
Abstract
Falcarindiol (FAD) is a natural polyacetylene compound found rich in many plants of the Umbelliferae family. Previously, we isolated FAD from the rhizome of Cnidium officinale Makino, which belongs to the Umbelliferae family and found it to have a significant inhibitory effect on lipopolysaccharide (LPS)-induced production of nitric oxide, a pro-inflammatory molecule in murine macrophage RAW 264.7 cells. In this study, we investigated its effect on the expression of other major pro-inflammatory molecules as well as the mechanism underlying these effects. Pre-treatment of RAW 264.7 cells with FAD suppressed LPS-stimulated mRNA expression of inducible nitric oxide synthase (iNOS), tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) and thereby reduced the respective protein levels. Mechanistic studies demonstrated that FAD attenuated the LPS-induced activation of JNK, ERK, STAT1, and STAT3 signaling molecules. Moreover, we found that FAD did not influence LPS-induced activation of p38 and NFκB signaling pathways. Collectively, this study provides evidence that FAD inhibits the production of major pro-inflammatory molecules in LPS-challenged murine macrophages via suppression of JNK, ERK, and STAT signaling pathways.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cnidium officinale Makino is a common flowering plant in the Umbelliferae family. In East Asian countries, including Korea, this plant has been traditionally used for treatment of various ailments [1]. Previous studies have shown that the extract of C. officinale Makino has a wide range of pharmacological properties including anti-inflammatory, anti-diabetic, anti-proliferative, apoptotic, antioxidant, immunomodulatory, and anti-angiogenic activity [2,3,4,5,6,7,8,9,10]. In addition, the extract of C. officinale Makino has been shown to have a significant inhibitory effect on retinal neovascularization in mice [11]. A recent study in experimental rats demonstrated that Hemomine, an herbal mixture consisting of five different herbal extracts including C. officinale Makino, can ameliorate subacute hemorrhagic anemia [12].
Moreover, the rhizome of C. officinale Makino is often considered an important source of various therapeutic phytochemicals [13,14,15]. Previously, we reported that the anticancer and anti-inflammatory activities of the rhizome of C. officinale Makino were related to the presence of major compounds, falcarindiol (FAD), 6-hydroxy-7-methoxy-dihydroligustilide, ligustilidiol, and senkyunolide H. Further, we found that comparatively, FAD exhibited a potent inhibitory effect on lipopolysaccharide (LPS)-induced production of nitric oxide (NO) in murine macrophages [16]. Kim et al. have also reported that FAD is a potent inhibitor of NO production in resident macrophages of brain tissues [17]. In addition to the attenuation of NO production, the inhibition of chronic overproduction of other major pro-inflammatory molecules such as tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) from acute inflammatory cells, especially macrophages, is often considered to be an important target in the treatment of the most prevalent inflammatory disorders, such as arthritis and atherosclerosis [18,19,20]. Therefore, the aim of this study was to determine the effects of FAD on the overproduction of TNFα, IL-6, and IL-1β, as well as the mechanism behind its effects on NO and cytokine production in LPS-challenged RAW 264.7 murine macrophage cells.
