjected to linkage analysis in L 4- and L 3-segregating populations together with previously developed markers. The 189D23M marker, which is known to co-segregate with L 3, was located on the opposite side of 087H3T7, about 0.7 cM away from L 4. This supports the idea that L 3 and L 4 may be different genes closely linked within the region instead of different alleles at the same locus. Finally, use of flanking markers in molecular breeding program for introgression of L 4 to elite germplasm against most aggressive tobamoviruses pathotype P1,2,3 is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Comparative genetic analysis of disease resistance in crop plants suggests that disease resistance genes (R genes) do not distribute randomly within a genome (Grube et al. 2000; Wisser et al. 2005, 2006). Clustering of R genes conferring resistance to several unrelated pathogen types often occurs and their syntenic positions across the genomes of tomato, potato, and pepper indicate that this clustering is conserved within the Solanaceae (Grube et al. 2000).
An earlier comparative genetic map had revealed the syntenic relationship between the I2 gene cluster in tomato and R3 gene complex in potato (Grube et al. 2000). In tomato, the I2C-1 gene has been cloned and demonstrated to confer partial resistance to race 2 of the soil-borne fungus Fusarium oxysporum f. sp lycopersici. The gene belongs to a multi-gene I2 complex (I2C) family and is located within the SL8D cluster on chromosome 11 in tomato. At least four other I2C homologs have been mapped and found to be distributed on three chromosomes in the tomato genome, of which the SL8E cluster has been mapped closely with SL8D. Genomic dissection of the SL8D and SL8E regions consequently led to the revelation of a microsyntenic relationship between the SL8E cluster in tomato and R3a cluster in potato (Huang et al. 2005; Ori et al. 1997). Based on colinearity, the R3a gene, which confers resistance to Phytophthora infestans in potato, has been isolated and cloned via a comparative genomics approach with the resistance gene analog of I2 (Huang et al. 2005). R7, which also confers late blight resistance and maps to the same locus, has been demonstrated to be allelic to the R3 gene complex (Huang et al. 2004, 2005). Sequence analysis also demonstrated that R3a is an I2 gene analog (GA) and shares 88% DNA sequence similarity (Huang et al. 2005). Furthermore, both I2C-1 and R3a are coiled-coil nucleotide-binding site leucine-rich repeat (CC–NBS–LRR) type R genes (due to the presence of a predicted nucleotide binding site and leucine-rich repeats with a coiled-coil region at the N-terminus) (Ballvora et al. 2002; Huang et al. 2005; Kuang et al. 2005; Song et al. 2003; van der Vossen et al. 2003, 2005), which is the most abundant type of R gene in plant genomes among the five classes of R genes responsible for disease resistance in plants (Dangl and Jones 2001; Meyers et al. 1999, 2003; Michelmore and Meyers 1998). The orthologous relationship, supported by syntenic positions and sequence similarity, indicate that the two genes belong to an ancient R gene locus prior to the divergence of tomato and potato.
In pepper, the syntenic position of the I2C and R3a loci contains an R-gene cluster having several quantitative trait loci (QTL) and also a dominant resistance gene, L (Grube et al. 2000). At the L locus, four sources of resistance genes have been found from cultivars and wild species of pepper including Capsicum annuum (L 1), Capsicum frutescens (L 2), Capsicum chinense (L 3), and Capsicum chacoense (L 4) (Berzal-Herranz et al. 1995; Boukema 1980; de la Cruz et al. 1997). Allelism of the four genes controlling resistance has been demonstrated in an early genetic study of TMV resistance (Boukema 1980). L has been demonstrated to confer resistance to several tobamoviruses infecting pepper, including tobacco mosaic virus (TMV), tomato mosaic virus (ToMV), paprika mild mottle virus (PaMMV) and pepper mild mottle virus (PMMoV) (Csillery et al. 1983; Pernezny et al. 2003; Wetter et al. 1984). These tobamoviruses have been classified into different pathotypes—P0, P1, P1,2, and P1,2,3—based on their pathogenicity and interaction with the counterpart resistance L genes (Alonso et al. 1991; Takeuchi et al. 2005). Several PMMoV isolates classified as P1,2,3 have been shown to be the most aggressive, all L gene-mediated resistance breaks down where systemic infection occurs with the exception of L 4 (Tsuda et al. 1998; Velasco et al. 2002; Matsunaga et al. 2003; Kim et al. 2008). L 4 confers resistance against most pathotypes of tobamoviruses by inducing hypersensitive response (HR). The coat protein of the tobamovirus has been demonstrated to elicit L gene-mediated resistance by inducing a distinctive HR (Berzal-Herranz et al. 1995; de la Cruz et al. 1997).
L has been mapped at the lower arm of pepper chromosome 11 (Lefebvre et al. 1995). Since then, several L-linked markers have been developed using various DNA markers and mapping populations. Random amplified polymorphic DNA (RAPD) markers, which are located at 4.0 cM from the L 3 locus, were developed by Sugita et al. (2004). Recently, more tightly linked markers to the L 3 locus (<0.1 cM away) were developed using bulked segregant analysis-amplified fragment length polymorphism (BSA-AFLP) and resistance gene analog (RGA) mapping (Tomita et al. 2008). Several L 4-linked markers were also developed. Matsunaga et al. (2003) reported L 4-linked RAPD markers. One of the RAPD markers (WA31-1500), located 1.5 cM from the L 4 locus, was converted to a SCAR marker. Another mapping study showed three AFLP markers linked to L 4 and the closest marker (L4SC340) was converted into a SCAR marker (Kim et al. 2008). When this marker was tested in two populations, the marker was mapped 0.9 and 1.8 cM away from L 4. Availability of these markers provides a great opportunity for fine mapping of the L 4 gene and for elucidating the allelic relationship between L 3 and L 4.
