
Abstract
Cerebral ischemia reperfusion (IR) is associated with neuronal death, which leads to disability and cognitive decline. The pathomechanism occurs because ischemia is exacerbated during the reperfusion period. Neuronal damage susceptibility depends on the affected brain areas and the duration of ischemia. Prevention and supplementation to neurons may help them endure during IR and further benefit them in rehabilitation. We investigated the protective effect of p-coumaric acid (PC) on cerebral IR injuries in mice. We randomly divided 30 male ICR mice into 3 groups of Sham (received vehicle and not induced IR), Control-IR (received vehicle and induced IR) and PC-IR (received 100 mg/kg PC and induced IR). We orally administered vehicle or 100 mg/kg of p-coumaric acid for 2 weeks before inducing the cerebral IR injuries by using 30 min of a bilateral common carotid artery occlusion followed by a 45-min reperfusion. We induced the IR condition in the Control-IR and PC-IR groups but not the Sham group, and only the PC-IR group received p-coumaric acid. After IR induction, we sacrificed all the mice and collected their brain tissues to evaluate their oxidative statuses, whole brain infarctions and vulnerable neuronal deaths. We studied the whole-brain infarction volume by 2, 3, 5-triethyltetrazoliumchloride staining of sections. We performed a histological investigation of the vulnerable neuronal population in the dorsal hippocampus by staining brain sections with 0.1% cresyl violet. The results indicated that IR caused significant increases in calcium and malondialdehyde (MDA) levels, whole brain infarction volume and hippocampal neuronal death. Pretreatment with p-coumaric acid significantly reduced MDA levels, whole-brain infarction volume and hippocampal neuronal death together and increased catalase and superoxide dismutase activities. We conclude here that pretreating animals with p-coumaric acid can prevent IR-induced brain oxidative stress, infarction size and neuronal vulnerability to death in cerebral IR injuries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral ischemia reperfusion (IR) injury is a serious clinical problem that results from many causes such as cardiac arrest, peripheral vascular insufficiency and stroke (Kalogeris et al. 2012). The pathomechanism involves many pathways, including free radical formation. In fact, damaging and protective mechanisms activate the cellular effects of both ischemia and reperfusion (Leker and Shohami 2002; White et al. 2000). Endogenous damaging mechanisms are triggered during ischemia and exacerbated during the reperfusion period. During ischemia, a variety of cellular metabolisms such as decreases of cellular oxidative phosphorylation, resulting in energy failure, as well as alterations of energy-dependent membranes for ionic transport, leading to ionic imbalance and further inducing water influx and causing edema are activated. The alterations of membranes potentially result in excitotoxicity and calcium overload which further activate intracellular calcium-dependent cascades, induce pro-inflammatory cytokines, inhibit anti-inflammatory cytokines, and increase hypoxanthine and free radical formation. These activities lead to an increase of neuronal susceptibility during reperfusion. Following a reperfusion period, excessive oxygen reacts with hypoxanthine, which was catalyzed by xanthine oxidase, and leads to the excessive formation of toxic hydroxyl radicals and nitric oxide-derived peroxynitrite. These potent oxidizing agents cause DNA damage, as well as protein oxidation and directly damage cellular membranes by lipid peroxidation (Hou et al. 2002; Jivad and Rabiei 2015; Shuaib and Breker-Klassen 1997; White et al. 2000).
Ischemia reperfusion not only activated endogenous damaging mechanism but also activated endogenous protective mechanism for reestablish homeostasis and mitigate tissue damage by increases arterial pressure and activates vasodilation for promoting the delivery of blood flow to ischemic area, induces antioxidant enzymes and anti-apoptotic proteins production and releases anti-inflammatory cytokines from surviving neurons to ischemic area (Ladecola and Anrather 2011). These mechanism have been helped neuronal survival during ischemia, however, they are exhausted during the severe stress of ischemia (Leker and Shohami 2002).