Materials and methods
Materials
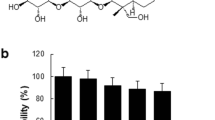
FAD was previously isolated from the rhizome of C. officinale Makino [16]. Figure 1 shows the structure of FAD. The purity of FAD is around 98%. The stock solution of FAD was prepared using DMSO (D8418) at a concentration of 50 mM. The solution was filter sterilized twice, using 0.2 µm DMSO-Safe Acrodisc® Syringe Filter (Catalog. No. 4433, Pall Corporation, NY, USA) and stored at − 20 °C for further use. LPS (L3012; E. coli, Serotype 0111:B4) and MTT (M2128) were purchased from Sigma-Aldrich (St. Louis, MO, USA). ELISA kits to assay TNFα (MTA00B), IL-6 (M6000B), and IL-1β (MLB00C) were purchased from R&D Systems (Minneapolis, MN, USA). Amersham™ Protran™ 0.2 µM nitrocellulose membranes (10600001) were purchased from GE Healthcare Life Sciences. AG490 (tlrl-ag4) was obtained from InvivoGen, San Diego, USA. Inhibitors SP600125 (sc-200635) and U0126 (sc-222395), antibodies against iNOS (sc-7271), NFκB (sc-372), Lamin-B (sc-374015) and β-actin (sc-47778), as well as HRP-conjugated secondary antibodies (sc-2004 and sc-2005) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Antibodies to MAPKs [P38, 9212; phospho-P38, 9211; JNK, 9252; phospho-JNK, 9251; ERK, 9102; phospho-ERK, 9101]; IκB, 4812; STAT3, 9139; phospho-STAT3 (Tyr705), 4113; STAT1, 9172; and phospho-STAT1 (Tyr701), 7649;] and HRP-conjugated secondary antibodies (7074 and 7076) were purchased from Cell Signaling Technology (Beverly, MA, USA). The primer sequences (Bioneer Corporation, Daejeon, 34302, Republic of Korea) used in the present study were as follows: iNOS, CAT GCT ACT GGA GGT GGG TG (forward), CAT TGA TCT CCG TGA CAG CC (reverse); TNFα, AGC ACA GAA AGC ATG ATC CG (forward), CTG ATG AGA GGG AGG CCA TT (reverse); IL-6, GAG GAT ACC ACT CCC AAC AGA CC (forward), AAG TGC ATC ATC GTT GTT CAT ACA (reverse); IL-1β, TGC AGA GTT CCC CAA CTG GTA CAT C (forward), GTG CTG CCT AAT GTC CCC TTG AAT C (reverse); and β-actin, ATC ACT ATT GGC AAC GAG CG (forward), TCA GCA ATG CCT GGG TAC AT (reverse).

Chemical structure of FAD
Cell culture
Murine macrophage RAW 264.7 cells (KCLB No. 40071), obtained from the Korea Cell Line Bank (Seoul, Korea), were cultured in DMEM (Hyclone, SH30022.01) containing 4.5 g/L glucose, 4 mM glutamine, 25 mM HEPES (15630080—Thermo Fisher Scientific), 10% heat-inactivated FBS (10082147—Thermo Fisher Scientific), and antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin sulfate and 0.25 µg/mL amphotericin B; 15240062—Thermo Fisher Scientific). Cells were seeded at a density of 1 × 106 cells/mL and maintained in DMEM supplemented with 2% FBS, before being treated with FAD and/or LPS.
MTT assay
The effect of FAD on the viability of RAW 264.7 cells was determined by the MTT colorimetric assay, as previously described. Briefly, 80 μL of cell suspension (1.25 × 106 cells/mL) per well was seeded into 96-well plates and incubated overnight, to which 20 μL of various concentrations (25, 50, 75, 100, and 125 µM) of FAD, prepared in fresh serum and phenol-red free DMEM, were added. The final concentrations of FAD were 5, 10, 15, 20, and 25 µM. After 24 h of treatment with FAD, medium was replaced with 100 μL of MTT (0.5 mg/mL) solution in phenol-red free DMEM and incubated for an additional 4 h. Thereafter, cell culture supernatants were discarded; formazan crystals were dissolved in 100 μL DMSO, and the optical density (OD) was measured at 570 nm, using a micro-plate reader (OPSYS MR Dynex Technologies).
Measurement of extracellular NO production
RAW 264.7 macrophage cells were pre-treated with FAD for 1 h and then stimulated with LPS (1 µg/mL) for 18 h. Afterwards, cell-free supernatants were collected by centrifugation at 3000 rpm and 4 °C for 5 min. NO levels were quantified using Griess reagent containing equal volumes of 1% sulfanilamide in 5% phosphoric acid and 0.1% N-(1-naphthyl)ethylenediamine. Each experiment was performed in triplicate. NO levels were expressed as number of folds versus control.