In this study, we have performed a comparative genomic study between the R3/R7 locus in potato, the I2 locus in tomato and the L locus in pepper to investigate the possible orthologous relationships among the R-gene clusters on chromosome 11. The goal of this study was to identify molecular markers, particularly Single nucleotide polymorphism (SNP) markers, closely linked to the L gene through comparative genomics of the syntenic regions among the three Solanaceous crops. We have also defined available markers and constructed a localized map flanking the L locus. Furthermore, we elucidated the allelic relationships between L 3 and L 4 using L-linked markers.
Materials and methods
Plant and virus materials
Capsicum annuum ‘Early California Wonder’ (ECW) and ‘NuMex RNaky’ (RN) were purchased from Asgrow Seed Co. (San Juan Bautista, CA, USA); ‘Special’ (SP) and ‘Cupra’ (CP) were obtained from Enza Zaden (Enkhuizen, The Netherlands); Myoung-Sung (MS) was purchased from Seminis Korea Inc. Hungnong Seeds. (Seoul, Korea). C. chinense PI159234 (234) and PI159236 (236) and C. chacoense PI260429 (429) were obtained from the USDA Southern Regional Plant Introduction Station (Experiment, GA, USA). C. chacoense PI260429 was used as a TMV resistant control, whereas ECW was the TMV susceptible control. CP had the L 3 allele, and SP and MS had the L 4 allele. For virus resistance and genotype analysis, F2-segregating populations were developed from the commercial F1 hybrid cultivars CP, SP, and MS. An F2 population derived from a cross between RN and 234 was used for mapping DNA markers.
Virus strains and resistance screening
Two different strains of TMV P0 and P1,2,3 were used for virus resistance screening: TMV P0 was provided by K. H. Paek (Korea University, Seoul, Korea), and TMV P1,2,3 was provided by K. H. Ryu (Seoul Women’s University, Seoul, Korea). The virus strains were maintained and multiplied on N. tabaccum var. ‘Samsun’. For virus inoculation, infected tobacco leaves were ground in chilled 50 mM potassium phosphate buffer (pH 7.5). Mechanical inoculation was carried out by applying virus inoculum onto the two oldest leaves of plants at the 5- to 6-leaf stage. Plants were monitored daily and observed for the presence of virus symptoms and hypersensitive response. Resistance to TMV was determined by the presence of hypersensitive response. In order to confirm HR response, virus accumulation was monitored in inoculated plants by the DAS-ELISA method.
BAC clone analysis and sequencing
The pepper BAC library was provided by J. Giovannoni (Cornell University, NY, USA). The BAC library was constructed using nuclear DNA from C. frutescens BG2816, which is known to contain the L 2 allele. The library is publically available at Arizona Genomics Institute (http://www2.genome.arizona.edu). The average insert size of this BAC library was estimated to be 125–130 kb. A total of 220,000 BAC clones were screened using the 3′ sequence of the tomato I2C-1 gene as a probe. Positive BAC clones were subjected to group using a BAC fingerprinting method. BAC fingerprinting was outsourced to Amplicon Express (Pullman, WA: http://www.genomex.com). After obtaining BAC fingerprints, we assembled BAC contigs with FPC program with tolerance setting of 3 and a cutoff of 1e-10. The 89 I2C-containing BAC clones also screened by PCR using a set of three primers: R7-1, R7-2, and LRR (Table S1). These primers were provided by B. Baker (U.C. Berkeley, CA, USA) and were designed to specifically amplify R3a homolog sequences. Amplified PCR fragments from 22 BAC clones using LRR primers were cloned and sequenced [Biotechnology Resource Center (BRC), Cornell University, NY, USA]. Sequences were analyzed and grouped using DNAstar and MegAlign program (DNASTAR, Inc., USA) to identify candidate BAC clones. The nine candidate BAC clones were sent to the Institute for Genomic Research (TIGR, MD, USA) for draft sequencing. After anchoring draft sequenced BAC clones (BAC clone 060I2 and 082F3) near the L locus, the remaining BAC clones were rescreened by PCR with primers (pepBAC060I2-H3) derived from BAC 060I2. BAC end sequences identified by pepBAC060I2-H3 were determined [National Instrumentation Center for Environmental Management (NICEM), Seoul National University, Korea], and primers were redesigned for the next round of screening. This screening for IC2-positive BAC clones was repeated until no further positive clones were obtained. After obtaining a BAC contig, three BAC clones—043M10, 158K24, and 268G7—were selected and sequences were determined by a shotgun sequencing method (NICEM, Seoul National University, Korea).
BAC sequence annotation
The FGENESH program (http://linux1.softberry.com/berry.phtml) was used to predict genes from the contig sequence based on tomato organism information. To search for reported proteins with similar predicted amino acid sequences, the BLASTP program (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) was used. BLASTP was also used to calculate similarity and identity. To identify coiled-coil domains, the MARCOIL program (http://www.isrec.isb-sib.ch/webmarcoil/webmarcoilC1.html) was used with default settings. Sequences having total coiled-coil probability over 90% were determined to code for a coiled-coil domain. Multiple sequence alignment was performed using ClustalW2 program (http://www.ebi.ac.uk/Tools/clustalw2/index.html). Global pairwise analysis was performed using Matcher program (http://bioweb.pasteur.fr/seqanal/interfaces/matcher.html) with default setting. Score of match was set 1, and gap open penalty and gap extend penalty were set 16 and 6 respectively. Neighbor-joining, minimum evolution, maximum parsimony, and UPGMA analysis were conducted using MEGA program version 4 (Tamura et al. 2007). Phylogenetic test was estimated using 1,000 bootstrap replicates and the other options follow default value.