The severity and duration of ischemia determine the balance between the endogenous damaging and protective mechanisms. The capability of an endogenous protective mechanism depends upon functional cells during IR. Ischemia limits cell functions by reducing oxygen, directly affecting energy and helping to overwhelm the damaging mechanism. Therefore, the prolonged endurance of cells may enhance the therapeutic window for cells to respond to treatment or increase their strength to cope with the reperfusion period.
As there are multiple pathomechanism of IR injuries, it difficult to treat them by using one substance to counteract the multiple damaging pathway. However, many substances can counteract more than one pathway, but their efficacy depends upon a variety of factors. Preventive therapy is gaining attention nowadays. Medicinal plants are major candidates for promoting cell strength through diet supplementation. There are many active ingredients in medicinal plants, and most of them belong to groups of phenolic compounds that have benefits as antioxidant agents and previously reported that food with phenolic compounds have increased due to their role of antioxidants and scavengers of free radicals and their implication in the prevention of many pathological diseases (Pandey and Rizvi 2009). Tran-4-hydroxycinnamic acid or p-coumaric acid (PC) are phenolic compound that is found ubiquitously in plants and mushrooms. It is synthesized from tyrosine by tyrosine ammonia-lyase through the shikimic acid pathway. The tmax and t1/2 values of PC vary according to their free or conjugated forms. They range from 3.72 to 10 min for tmax and 15.9 min to 1.3 h for t1/2 and exhibit low toxicity by LD50 at about 2850 mg/kg in mice (Pei et al. 2015). PC that have been depicted as having potent antioxidant properties include the scavenging free radicals, up-regulate of endogenous antioxidant enzymes and furthermore studies of PC found that PC could decreases apoptosis proteins and increases nuclear respiratory factors 1 (NRF-1) that could be improve mitochondrial function in stress condition (Guven et al. 2015; Mehta et al. 2012). Antidiabetic, antihyperlipidemic, anticancer, antimicrobial, anti-inflammation, anti-ulcer, anxiolytic, antipyretic, analgesic, anti-arthritic, antioxidant and neuroprotective effects as well as effects regarding the prevention of platelet aggregation have been reported (Abdel-Wahab et al. 2003; Amalan et al. 2016; Guven et al. 2015). Using PC acid to prevent multiple pathomechanisms of IR injury is interesting. Therefore, the present aim is to investigate the protective effect of PC on cerebral IR injuries.
Material and methods
Chemicals and reagents
P-Coumaric acid and other biochemical analysis reagents use in the present study were purchased from Chemical express Co., Ltd. Merck Millipore (Samutprakarn, Thailand).
Animals
We obtained thirty 12-week-old male ICR mice from the National Laboratory Animal Center, Mahidol University (Nakornprathom, Thailand). We fed all animals with a standard diet (mouse diet food No.082G) and RO water. We maintained the room temperature at 23 ± 2 °C with a 12 h-light/ 12 h-dark cycle. All of the experiments were performed in full compliance with the guidelines of the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals approved by the National Institutes of Health (NIH Publication No. 85–23, revised 1996) The Animal Ethic Committee, Kasetsart University, approved the experimental protocol (ACKU#02756).
Pretreatment and IR induction
We randomly divided the mice into 3 groups: Sham, Control-IR and PC-IR. We gave the vehicle (10% Tween 80) to the Sham and Control-IR groups. We gave 100 mg/kg of p-coumaric acid (dissolved in 10% Tween 80) to the PC-IR group. We gave both the vehicle and p-coumaric acid for 2 weeks before IR induction. On the operation day, all mice fasted for 2 h before surgery. We anesthetized each mouse through an intraperitoneal injection of 45 mg/kg of sodium pentobarbital. We made a midline incision on the ventral side of the neck and transiently occluded both common carotid arteries for 30 min. This was followed by 45 min of reperfusion. We induced the IR in the Control-IR and PC-IR groups but not the Sham group. After the completion of the IR, we decapitated all of the mice and quickly removed their brains for biochemical analyses of their oxidative statuses and whole brain infarction volumes as well as histological studies.