Measurement of intracellular NO production
Cells were pre-treated with FAD for 1 h and then challenged with LPS (1 µg/mL) for 12 h. Thereafter, cells were washed with Hank’s Balanced Salt Solution (HBSS) and incubated with 5 µM DAF-FM DA (D-23844, Thermo Fisher Scientific) for 1 h. After the removal of excess probe, cells were incubated in fresh HBSS for an additional 15 min. Cells were then collected, and fluorescence intensity was analyzed using flow cytometer (BD Biosciences).
Measurement of extracellular cytokines production
The levels of cytokines TNFα, IL-6, and IL-1β in cell culture supernatants of FAD and/or LPS-treated groups were measured using commercial ELISA kits (Mouse TNFα Immunoassay, MTA00B; Mouse IL-6 Immunoassay, M6000B; Mouse IL-1β Immunoassay, MLB00C), according to the manufacturer’s instructions. At least three replicates were fixed for each of the three independent experiments.
Quantitative real-time-polymerase chain reaction (qRT-PCR)
Cells were treated with FAD for 1 h and then challenged with LPS for 6 h. Total cellular RNA was isolated by Trizol reagent (Invitrogen, 15596026) according to the manufacturer’s instructions. From each sample group, 1 µg of total RNA was utilized to synthesize cDNA using an iScriptTM cDNA Synthesis kit (170–8891) from Bio-rad. Then qRT-PCR analysis was performed using the customer iQTM SYBR Green Super mix (Bio-rad, 170-8880AP) and specific primer sequences. The PCR reaction conditions were as follows: 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s, 55–60 °C for 30 s, and 72 °C for 30 s. In addition, a melt curve analysis was performed to exclude non-specific signals. The mRNA expression levels of iNOS, TNFα, IL-6, and IL-1β were normalized with the mRNA levels of a control gene, β-actin. Three independent experiments were carried out in triplicate. The expression level of each gene is presented as the relative number of folds versus control gene.
Western blot analysis
After treatment with FAD and/or LPS, cells were washed twice with ice-cold phosphate-buffered saline (PBS), and whole cell lysates were prepared using RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease and phosphatase inhibitors (04693159001 and 04906837001—Roche). Cytoplasmic and nuclear fractions were prepared according to our previous study [21]. The protein concentration of the lysates was determined using the BCA protein assay reagent according to the manufacturer’s instructions. Lysates were resolved on 10% gels, and proteins were blotted onto a nitrocellulose membrane. The membranes were blocked with either 5% (w/v) non-fat milk or 5% (w/v) BSA for 1 h, and target proteins were conjugated with specific primary antibodies at 4 °C overnight. Then, the membranes were incubated with their respective HRP-conjugated secondary antibody at room temperature for 1 h. Target signals were developed using SuperSignal™ West Pico Chemiluminescent Substrate (34577—Thermo Fisher Scientific). The signals were recorded with a LI-COR image processing system and analyzed with a gel imaging software.
Statistical analysis
All experiments were performed three independent times (N = 3). A minimum of three replicates (n = 3) in each sample group was used in each single experimental assay of MTT, NO quantification, ELISA, and PCR assays. Data were subjected to One-Way Analysis of Variance (ANOVA) followed by Dunnett’s multiple comparisons test to measure the significant difference between the sample groups. The data were given as the mean ± SEM. A p value of less than 0.05, 0.01, or 0.001 was considered statistically significant. Analysis was performed using the GraphPad Prism 7.02 Software (San Diego, CA).