Development of markers linked to the L locus based on BAC sequences
Simple sequence repeat marker
Simple sequence repeat markers were designed from the SSR Discovery program (PGG Bioinformatics, http://hornbill.cspp.latrobe.edu.au/ssrdiscovery.html) based on the draft sequences of the nine selected BAC clones. In order to obtain markers with a higher chance of polymorphism, only primers flanking putative 5× trinucleotide repeats or 7× binucleotide repeats were used. Three to six SSR markers were designed for each selected BAC clone and a total of 40 SSR markers were developed and tested. SSR marker analysis was conducted according to Yi et al. (2006).
Single nucleotide polymorphism marker
Single nucleotide polymorphism markers linked to the L loci were developed from the end sequences of BAC clones. To find SNPs, sequences of PCR products amplified using BAC-derived primers were compared between C. chacoense (L 4) and C. annuum (L 0), resistant and susceptible plants from L-segregating populations respectively. SNPs were converted to cleaved amplified polymorphic sequence (CAPS) or high resolution melting (HRM) markers. The CAPS finder program (www.sgn.cornell.edu) was used to identify restriction enzymes (RE) cutting potential SNPs. SspI (087H3T7 CAPS marker) was used according to protocols provided by the manufacturers. Cut DNA products were electrophoresed on 1.5% agarose gels. For HRM analysis, PCR was performed using a Rotor-Gene™ 6000 (Corbett, Australia). PCR was carried out in a 20 μl volume containing 60 mM KCl, 10 mM Tris–Cl, 2.5 mM MgCl2, 0.25 mM each dNTP, 5 pmol each primer, 1 unit Taq polymerase, 1.25 μM Syto9, and 50 ng gDNA stock. After the PCR, HRM was performed each 0.1°C from 65 to 90°C.
Linkage analysis of molecular markers
Linkage analysis of markers developed in this study and previously was performed using AC99, SP, CP, and MS F2 populations. Linkage analysis was performed using CarthaGene software (de Givry et al. 2005) with a LOD score threshold of 4.0 and a maximum distance of 30 cM.
Results
Identification of BAC clones cross-hybridized with tomato I2C-1 gene and potato R3/R7
To identify pepper I2 homologs, a pepper BAC library containing 221,184 clones was screened with a probe developed from the 3′ sequence of the I2C-1 gene. A total of 89 positive BAC clones were obtained. In the R3/R7 region in potato, 17 copies of RFLP markers GP285 and TG105 are tightly linked to the I2 homolog. Therefore, we attempted to hybridize the 89 BAC clones with the two RFLP markers to identify BAC clones containing sequences related to the R3/R7 region. However, none of the 89 clones were cross-hybridized with the two RFLP markers. This demonstrates that potato and pepper have different genomic structures in this region.
To identify clones simultaneously homologous to potato R3, the 89 I2-positive BAC clones were screened using primer sets developed from potato genomic sequences. The three primer sets—R7-1, R7-2, and LRR—designed from sequence near R3a on chromosome XI in potato, amplified 1.5, 0.8–1.3, and 1.2 kb bands from 37, 52, and 22 BAC clones in pepper respectively (Table S1). Twelve BAC clones containing LRR priming sequences also contained both R7-1 and R7-2 priming sequences, while the other ten clones contained either R7-1 or R7-2 priming sequences (data not shown). In order to pool the BAC clones into groups, the resulting 22 PCR fragments amplified with LRR primers were cloned and sequenced. Sequence similarity analysis revealed that the 22 clones could be grouped into nine different sequence groups. Based on these results, the BAC clones from which both the tomato I2C-1 gene and the potato R3a gene could be amplified were chosen as candidate clones.
Mapping of the candidate BAC clones
The I2 gene family in tomato is distributed at least at five genomic locations across three chromosomes. It is hard to estimate how many copies there might be for the L gene or gene family in pepper. Nevertheless, L was previously mapped 5.2 cM away from RFLP marker TG36 on chromosome 11 (Paran et al. 2004), this region of the pepper genome is syntenic to the R3/R7 region in potato and the I2 region in tomato. We therefore focused our study on the candidate BAC clones mapped in the target region. To locate the candidate BAC clones on the pepper genetic map, SSR markers were developed using the nine draft BAC clone sequences. We successfully anchored four BAC sequences using six SSR markers on our reference mapping population AC99 (Livingstone et al. 1999) (Table S1). One SSR marker, pepBAC082F3-5 (developed from BAC clone 082F3), was cosegregated with TG36 on chromosome 11 (syntenic with I2/R3 loci) (Fig. 1). Another marker representing BAC clone 337L21 was mapped on the same chromosome but near TG105. Other markers mapped outside of the target syntenic region and therefore were discarded for the following analysis (data not shown).

Comparative map showing positions of resistance genes on the lower arms of chromosome 11 of potato, tomato, and pepper. Marker order close to syntenic gene group including L, I2, and R3 was conserved well except TG105 marker
Based on the BAC fingerprinting results, BAC clone 060I2 was closely related to BAC clone 082F3 (data not shown). Therefore, we also mapped BAC clone 060I2. Since no polymorphic SSR markers had been developed for this BAC, six markers were developed from the BAC end sequences of BAC clone 060I2. One of which, pepBAC060I2-H3, polymorphic marker was cosegregated with pepBAC082F3-5 and mapped near TG36 on chromosome 11 (Fig. 2a).