Measurement of oxidative status
We washed the brains in a cool 0.9% normal saline solution and homogenized them in a potassium-phosphate buffer (0.05 M, pH 7.4). We kept the homogenates to estimate the lipid peroxidation (malondialdehyde; MDA) and centrifuged them at 10,000×g at 4 °C for 10 min. We collected the supernatants and used them for the SOD, CAT and calcium estimations.
Lipid peroxidation
We mixed 0.2 ml of brain homogenate, 0.2 ml of 4% sodium dodecyl sulfate, 1.5 ml of 20% acetic acid and 1.5 ml of 0.5% thiobarbituric acid and heated the mixture at 95 °C for 1 h. After centrifugation at 3500 rpm for 10 min, we read the supernatant at 532 nm. We calculated the concentration of MDA from the standard curve of the MDA concentration: 0, 7.80, 15.62, 62.50, 93.75, 156.25 and 312.50 μM (y = 0.0057× + 0.0547; r2 = 0.9907). We expressed the MDA content as mmol/mg of protein.
Calcium
The stock color reagent was 40 mg murexide (ammonium purpurate), which we dissolved in 5 ml dH2O and diluted immediately to 500 ml with propylene glycol. The working color reagent was a 50-ml stock color reagent, to which we added 1.5 ml of 3.75 N NaOH. The stock standard calcium solution was 2.497 g of pure, dry CaCO3 in a 5-ml HCl and diluted to 1 L with dH2O (1 mg/ml). We diluted the working standard calcium solution from the stock calcium solution by using 1 in 10 parts with dH2O (0.1 mg/ml).
We prepared the blank (2.55 ml dH2O + 1.5 ml working color reagent), standard (2.5 ml dH2O + 0.05 ml working standard calcium solution +1.5 ml working color reagent) and test (2.5 ml dH2O + 0.05 ml supernatant +1.5 ml working color reagent) and incubated them for 5 min at room temperature. We then read them within 10 min at 490 nm. We expressed the calcium concentration as mEq/L by using the formula (test O.D./standard O.D.) ×5 = mEq/L (Spare 1964).
Catalase
Supernatant 50 μl had volumes of up to 3 ml with PBS (50 mM, pH 7.4) containing H2O2 (20 mM), and we read the supernatant at 240 nm for 3 min (30-s interval). We expressed the catalase activity as U/mg of protein by using the extinction coefficient of H2O2 43.6 M−1 cm−1 (Hadwan and Abed 2016).
Superoxide dismutase
Supernatant 0.1 ml, 0.1 ml of EDTA (0.0001 M), 0.5 ml of carbonate buffer (pH 9.7) and 1 ml of epinephrine (0.003 M). We read the mixture at 480 nm for 3 min (30-s interval). We expressed the enzyme activities of SOD as U/mg of protein by using the standard curve of the SOD concentration: 0, 0.035, 0.180, 0.719 and 1.80 μg/ml (y = 0.0015× + 0.0001; r2 = 0.938). The enzyme activity was 6150 U/mg, Merck, Germany).
Brain-infarction volume
We cut the brains into serial coronal sections (2-mm thick) using mouse templates and stained them with 2% of 2, 3, 5-triphenyltetrazolium chloride (TTC) at 37 °C for 10 min. We then kept them in a 10% neutral buffer formalin for 24 h. We captured the brain images and analyzed them for the infarct volume by NIH ImageJ. We represented the data as the percentage of infarction (% infarction).
Histological study
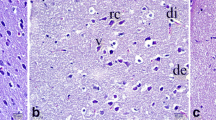
We further processed the brain sections from the TTC staining for the histological study of the dorsal hippocampi. We embedded the brains in paraffin and sectioned them so that they had 5 μm of thickness. We picked 5 brain slides from each mouse, and the slides had 125-μm intervals starting from bregma −1.94 (Paxinos and Franklin 2008), which covered the area of interest (Fig. 1b). We stained these brain slides with 0.1% cresyl violet. In brief, we deparaffinized and rehydrated the slides via serial xylene and ethanol (EtOH; 100, 95, 80 and 70%, respectively), soaked them in distilled water for 5 min and stained them with 0.1% cresyl violet for 30 s. We then dehydrated the brain slides via serial EtOH (70, 80, 95 and 100%, respectively) and xylene before sealing them with a glass cover.