Results
Effect of FAD on RAW 264.7 cell viability
Prior to determining the effects of FAD on the expression of pro-inflammatory molecules, cytotoxicity assays were performed using MTT reagent to assess the concentration-dependent effect of FAD on RAW 264.7 cell viability. As shown in Fig. 2, FAD showed no significant toxic effects on RAW 264.7 cells up to a concentration of 20 µM, whereas FAD at the 25 µM concentration showed a significant (p < 0.05) cytotoxic effect against RAW 264.7 cells. Hence, the concentration of FAD was limited to 20 µM for further experimental analysis.

Cytotoxicity of FAD in RAW 264.7 cells. Cells were treated with FAD at 5–25 μM and tert-Butyl hydroperoxide (TBHP) at 200 μM for 24 h. Then, cell viability was determined by the MTT assay which was conducted three independent times with triplicates (n = 3) in each group. The viability of cells untreated with FAD was taken as the control. *p < 0.05, ***p < 0.001 in comparison to the control group
Effect of FAD on LPS-stimulated NO production
Although previous studies of ours, as well as those of other researchers, have already demonstrated FAD to be a potent inhibitor of NO production [16, 17], we further confirmed its effect on NO production in LPS-challenged RAW 264.7 cells. Figure 3 shows the dose-dependent effect of FAD on NO production. FAD at 15 and 20 µM concentrations showed a maximum inhibitory effect without further influencing the LPS-induced cytotoxicity. NO levels in the LPS (1 µg/mL)-treated group were 13.63 ± 0.59 fold higher than vehicle-treated control cells. However, in the presence of FAD at the 5, 10, 15, and 20 µM concentrations, NO levels were 12.96 ± 0.72, 8.67 ± 0.56, 4.66 ± 0.11, and 2.83 ± 0.37 fold higher when compared to the control group, respectively. Flow cytometric analysis of intracellular NO production, using DAF-FM DA, further demonstrated that FAD has a significant inhibitory effect on NO production. The results of both assays indicate that FAD at concentrations greater than or equal to 10 µM induces a significant inhibitory effect on LPS (1 µg/mL)-induced NO production from RAW 264.7 murine macrophage cells. Further, it is evident that there was no significant difference between the inhibitory effects for FAD concentrations of 15 and 20 µM. Hence, the concentration of FAD in subsequent experiments was limited to 15 µM.

Effect of FAD on LPS-induced NO production in RAW 264.7 cells. Cells were pre-treated with FAD at 0–20 μM for 1 h and then stimulated with LPS for 18 h. The amount of NO (in the form of nitrite) in cell-free supernatant was quantified by the Griess reaction. For the intracellular NO production assay using DAF-FM DA, cells were pre-treated with FAD at 0–20 μM for 1 h and then stimulated with LPS for 12 h. Both the assays were performed three separate times with triplicates (n = 3) in each sample group. ***p < 0.001 in comparison to the control group; ns: non-significant, ###p < 0.001 in comparison to the LPS-stimulated group
Effect of FAD on mRNA and protein expression of iNOS, TNFα, IL-6, and IL-1β
To further study the anti-inflammatory effect of FAD in LPS-challenged RAW 264.7 cells, mRNA and protein levels of iNOS, TNFα, IL-6, and IL-1β were determined by qRT-PCR and immunoblotting or ELISA methods, respectively. FAD dose-dependently decreased the LPS-induced expression of the above-mentioned pro-inflammatory molecules in RAW 264.7 cells (Fig. 4). When compared to the vehicle-treated control group, mRNA expression levels in the presence of FAD at concentrations 0, 5, 10, and 15 µM were increased, for iNOS: 45.37 ± 3.77, 41.02 ± 3.76, 28.51 ± 3.62, and 16.86 ± 1.29 fold; for TNFα: 37.64 ± 4.95, 35.14 ± 4.97, 22.69 ± 3.20, and 17.00 ± 1.43 fold; for IL-6: 68.22 ± 7.63, 63.80 ± 7.97, 38.97 ± 3.43, and 26.94 ± 1.16 fold; for IL-1β: 315.10 ± 42.46, 285.66 ± 28.27, 206.65 ± 18.81, and 119.05 ± 9.13 fold, respectively. Protein levels were increased, for iNOS: 11.10, 10.20, 7.18, and 4.82 fold; for TNFα: 58.47 ± 6.22, 53.24 ± 8.92, 34.83 ± 5.79, and 25.23 ± 2.05 fold; for IL-6: 33.81 ± 2.92, 29.67 ± 3.21, 22.30 ± 1.99, and 15.11 ± 1.22 fold; for IL-1β: 53.02 ± 3.23, 47.57 ± 1.97, 30.86 ± 2.12, and 20.17 ± 1.01 fold, respectively. The data represent that FAD, at concentrations of 10 and 15 μM, had a significant inhibitory effect on both mRNA and protein expression of iNOS, TNFα, IL-6, and IL-1β, in LPS-challenged RAW 264.7 macrophage cells.