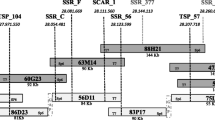
Partial contig of 13 BAC clones. a, b Partial contig was constructed using six primers (under dotted lines). Dark grey rectangles are located based on sequence information and light grey rectangles are located based on PCR information of six primers. About 224 kb DNA sequences were obtained by assembly of four BAC clones (158K24, 060I2, 043M10, and 268G7). c Gene prediction conducted by FGENESH program. Three R3a, I2 family gene analogs (RIGA, arrows) and five transposable elements with long terminal repeats (LTRs, thick bars) were predicted
Construction of a partial contig
The BAC contig assembled with the FPC program was re-evaluated by PCR using primers derived from BAC clone 060I2. However, there were some discrepancies between PCR analysis and the BAC fingerprinting: some BAC clones predicted to have overlapping sequences by BAC fingerprints turned out to be false positive in PCR analysis. This may be due to repetitive nature of R gene homologs. Therefore, we attempted to construct a new contig by PCR using BAC end primers. Six primers were designed from four BAC clones (087H3, 158K24, 207E13, and 290J13), which had strong positive signals by PCR using primer pepBAC060I2-E4, were selected to rescreen 89 BAC clones. We were able to obtain an additional 12 BAC clones. A new contig was constructed by presence/absence of amplicons giving overlap order (Fig. 2b). Three BAC clones (043M10, 158K24, and 268G7) in the contig were selected and fully sequenced.
Development of SNP markers linked to the L alleles
We developed L-linked markers by making use of the BAC contig information (Fig. 2b). It is known that the L 4 gene originated from C. chacoense and had been introgressed into the C. annuum genome during the breeding process. Since the sequence information in the vicinity of the L 2 allele is available, it may be possible to develop markers linked to L 4 by simply comparing syntenic sequences of resistant plants containing L 4 and susceptible plants containing L 0.
To develop SNP markers, a total of 16 primer pairs were designed using BAC sequences from the contig (Table S1). PCR using nine primer pairs (pepBAC060I2-H1, pepBAC060I2-H3, pepBAC060I2-E1, pepBAC060I2-E2, pepBAC060I2-E3, 060I2END-1, K24D2SP6, K24D2T7, 207E13T7) resulted in multiple bands for both C. annuum ECW and C. chacoense 429. The 087H3T7 and 207E13SP6 primers amplified single bands for both genotypes. These single bands were sequenced and polymorphisms were surveyed. Nine and five SNPs were discovered for 087H3T7 (420 bp) and 207E13SP6 (422 bp) sequences respectively. For HRM analysis, primers were redesigned to amplify smaller fragments containing one or two SNPs. When HRM analyses were performed, we were able to distinguish SNPs between ECW and 429 for only 087H3T7 (Fig. 3a, d). The pepBAC060I2-H2 and 290J13SP6 primers amplified very faint single bands. The 087H3SP6, 290J13T7, and 060I2END primers amplified a 700 bp single band for ECW, while no PCR products were obtained for 429. A dominant SCAR marker was developed for 060I2END based on the presence or absence of the 700 bp fragment (Fig. 3c).

Analysis of molecular markers developed in this study. a, b HRM marker analysis using two markers 087H3T7HRM and 158K24HRM. There were three distinct melting curves that distinguish three genotypes. c, d Electrophoresis gels of 060I2END-2 and 087H3T7 markers that were stained by EtBr. ‘R’ means L 4 /L 4 genotype, ‘H’ means L 4 /L 0 genotype, ‘S’ means L 0 /L 0 genotype, and ‘E’ means both L 0 /L 0 and L 4 /L 0 genotype
To test if the markers, which were polymorphic between ECW and 429 could be used for an L 4-segregating population, polymorphism and segregation of 087H3T7HRM and 060I2END markers were investigated using an SP F2 population (Table 1). These markers showed polymorphism and segregated as expected (Fig. 3a, c). However, the melting curves for 087H3T7HRM between resistant plants and 429 were slightly different, indicating sequence differences.
After confirming that the BAC contig sequences could be utilized for L-linked marker development, we directly compared sequences of resistant and susceptible F2 plants and developed markers. The 158K24HRM marker was developed using the left end sequences of the BAC contig from resistant and susceptible plants in the L 4-segregating population (Fig. 3b, Table 1). To test cosegregation of putative L-linked markers with the resistance phenotype, three L segregation populations were evaluated. In the L 3 allele segregation population, there were 189 resistant and 54 susceptible plants, which fitted to an expected 3:1 model (P value = 0.3173). The phenotype segregation ratio was observed in two L 4 allele segregation populations—the resistance/susceptible ratios were 537:109 and 504:341 in SP and MS populations respectively. The segregation ratios of both populations were significantly deviated from the expected model of 3:1 segregation ratio (P values < 0.0001). When we tested the 087H3T7HRM marker in the L 3 population, the genotype ratio of L 3 /L 3:L 3 /L 0:L 0 /L 0 was 57:128:58. Among the individuals, three plants were recombinant. The genotype ratio of L 4 /L 4:L 4 /L 0:L 0 /L 0 was 222:315:107 in SP populations and the genotype ratio of L 4 /L 4:L 4 /L 1:L 1 /L 1 was 159:299:341 in MS populations. Among them, 5 and 11 plants were recombinants in SP and MS populations, respectively.
Linkage analysis and allelic relationship of L-linked markers
In order to have an overview of the recently published L-linked markers from different studies, we have mapped all the reported markers (Kim et al. 2008; Tomita et al. 2008) together with our newly developed SNP marker, 087H3T7 on a single population. To determine the relative positions of the L 4-linked markers, linkage analysis was performed using MS F2 population. L4SC340 and L2kstd (Kim et al. 2008) are dominant SCAR markers, which were reported to locate about 0.9–1.8 cM away from L 4. When the two markers were tested in the MS F2 population together with 087H3T7, 11, 14, and 16 recombinants were detected for 087H3T7, L4SC340, and L2kstd respectively.