The experimental protocol. a. A histological study of the dorsal hippocampus started from bregma −1.94 b. Areas of interest such as CA1, CA3 and DG. Each of these areas was captured for three images and analyzed for viable and dead cells (arrows)
In each hemisphere, we captured 3 photomicrographs of the dorsal hippocampus subregion’s cornus ammonis (CA) 1, 3 and dentate gyrus (DG), and we counted for viable and dead cells (Fig. 1b). A viable cell was characterized by a round light-purple cell with a visible nucleus and nucleolus. A dead cell was characterized by dark purple shrinkage with a vacuole around cell (Thong-asa and Tilokskulchai 2014). We represented the data as viable or dead cells and included the percentage of dead cells in each subregion and in the total area of the dorsal hippocampus.
Statistical analysis
We analyzed all data through a one-way analysis of variance followed by Fisher’s post hoc test, and we represented as mean ± standard error of mean (S.E.M.). We indicated the statistical significance by p-value <0.05.
Results
Oxidative status
We induced a cerebral IR injury through a 30-min bilateral common carotid artery occlusion followed by 45 min of reperfusion, which significantly increased calcium as well as MDA and significantly reduced catalase levels (p = 0.0029, 0.0092 and 0.0001, respectively; compared Control-IR to Sham; Fig. 2a–c). Pretreatment with 100 mg/kg of p-coumaric acid for 2 weeks reduced calcium and MDA levels, but only MDA was significantly reduced (p = 0.0063; compared Control-IR to PC-IR; Fig. 2b). Antioxidant indices such as CAT and SOD were significantly increased in PC-IR (p = 0.0017 and 0.0246, respectively; compared to Control-IR; Fig. 2c, d). The results of the oxidative statuses suggested that cerebral IR induced by 30-min bilateral common carotid artery occlusion followed by 45 min of reperfusion significantly induced brain oxidation and that pretreatment with 100 mg/kg of p-coumaric acid improved brain oxidation by activating intrinsic antioxidants such as CAT and SOD.

A histogram showing brain tissue oxidative status. Calcium (a), MDA (b), CAT (c) and SOD (d). *indicated significant difference compared to Sham. #indicated significant difference compared to Control-IR
Infarction volume
We show the whole-brain infarction volume with TTC staining in Fig. 3. The percentage of infarction in the Control-IR group was significantly higher than that of the Sham group (p = 0.0015), and pretreatment with 100 mg/kg of p-coumaric acid significantly reduced the infarction (p = 0.0097; compared PC-IR to Control-IR; Fig. 3).

A photomicrograph of brain tissue stained with 2% 2, 3, 5-triethyltetrazoliumchloride (TTC). The scale bar indicates a distance of 1 cm. The histogram shows the whole brain percentage of infarction (% infarction). *indicated significant difference compared to Sham. #indicated significant difference compared to Control-IR (below)
Histological changes
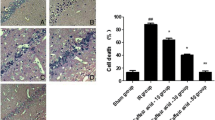
We show the viable and dead cells in each area of interest—such as CA1, CA3 and DG—and in the total area of the dorsal hippocampus (CA1 + CA3 + DG) in Table 1. Viable cells in CA1, CA3, DG and the total area of the dorsal hippocampus were significantly reduced in Control-IR group (p = 0.0021, 0.0001 and 0.0002, respectively). Pretreatment with p-coumaric acid significantly prevented viable-cell reduction in the PC-IR group (p = 0.0129, 0.0323 and 0.0044, respectively). Dead cells were significantly increased in the CA1 area (p = 0.0408) but not in the CA3, DG and total areas of the dorsal hippocampus in the Control-IR group. On the contrary, pretreatment with p-coumaric acid significantly reduced the dead cells in the CA1 area (p = 0.0129). In the DG area, dead cells were significantly reduced by pretreatment with p-coumaric acid compared to the Sham group (p = 0.0368). The dead cells in the total area of the dorsal hippocampus were significantly reduced in the PC-IR group (p = 0.0428).