a Effect of FAD on LPS-induced mRNA expression of iNOS and cytokines in RAW 264.7 cells. Cells were pre-treated with FAD at 0–15 μM for 1 h, followed by LPS stimulation for 6 h. The mRNA levels were determined by qRT-PCR as described in “materials and methods.” The graphical figures represent the change in mRNA expression of inflammatory mediators normalized against actin. ***p < 0.001 in comparison to the control group; ns non-significant, #p < 0.05, ##p < 0.01, ###p < 0.001 in comparison to the LPS-stimulated group. b Effect of FAD on LPS-induced iNOS protein expression in RAW 264.7 cells. The cells were pre-treated with FAD at 0–15 μM for 1 h, followed by LPS stimulation for 18 h. Then, whole cell lysates were prepared and used to quantify iNOS levels by the immunoblotting assay method. Western blotting was performed three separate times. The graphical figure represents the change in the ratio of iNOS to actin levels. ***p < 0.001 in comparison to the control group; ns non-significant, #p < 0.05, ##p < 0.01, ###p < 0.001 in comparison to the LPS-stimulated group. c Effect of FAD on LPS-induced cytokine production in RAW 264.7 cells. The cells were pre-treated with FAD at 0–15 μM for 1 h, followed by LPS stimulation for 18 h. The cell-free supernatant was subjected to quantify TNFα, IL-6, and IL-1β levels, using ELISA kits, according to manufacturer’s protocol. The graphical figures represent the change in cytokine levels relative to the control group. ***p < 0.001 in comparison to the control group; ns non-significant, #p < 0.05, ##p < 0.01, ###p < 0.001 in comparison to the LPS-stimulated group. Both qRT-PCR and ELISA assays were conducted three independent times with triplicates (n = 3) in each sample group
Effect of FAD on LPS-induced activation of MAPKs, NFκb, and JAK-STAT signaling pathways
Figure 5 shows the dose-dependent effect of FAD on LPS-induced activation of p38, JNK, and ERK, IκB, NFκB p65, STAT1, and STAT3 signaling molecules in RAW 264.7 cells. FAD significantly reduced the LPS (1 µg/mL)-induced phosphorylation of JNK, ERK, STAT1, and STAT3 proteins. However, FAD did not affect the activation of p38, IκB, and NFκB p65 signaling molecules. Moreover, LPS (1 µg/mL)-induced nuclear translocation of NFκB p65 was not affected by FAD.