In order to determine the allelic relationship of L 3 and L 4 alleles, seven L 3-linked markers (A339, YB2A25, IH1-04, 189D23M, 197AD5R, 253A1R, and 213E3R) reported by Tomita et al. (2008) were also tested in the MS F2 population using HRM analysis. One marker, 189D23M, demonstrated to be completely linked to L 3 in tested 2,016 individuals of an interspecific F2 population and one recombination out of 3,391 individuals of an intraspecific F2 population respectively (Tomita et al. 2008). Two primers sets were reported for the 189D23M marker and both were polymorphic in our mapping population. 189D23M showed six recombinants in 858 individuals of MS F2 population. Occurrence of recombination of 189D23M was significantly higher in our L 4-segregating population than that reported in L 3 indicates that L 3 and L 4 might not be allelic. This was further confirmed with SP F2 population, in which 189D23M showed five recombinants out of 631 individuals.
Taken together, we were able to construct a linkage map around the L locus using four tested markers: L4SC340, L2kstd, 087H3T7, and 189D23M. Our results showed that the nearest markers were 087H3T7 and 189D23M located on opposite sides of the L locus at a distance of 1.2 and 0.8 cM, respectively (Fig. 4, Table 2).

Genetic maps of four molecular markers (087H3T7, L4SC340, 189D23M, and L2kstd) around the L 4 locus. A linkage map of AC99 around the TG36 marker is located in the middle with Kosambi distances (black). A linkage map of MS around the L 4 locus is in the right side (gray). The nearest markers 189D23M and 087H3T7 were located in opposite sides of the L 4 locus
Gene annotation from contig sequence and the genomic relationships of RGAs
Shotgun sequence analysis revealed the insert size of BAC clones as 90,162, 105,856, and 114,328 bp for 043M10, 158K24, and 268G7, respectively, which overlapped with 060I2 draft sequence. Taken together, these three BAC clones makes a contig sequence of 223,989 bp. In order to predict the open reading frames, we used FGENESH program. A total of 42 genes were mined and were refined with the BLASTP program. Finally, 19 putative genes were predicted and were classified into seven groups (Table 3). Six putative genes were CC–NBS–LRR class resistance gene analogs (RGAs). Five out of six RGAs aligned very well with R3a, I2, and homologs of potato and tomato in BLAST search, but one gene containing an NBS domain aligned with very low significance relatively than other five putative genes (data not shown). The five RGAs were designated RIGAs (R3a and I2 family Gene Analogs). Three RIGAs, contained CC, NBS, and LRR domains, were denoted RIGA-1, -2, and -3 (Fig. 2c), but the other two RIGAs, contained only truncated CC or NBS domains, were denoted RIGA-4 and RIGA-5.
Except for truncated region, various motives (hhGRExE, P-loop, Walker B, RNBS-A to D, GLPL, and MHD) were conserved well in the NBS domain of RIGAs showing high homology with I2 and R3a even inter region among conserved domains (van Ooijen et al. 2008). But, multiple nucleotide substitution, deletions, and insertions were existed in the whole amino acid sequences when compared to that of R3a and I2. Short deletion of RIGA-1 and RIGA-2 were existed in the NBS sequence even in the conserved motives. LRR domains were consisted of 27 subunits as I2. Interestingly, distinct amino acid differences starting from the 13th LRR domain: insertions at 13th LRR, deletions at 25th LRR regions, and many amino acid substitutions (Fig S1).
Global pairwise alignment analysis among R3a, I2, and five RIGAs was performed using Matcher program. DNA sequences of the five RIGAs shared high identities with each other (74.9–82.1% identity), and identity between R3a and I2 was 83.4% with the least identities among RIGAs and R3a or I2 were 68.4–77.2%. Homologies among RIGAs, R3a, and I2 were higher than other resistance gene (data not shown). Identities among RIGAs were higher than those between RIGAs and each of R3a or I2, but lower than those among R3a and I2.
In order to evaluate genetic relationship between RIGAs, R3a, and I2, phylogenetic analysis was conducted based on NBS region, from the P-loop motif to the GLPL motif, with other cloned resistance genes that were classified to CC–NBS–LRR in Solanaceae. Phylogenetic analysis using neighbor-joining algorithm revealed RIGAs, R3a, and I2 fall under the same clade, which shows the orthologous relationship among RIGAs, R3a, and I2 (Fig. 5). Other algorithms including minimum evolution, maximum parsimony, and UPGMA also confirmed that RIGAs, R3a, and I2 are classified as one clade although the positions and patterns in the tip of branches in the phylogenetic clade were diverged (data not shown).

Phylogenetic analysis of RIGAs, R3a, and I2 with other resistance genes in Solanaceae by neighbor-joining algorithm. Bs4 is the resistance gene belonging to TIR–NBS–LRR class and was used as an out-group
Discussion
Based on the hypothesis that the L gene in pepper conferring tobamovirus resistance is an ortholog of the R3 gene in potato and the I2 gene in tomato conferring resistance against Phytophthora infestans and Fusarium wilt, respectively, molecular markers closely linked to the L locus were developed in this study by identification of R3/I2 orthologs in pepper.
L 4 confers resistance to the most aggressive PMMoV pathotype P1,2,3. Development of reliable markers is important for introgression of L 4 to elite germplasm through marker-assisted-selection (MAS)/marker-assisted-backcrossing (MAB). Therefore, we have defined recently published markers linked to L locus together with our newly developed SNP markers (Tomita et al. 2008; Kim et al. 2008) to construct a localized map around the L locus. Interestingly, we found that molecular markers developed for L 3 (C. chinense) (Tomita et al. 2008) are also applicable to L 4, although L 4 and L 3 may not be allelic. We have identified new markers specifically linked to L 4, 189D23M (Tomita et al. 2008) and our SNP marker, 087H3T7HRM, flanks the L 4 locus. Use of two markers simultaneously in a molecular breeding program is recommended; successful rate of introgression of L 4 is expected to improve when compared to the previous system using a single SCAR marker (Kim et al. 2008). In addition, our SNP markers have an additional advantage, i.e., capable of differentiating L 3 and L 4; breeders could introgress the two resistance genes to different lines and pyramid the two genes by making F1 hybrids.