In our interpretation of the percentage of dead cells, we found that IR significantly increased the percentage of dead cells in CA1 and CA3 (p = 0.0188 and 0.0205, respectively, Fig. 4) but not in DG. Pretreatment with p-coumaric acid ameliorated the increase of the percentage of dead cells in CA1 and DG (p = 0.0082 and 0.0486, respectively; Fig. 4) but not in CA3. Our total-area interpretation revealed significantly increased percentages of dead cells in the Control-IR group (p = 0.0244), and pretreatment with p-coumaric acid significantly reduced the percentage of dead cells in PC-IR (p = 0.0044).

A photomicrograph of the dorsal hippocampal (HP) subregion of CA1, CA3 and DG with 0.1% cresyl violet staining. Images were captured at 200× of magnification and the scale bar indicates a distance of 50 μm. Viable cells were indicated by light purple staining of cresyl violet as well as visibility of nucleus and nucleolus. Dead cells were indicated by dark purple staining as well as the disappearance of nucleus and nucleolus and the appearance of vacuoles around cells. The histogram shows the percentage of dead cells in the dorsal hippocampus subregion of CA1, CA3, DG and total HP (below). *indicated significant difference compared to Sham. #indicated significant difference compared to Control-IR
Discussion
It is well known that IR injury causes neurological deficits and disabilities. The deteriorative effects of IR depend on the severity and duration of ischemia, which further reflect the neuronal endurance during reperfusion. In most cases, if ischemia persists for a long period of time, it will exacerbate the damage during reperfusion and cause more disabilities, which will lead to difficulties during rehabilitation. There are many ways—such as ischemic preconditioning, brain cooling (hypothermia), antioxidant therapy, calcium antagonist, anti-cytokines, anti-leukocyte adhesion molecules and so on—to prevent an exacerbation of an IR injury (Collard and Gelman 2001; Pan et al. 2007). In the present study, we used an antioxidant to prevent a cerebral IR injury. We used p-coumaric acid, the phenolic compound that has a low level of toxicity with rapid tmax and t1/2 (Pei et al. 2015). We used a dose of 100 mg/kg, which is suggested to have an active biological effect on embolic cerebral ischemia (Guven et al. 2015). The IR-injury induction in the present study was a 30-min bilateral common carotid artery occlusion followed by a 45-min reperfusion (Raghavendra et al. 2009; Iwasaki et al. 1989), which exhibited significant oxidative outcomes—such as a significant increase of calcium and MDA levels as well as a reduction of SOD and CAT activities. The infarction area and neuronal damage in the dorsal hippocampus were also significantly increased by IR induction. Pretreatment with 100 mg/kg of p-coumaric acid for 2 weeks significantly reduced oxidative stress by ameliorating the MDA level, and it increased SOD and CAT activities. Significant reductions to the infarction area and hippocampal neuronal damage were also revealed.
The antioxidant properties of p-coumaric acid exert themselves as reducing agents by activating of endogenous antioxidant enzymes such as SOD and CAT. Increases of lipid peroxidation during IR are caused by a polyunsaturated fatty-acid peroxide radical with monocyclic peroxide or bicyclic endoperoxide by lipid hydroperoxide and oxy radical (Ayala et al. 2014). Once MDA has formed, biomolecular damage and cell death are activated by MDA-protein and MDA-DNA adducts. Normally MDA can be enzymatically metabolized into a nontoxic form via enzyme cascades—such as aldehyde dehydrogenase, decarboxylase, acetyl CoA synthase and a tricarboxylic-acid cycle. The amelioration of lipid peroxidation and increasing of SOD activity by p-coumaric acid during IR injury in the present study were similar to results in a previous report (Guven et al. 2015). Additionally, we showed that p-coumaric acid increases CAT activity, which supports the enzymatic activity of SOD. SOD detoxifies O2− into H2O2, and CAT further converts H2O2 into H2O and O2. We found that SOD and CAT activities were reduced during IR, which indicates that excessive O2− leads to an increase of lipid peroxidation and is reflected by high MDA levels in the present study. Pretreatment with p-coumaric acid ameliorated brain oxidative stress by reducing MDA while increasing CAT and SOD as well.
The infarction area was significantly increased following IR, and pretreatment with p-coumaric acid can prevent the increase of infarction volume. The evaluation of infarction area in the present study used the reaction of TTC with NADH. As a marker of aerobic respiration, NADH represents viable tissue. Evidence showed that p-coumaric acid helped increase mitochondrial oxidative phosphorylation, which ameliorates the suppression of ischemia-induced respiratory complex activities. Due to increases of nuclear respiratory factor-1 and nuclear gene-encoding respiration proteins, p-coumaric acid helps prevent energy failure during ischemia, maintaining the neuronal function and increasing neuronal strength against reperfusion injury (Guven et al. 2015).
Calcium overload is clearly revealed in IR injury. During ischemia, energy failure occurs and leads to many conditions that induce cytosolic calcium overload. The maintenance of ionic homeostasis via Na+/H+ exchangers counteracts the drop of cytosolic pH, which in turn increases Na+/Ca2+ exchangers and Ca2+ reuptake into the ER/SR by SERCA. IR impairs ATPase. The enhancement of Na+/Ca2+ exchangers together with the release of calcium through the ryanodine receptors exacerbate the lethal elevation of cytosolic calcium and further activate a variety of calcium-dependent cascades—such as calcium-dependent proteases, mitochondrial permeability transition-pore openings, inflammatory and prothrombogenic cascades, free radical formation and apoptotic cell death—that contribute to cell death following IR (Kalogeris et al. 2012). Calcium levels in brain tissues significantly increased following IR in the present study. Pretreatment with p-coumaric acid tended to reduce calcium levels but did not lead to a significant difference. It may be that p-coumaric acid did not directly act on cytosolic calcium controllers but rather helped prevent neuronal damage via other pathways, such as the activation of the antioxidant system.
In the present study, we observed neuronal damage in the vulnerable brain area such as the dorsal hippocampus following IR. During ischemia, excitotoxicity mediated by glutamate receptors, such as the NMDA receptor, is activated. Energy failure causes excessive glutamate release and leads to an influx of calcium through this channel (Arundine and Tymianski 2003). As a key mediator, calcium overload further triggers many distinct cascades that lead to neuronal death (Kalogeris et al. 2012; Martin et al. 1998). Vulnerable brain regions are characterized by vascular distribution and intrinsic factors. An abundance of glutamate receptors and high intrinsic oxidative stress are indicated in vulnerable neurons such as the CA1 of the dorsal hippocampus (Davolio and Greenamyre 1995; Wang et al. 2005). The present study found significant damage to the CA1 and CA3 but not the DG following IR; this is because of the difference in the areas’ susceptibility. In the pattern of selective vulnerability, DG was less than CA3, which was less than CA1 (Mattson and Kater 1989). Pretreatment with p-coumaric acid significantly reduced the damage to CA1 but not CA3 areas. The inhibition of mitochondrial-associated proapoptotic proteins, such as apoptosis signal-regulating kinase 1 (ASK1), and caspase activity (caspase-3 and caspase-9) were involved (Guven et al. 2015). Interestingly, p-coumaric acid significantly reduced the percentage of dead cells in the DG even though this area has less susceptibility and displayed no significant damage following IR in the present study. This may be due to the involvement of p-coumaric acid in neurogenesis within this region. It is widely known that adult neurogenesis can be found in two distinct brain regions, the subventricular zone and the DG of the dorsal hippocampus (Taupin 2006). Evidence showed that p-coumaric acid exerts an effect on the inhibition of NF-kB signaling pathways, a transcription factor that plays a key role in gene regulation for both damaging and protecting mechanisms. Activation by some activators of this signaling pathway can lead to neurotoxicity (Yoon et al. 2014; Zhang and Hu 2012). The signaling of NF-kB regulates many genes that are involved in immunity, inflammation, neural plasticity, cell survival and neurogenesis (Zhang and Hu 2012). In the present study, the survival of neurons in CA1 and DG but not CA3 by p-coumaric acid may depend on a variety of factors. This indicates a double-edged sword in regard to the action of NF-kB signaling in neurodegenerative disease. The outcome depends on varieties of factors (Zhang and Hu 2012). The difference of susceptibility and cell type properties of CA1, CA3 and DG neurons may lead to a different outcome as showed in the present study. As with all the results, p-coumaric acid exhibits a neuroprotective effect via amelioration of oxidative stress, infarction and vulnerable neuronal damage.