Earlier comparative genetic studies revealed the colinearity between the R3 late blight resistance locus in potato and the I2 locus in tomato. The perfect microsyntenic relationship between tomato and potato on chromosome 11 allowed the isolation of the potato R3a gene by a comparative genomics approach using the resistance gene analog of I2. A macrosyntenic comparison between pepper and potato/tomato illustrated a potential orthology of L and R3/I2. However, since the syntenic region on pepper chromosome 11 has been demonstrated to be one of the 22 genome rearrangements distinguishing tomato and pepper (Livingstone et al. 1999), it was less likely that the L region could be characterized by examining microsynteny and comparative gene content with the corresponding regions. Because of the complexity, instead of using the tomato and potato physical maps for fine mapping and physical mapping of L directly, we prioritized and employed PCR approaches using portions of the R3 gene to identify R3/I2 orthologs at the target syntenic region near TG36 in pepper (where L was previously mapped near to). With this approach, we successfully found BAC clones closely linked to L, other clones identified but mapped elsewhere in the genome are considered as paralogs.
Mazourek et al. (2009) demonstrated the association between R-genes with chromosome breakpoints of genome rearrangement, characteristics of the syntenic region of chromosomes 11 in pepper, tomato, and pepper further supports this observation. Tomato and potato are collinear throughout the lower arm of the chromosome 11. I2 in tomato is located between TG36 and TG105, and R3a is located near TG105 with TG105 nearer to the telomere (Fig. 1). Pepper is collinear with both tomato and potato in this region except TG105 is centromeric in pepper with TG36 facing the telomere (Paran et al. 2004) (Fig. 1). L together with disease resistance QTL against cucumber mosaic virus and Phytophthora spp. mapped in cluster near TG36 toward the telomere of chromosome 11 in pepper (Grube et al. 2000; Livingstone et al. 1999), apparently associates with (or located at or near) the breakpoint of chromosomal rearrangement.
We have obtained 224 kb sequences from four BAC clones, and only five R3/I2 RGAs were predicted. By comparison, at the syntenic region in potato, five I2 RGAs were identified within one BAC clone where the R3a gene lies (Huang et al. 2005). Three resistance gene analogs (RGAs) of R3/I2 with CC, NBS, and LRR domains were predicted and located at the syntenic region in pepper. Alignment of the three RGA proteins showed high similarity, indicating that the RGAs are paralogs. The roles of these RGAs are not clear. Base on our phenotypic and genetic analysis, recombination occurs between L resistance and our markers, indicating that the RGAs found in this study might not be candidate genes for L. However, we cannot totally rule out a possible connection to resistance against tobamoviruses. In fact, I2C-1 has been shown to contribute partial resistance against Fusarium oxysporum in tomato (Ori et al. 1997). Alternatively, these three RGAs might be functional candidates for other disease resistance genes or quantitative trait loci (QTL) associated with disease resistance. This is not surprising as it has been known that L is located in a R-gene cluster where disease resistance QTLs for potyvirus, cucumber mosaic virus (CMV), tomato spotted wilt virus (TSWV), and Phytophthora capsici have been reported (Lefebvre 2004).
Genetic inheritance studies have demonstrated the allelic relationship of L 1, L 2, L 3, and L 4. However, our study did not confirm the expected classical allelic relationship at the molecular level. We found consistent contradiction between our study and a recent study on fine mapping of L 3. Tomita et al. (2008) also found the presence of I2 homologs linked to L 3. However, surprisingly, a genetic distance of 0.83 cM from L was obtained when we tested Tomita’s L 3-linked markers in our L 4-segregating populations, an almost 1.4-fold difference from what Tomita et al. (2008) found. However, it might be possible that the difference in genetic distance between L 3 and L 4 was due to occurrences of unequal cross-over in interspecific crosses between C. annuum/C. chacoense and C. annuum/C. chinense that might have obscured the allelic relationship. In addition, use of different segregating populations and sampling error might have played a role in the differences. Alternatively, L 1, L 2, L 3, and L 4 might be different genes closely linked within the region instead of different alleles at the same locus. In fact, two closely linked genes with distinct specificities have been found for R3 resistance to late blight in potato including the cloned R3a (Huang et al. 2004, 2005). Furthermore, a BAC library was developed from C. frutescens in this study; however, gene content and/or genome structure might be different between C. chinense (L 3) and C. chacoense (L 4), as was demonstrated for the R1 contig conferring resistance to Phytophthora infestans on chromosome V in potato, where gene content and genome structure varied between different haplotypes (Ballvora et al. 2007).
Different segregation ratios were obtained for the two L 4-segregating populations. In both populations, distorted 3:1 segregation ratios were obtained, indicating preferential segregation has occurred in the region and resulting in segregation distortion. It is known that segregation distortion commonly occurs in interspecific crosses. For instance, segregation distortion has been found in other regions of chromosome 11 for crosses between C. annuum and C. chinense (L 3). However, interspecific crosses between C. annuum and C. chacoense (L 4) have not been studied. By comparison, such segregation distortion in this region is not seen in the C. annuum/C. chinense (L 3)-segregating population, as the marker segregated according to the Mendelian pattern.