Conclusion
Pretreatment with p-coumaric acid ameliorates brain oxidative stress, infarction and vulnerable neuronal damage in cerebral IR injury. There is an indication of p-coumaric acid’s ability to maintain neuronal function and increase neuronal strength against IR injury.
References
Abdel-Wahab MH, El-Mahdy MA, Abd-Ellah MF, Helal GK, Khalifa F, Hamada FM (2003) Influence of p-coumaric acid on doxorubicin-induced oxidative stress in rat’s heart. Pharmacol Res 48:461–465
Amalan V, Vijayakumar N, Indumathi D, Ramakrishnan A (2016) Antidiabetic and antihyperlipidemic activity of p-coumaric acid in diabetic rats, role of pancreatic GLUT 2: In vivo approach. Biomed Pharmacother 84:230–236. https://doi.org/10.1016/j.biopha.2016.09.039
Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34(4-5):325–337. https://doi.org/10.1016/S0143-4160(03)00141-6
Ayala A, Munoz MF, Arguelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med Cell Longev 2014:360438
Collard CD, Gelman S (2001) Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology 94(6):1133–1138. https://doi.org/10.1097/00000542-200106000-00030
Davolio C, Greenamyre JT (1995) Selective vulnerability of the CA1 region of hippocampus to the indirect excitotoxic effects of malonic acid. Neurosci Lett 192(1):29–32. https://doi.org/10.1016/0304-3940(95)11600-2
Guven M, Aras AB, Akman T, Sen HM, Ozkan A, Salis O, Sehitoglu I, Kalkan Y, Silan C, Deniz M, Cosar M (2015) Neuroprotective effect of p-coumaric acid in rat model of embolic cerebral ischemia. Iran J Basic Med Sci 18(4):356–363
Hadwan MH, Abed HN (2016) Data supporting the spectrophotometric method for the estimation of catalase activity. Data Brief 6:194–199. https://doi.org/10.1016/j.dib.2015.12.012
Hou ST, MacManus JP, Kwang WJ (2002) Molecular mechanisms of cerebral ischemia-induced neuronal death. Int Rev Cytol 221. Academic Press, pp 93–148
Iwasaki Y, Ito S, Suzuki M, Nagahori T, Yamamoto T, Konno H (1989) Forebrain ischemia induced by temporary bilateral common carotid occlusion in normotensive rats. J Neurol Sci 90(2):155–165. https://doi.org/10.1016/0022-510X(89)90098-1
Jivad N, Rabiei Z (2015) Review on herbal medicine on brain ischemia and reperfusion. Asian Pac J Trop Biomed 5(10):789–795. https://doi.org/10.1016/j.apjtb.2015.07.015
Kalogeris T, Baines CP, Krenz M, Korthuis RJ, Kwang WJ (2012) Chapter six - cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298. Academic Press, pp 229–317. https://doi.org/10.1016/B978-0-12-394309-5.00006-7
Ladecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14(11):1363–1368. https://doi.org/10.1038/nn.2953
Leker RR, Shohami E (2002) Cerebral ischemia and trauma different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Rev 39(1):55–73. https://doi.org/10.1016/S0165-0173(02)00157-1
Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C (1998) Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull 46(4):281–309. https://doi.org/10.1016/S0361-9230(98)00024-0
Mattson MP, Kater SB (1989) Development and selective neurodegeneration in cell cultures from different hippocampal regions. Brain Res 490(1):110–125. https://doi.org/10.1016/0006-8993(89)90436-8
Mehta SL, Kumari S, Mendelev N, Li PA (2012) Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci 13(1):79. https://doi.org/10.1186/1471-2202-13-79
Pan J, Konstas AA, Bateman B, Ortolano GA, Pile-Spellman J (2007) Reperfusion injury following cerebral ischemia: pathophysiology, MR imaging, and potential therapies. Neuroradiology 49(2):93–102. https://doi.org/10.1007/s00234-006-0183-z
Pandey KB, Rizvi SI (2009) Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Med Cell Longev 2(5):270–278. https://doi.org/10.4161/oxim.2.5.9498
Paxinos G, Franklin K (2008) The mouse brain in stereotaxic coordinates. 3rd edn. Academic Press, San Diego
Pei K, Ou J, Huang J, Ou S (2015) p-Coumaric acid and its conjugates: dietary sources, pharmacokinetic properties and biological activities. J Sci Food Agric 96(9):2952–2962. https://doi.org/10.1002/jsfa.7578
Raghavendra M, Rituparna M, Shafalika K, Anshuman T, Sumit M, S A (2009) Role of Centella asiatica on cerebral post-ischemic reperfusion and long-term hypoperfusion in rats. Int J Green Pharm 3:88–96, https://doi.org/10.4103/0973-8258.54893
Shuaib A, Breker-Klassen MM (1997) Inhibitory mechanisms in cerebral ischemia: a brief review. Neurosci Biobehav Rev 21(2):219–226. https://doi.org/10.1016/S0149-7634(96)00012-7
Spare PD (1964) A stable murexide reagent for the estimation of calcium in micro quantities of serum. Clin Chem 10:726–729
Taupin P (2006) Neurogenesis in the adult central nervous system. C R Biol 329(7):465–475. https://doi.org/10.1016/j.crvi.2006.04.001
Thong-asa W, Tilokskulchai K (2014) Neuronal damage of the dorsal hippocampus induced by long-term right common carotid artery occlusion in rats. Iran J Basic Med Sci 17(3):220–226
Wang X, Pal R, Chen XW, Limpeanchob N, Kumar KN, Michaelis EK (2005) High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region. Brain Res Mol Brain Res 140(1-2):120–126. https://doi.org/10.1016/j.molbrainres.2005.07.018
White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 179(1-2):1–33. https://doi.org/10.1016/S0022-510X(00)00386-5
Yoon JH, Youn K, Ho CT, Karwe MV, Jeong WS, Jun M (2014) p-Coumaric acid and ursolic acid from Corni fructus attenuated beta-amyloid(25-35)-induced toxicity through regulation of the NF-kappaB signaling pathway in PC12 cells. J Agric Food Chem 62(21):4911–4916. https://doi.org/10.1021/jf501314g
Zhang Y, Hu W (2012) NFkappaB signaling regulates embryonic and adult neurogenesis. Front Biol (Beijing) 7(4):277–291. https://doi.org/10.1007/s11515-012-1233-z
Acknowledgements
We would like to thank the Animal Toxicology and Physiology Specialty Research Unit (ATPRS). The present study was supported by grant from the Department of Zoology (Project in Zoology of Graduate Program), Faculty of Science, Kasetsart University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that no conflict of interest.
Rights and permissions
About this article

Cite this article
Sakamula, R., Thong-asa, W. Neuroprotective effect of p-coumaric acid in mice with cerebral ischemia reperfusion injuries. Metab Brain Dis 33, 765–773 (2018). https://doi.org/10.1007/s11011-018-0185-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-018-0185-7