References
Alonso E, Garcia-Luque I, de la Cruz A, Wicke B, Avila-Rincon MJ, Serra MT, Castresana C, Diaz-Ruiz JR (1991) Nucleotide sequence of the genomic RNA of pepper mild mottle virus, a resistance-breaking tobamovirus in pepper. J Gen Virol 72:2875–2884. doi:10.1099/0022-1317-72-12-2875
Ballvora A, Ercolano MR, Weiss J, Meksem K, Bormann CA, Oberhagemann P, Salamini F, Gebhardt C (2002) The R1 gene for potato resistance to late blight (Phytophthora infestans) belongs to the leucine zipper/NBS/LRR class of plant resistance genes. Plant J 30:361–371. doi:10.1046/j.1365-313X.2001.01292.x
Ballvora A, Jöcker A, Viehöver P, Ishihara H, Paal J, Meksem K, Bruggmann R, Schoof H, Weisshaar B, Gebhardt C (2007) Comparative sequence analysis of Solanum and Arabidopsis in a hot spot for pathogen resistance on potato chromosome V reveals a patchwork of conserved and rapidly evolving genome segments. BMC genomics 8:112–124
Berzal-Herranz A, de la Cruz A, Tenllado F, Diaz-Ruiz JR, Lopez L, Sanz AI, Vaquero C, Serra MT, Garcia-Luque I (1995) The Capsicum L3 gene-mediated resistance against the tobamoviruses is elicited by the coat protein. Virology 209:498–505. doi:10.1006/viro.1995.1282
Boukema IW (1980) Allelism of genes controlling resistance to TMV in Capsicum L. Euphytica 29:433–439. doi:10.1007/BF00025143
Csillery G, Tobias I, Rusko J (1983) A new pepper strain of tomato mosaic virus. Acta Phytopathol Acad Sci Hung 18:195–200
Dangl JL, Jones JD (2001) Plant pathogens and integrated defence responses to infection. Nature 411:826–833. doi:10.1038/35081161
de Givry S, Bouchez M, Chabrier P, Milan D, Schiex T (2005) Cartha gene: multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics 21:1703–1704
de la Cruz A, Lopez L, Tenllado F, Diaz-Ruiz JR, Sanz AI, Vaquero C, Serra MT, Garcia-Luque I (1997) The coat protein is required for the elicitation of the Capsicum L2 gene-mediated resistance against the tobamoviruses. Mol Plant Microbe In 10:107–113. doi:10.1094/MPMI.1997.10.1.107
Grube RC, Radwanski ER, Jahn M (2000) Comparative genetics of disease resistance within the solanaceae. Genetics 155:873–887
Huang S, Vleeshouwers VG, Werij JS, Hutten RC, van Eck HJ, Visser RG, Jacobsen E (2004) The R3 resistance to Phytophthora infestans in potato is conferred by two closely linked R genes with distinct specificities. Mol Plant Microbe In 17:428–435. doi:10.1094/MPMI.2004.17.4.428
Huang S, van der Vossen EA, Kuang H, Vleeshouwers VG, Zhang N, Borm TJ, van Eck HJ, Baker B, Jacobsen E, Visser RG (2005) Comparative genomics enabled the isolation of the R3a late blight resistance gene in potato. Plant J 42:251–261. doi:10.1111/j.1365-313X.2005.02365.x
Kim HJ, Han JH, Yoo JH, Cho HJ, Kim BD (2008) Development of a sequence characteristic amplified region marker linked to the L 4 locus conferring broad spectrum resistance to tobamoviruses in pepper plants. Mol Cells 25:205–210
Kuang H, Wei F, Marano MR, Wirtz U, Wang X, Liu J, Shum WP, Zaborsky J, Tallon LJ, Rensink W, Lobst S, Zhang P, Tornqvist CE, Tek A, Bamberg J, Helgeson J, Fry W, You F, Luo MC, Jiang J, Buell CR, Baker B (2005) The R1 resistance gene cluster contains three groups of independently evolving, type I R1 homologues and shows substantial structural variation among haplotypes of Solanum demissum. Plant J 44:37–51. doi:10.1111/j.1365-313X.2005.02506.x
Lefebvre V (2004) Molecular markers for genetics and breeding: development and use in pepper (Capsicum spp.). In: Lo¨rz H, Wenze G (eds) Biotechnology in agriculture and forestry molecular marker systems. Springer, Heidelberg, pp 189–214
Lefebvre V, Palloix A, Caranta C, Pochard E (1995) Construction of an intra-specific integrated linkage map of pepper using molecular markers and doubled-haploid progenies. Genome 38:112–121
Livingstone KD, Lackney VK, Blauth JR, van Wijk R, Jahn MK (1999) Genome mapping in capsicum and the evolution of genome structure in the solanaceae. Genetics 152:1183–1202
Matsunaga H, Saito T, Hirai M, Nunome T, Yoshida T (2003) DNA markers linked to Pepper mild mottle virus (PMMoV) resistant locus (L 4) in Capsicum. J Jpn Soc Hortic Sci 72:218–220
Mazourek M, Cirulli ET, Collier SM, Landry LG, Kang BC, Quirin EA, Bradeen JM, Moffett P, Jahn M (2009) The fractionated orthology of Bs2 and Rx/Gpa2 supports shared synteny of disease resistance in the Solanaceae. Genetics (in press) doi:10.1534/genetics.109.101022
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332. doi:10.1046/j.1365-313X.1999.t01-1-00606.x
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834. doi:10.1105/tpc.009308
Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8:1113–1130
Ori N, Eshed Y, Paran I, Presting G, Aviv D, Tanksley S, Zamir D, Fluhr R (1997) The I2C family from the wilt disease resistance locus I2 belongs to the nucleotide binding, leucine-rich repeat superfamily of plant resistance genes. Plant Cell 9:521–532
Paran I, van der Voort JR, Lefebvre V, Jahn M, Landry L, van Schriek M, Tanyolac B, Caranta C, Chaim AB, Livingstone K, Palloix A, Peleman J (2004) An integrated genetic linkage map of pepper (Capsicum spp.). Mol Breeding 13:251–261
Pernezny K, Roberts PD, Murphy J, Goldberg NP (2003) Compendium of pepper diseases. APS Press, St. Paul
Song J, Bradeen JM, Naess SK, Raasch JA, Wielgus SM, Haberlach GT, Liu J, Kuang H, Austin-Phillips S, Buell CR, Helgeson JP, Jiang J (2003) Gene RB cloned from Solanum bulbocastanum confers broad spectrum resistance to potato late blight. Proc Natl Acad Sci USA 100:9128–9933. doi:10.1073/pnas.1533501100
Sugita T, Yamaguchi K, Sugimura Y, Nagata R, Yuji K, Kinishita T, Todoroki A (2004) Development of SCAR markers linked to L3 gene in Capsicum. Breed Sci 54:111–115. doi:10.1270/jsbbs.54.111
Takeuchi S, Hamada H, Toyoda K, Suzuki K, Kiba A, Hikichi Y, Okuno T (2005) Discrimination between tobamoviruses and their pathotypes for L-gene-mediated resistance in green pepper (Capsicum annuum L.) by reverse transcription-polymerase chain reaction. J Gen Plant Pathol 71:60–67. doi:10.1007/s10327-004-0161-4
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi:10.1093/molbev/msm092
Tomita R, Murai J, Miura Y, Ishihara H, Liu S, Kubotera Y, Honda A, Hatta R, Kuroda T, Hamada H, Sakamoto M, Munemura I, Nunomura O, Ishikawa K, Genda Y, Kawasaki S, Suzuki K, Meksem K, Kobauashi K (2008) Fine mapping and DNA fiber FISH analysis locates the tobamovirus resistance gene L 3 of Capsicum chinense in a 400-kb region of R-like genes cluster embedded in highly repetitive sequences. Theor Appl Genet 117:1107–1118. doi:10.1007/s00122-008-0848-6
Tsuda S, Kirita M, Watanabe Y (1998) Characterization of a pepper mild mottle tobamovirus strain capable of overcoming the L 3 gene-mediated resistance, distinct from the resistance-breaking Italian isolate. Mol Plant Microbe In 11:327–331. doi:10.1094/MPMI.1998.11.4.327
van der Vossen E, Sikkema A, Hekkert BL, Gros J, Stevens P, Muskens M, Wouters D, Pereira A, Stiekema W, Allefs S (2003) An ancient R gene from the wild potato species Solanum bulbocastanum confers broad-spectrum resistance to Phytophthora infestans in cultivated potato and tomato. Plant J 36:867–882. doi:10.1046/j.1365-313X.2003.01934.x
van der Vossen EAG, Gros J, Sikkema A, Muskens M, Wouters D, Wolters P, Pereira A, Allefs S (2005) The Rpi-blb2 gene from Solanum bulbocastanum is an Mi-1 gene homolog conferring broad-spectrum late blight resistance in potato. Plant J 44:208–222. doi:10.1111/j.1365-313X.2005.02527.x
van Ooijen G, Mayr G, Kasiem MMA, Albrecht M, Cornelissen BJC, Takken FLW (2008) Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J Exp Bot 59:1383–1397. doi:10.1093/jxb/ern045
Velasco L, Janssen D, Ruiz-Garcia L, Segundo E, Cuadrado IM (2002) The complete nucleotide sequence and development of a differential detection assay for a pepper mild mottle virus (PMMoV) isolate that overcomes L 3 resistance in pepper. J Virol Methods 106:135–140. doi:10.1016/S0166-0934(02)00144-1
Wetter C, Conti M, Altschuh D, Tabillion R, Van Regenmortel M (1984) Pepper mild mottle virus, a tobamovirus infecting pepper cultivars in Sicily. Phytopathology 74:405–410. doi:10.1094/Phyto-74-405
Wisser RJ, Sun Q, Hulbert SH, Kresovich S, Nelson RJ (2005) Identification and characterization of regions of the rice genome associated with broad-spectrum, quantitative disease resistance. Genetics 169:2277–2293. doi:10.1534/genetics.104.036327
Wisser RJ, Balint-Kurti P, Nelson RJ (2006) The genetic architecture of disease resistance in maize: a synthesis of published studies. Phytopathology 96:120–129. doi:10.1094/PHYTO-96-0120
Yi G, Lee JM, Lee S, Choi D, Kim BD (2006) Exploitation of pepper EST-SSRs and an SSR-based linkage map. Theor Appl Genet 114:113–130. doi:10.1007/s00122-006-0415-y
Acknowledgments
This work was supported by a grant (Code20070401034028) from the BioGreen21 program, Rural Development Administration, Republic of Korea and by a grant (GC1132-1) from the Crop Functional Genomics Center of the 21st Century Frontier Research Program, funded by the Ministry of Education, Science and Technology (MEST) of The Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Hee-Bum Yang and Wing Yee Liu contributed equally to this work.
Sequence data from this article have been deposited with the EMBL/GenBank data libraries under accession numbers: DQ913808, DQ913809, DQ913810, DQ913811, DQ913812, DQ913813, DQ913814, DQ913815, DQ913816, DQ913817, FJ597539, FJ597540, and FJ597541.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yang, HB., Liu, W.Y., Kang, WH. et al. Development of SNP markers linked to the L locus in Capsicum spp. by a comparative genetic analysis. Mol Breeding 24, 433–446 (2009). https://doi.org/10.1007/s11032-009-9304-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-009-9304-9