a Effect of FAD on the MAPK pathway in RAW 264.7 cells. Cells were pre-treated with FAD at 0–15 μM for 1 h, followed by LPS stimulation for 30 min. Then, whole cell lysates were prepared and used to quantify the levels of phosphorylated and total p38, JNK, and ERK by the immunoblotting method. The graphical figure represents the change in the ratio of their phosphorylated protein level to their respective total protein level. ***p < 0.001 in comparison to the control group; ns non-significant, #p < 0.05, ##p < 0.01, ###p < 0.001 in comparison to the LPS-stimulated group. b Effect of FAD on NFκB pathway in RAW 264.7 cells. Cells were pre-treated with FAD at 0–15 μM for 1 h, followed by LPS stimulation for 30 min. Cytosolic and nuclear fractions were prepared and used to quantify the levels of IκB and NFκB p65 by the western blot method. The graphical figure represents the change in the ratio of IκB to actin in the cytosol and NFκB p65 to actin in the cytosol or to Lamin B in the nucleus. ***p < 0.001 in comparison to the control group; ns non-significant in comparison to the LPS-stimulated group. c Effect of FAD on LPS-induced activation of the JAK-STAT pathway in RAW 264.7 cells. Cells were pre-treated with FAD at 0 or 15 μM for 1 h, followed by LPS stimulation for 120–240 min. Then, whole cell lysates were prepared and used to determine the total levels as well as phosphorylated levels of STAT1 and STAT3 by the western blot method. Western blotting was performed three separate times. The graphical figure represents the change in the ratio of their phosphorylated protein level to their respective total protein level. ***p < 0.001 in comparison to the control group; ###p < 0.001 in comparison to the LPS-stimulated group
Effect of FAD, SP600125, U0126, and AG490 on LPS-stimulated production of inflammatory molecules
Specific inhibitors for JNK (SP600125), ERK (U0126) and JAK-STAT (AG490) pathways were used to further confirm that the effect of FAD on the LPS-induced NO, TNFα, IL-6, and IL-1β production in RAW 264.7 murine macrophage cells involves the MAPK and JAK-STAT signaling pathways. As shown in Fig. 6, both FAD and JNK inhibitors (SP600125) significantly suppressed the LPS (1 µg/mL)-stimulated increased levels of NO, TNFα, IL-6, and IL-1β. Though the ERK inhibitor (U0126) significantly decreased the levels of TNFα, IL-6, and IL-1β, it showed a stimulatory effect on LPS (1 µg/mL)-induced NO production. JAK2 is an upstream activator of both STAT1 and STAT3 in JAK-STAT pathway. AG490, which inhibits JAK2, significantly decreased the levels of NO, IL-6, and IL-1β but not TNFα. Based on these data, it seems reasonable to say that FAD may suppress the production of NO, TNFα, IL-6, and IL-1β from murine macrophage RAW 264.7 cells by inhibiting the LPS-induced activation of MAPK and JAK-STAT signaling pathways.

Effect of FAD, SP600125 (JNK inhibitor), U0126 (ERK inhibitor), and AG490 (JAK2 inhibitor) on LPS-induced NO and cytokine production in RAW 264.7 cells. The cells were pre-treated with FAD (15 μM), SP600125 (20 μM), U0126 (20 μM), or AG490 (30 μM) for 1 h, followed by LPS stimulation for 18 h. The cell-free supernatant was subjected to quantify NO, TNFα, IL-6, and IL-1β levels. The assay was performed three independent times with triplicates (n = 3) in each group. The graphical figures represent the change in NO or cytokine levels relative to the control group. ***p < 0.001 in comparison to the control group; ##p < 0.01, ###p < 0.001 in comparison to the LPS-stimulated group
Discussion
Inflammation is a normal immunological defense response of organisms to invading foreign pathogens and various internal or external harmful stimuli that induce systemic tissue damage [22]. In the immunological defense mechanism, macrophages are considered primary acute inflammatory cells as they initiate, as well as modulate, the host inflammatory response [23, 24]. They produce a variety of inflammatory mediators which can amplify the response to eliminate pathogens and remodel damaged tissues [25]. However, prolonged activation of macrophages and increased production of inflammatory mediators are reported to contribute to the development of many disease complications [26]. For instance, macrophages in atherosclerotic lesions initiate and maintain the local inflammatory response by continuously releasing pro-inflammatory mediators. This eventually leads to the failure of inflammation resolution and results in the progression of the lesion into a complicated atherosclerotic plaque [19]. Hence, inhibition of macrophage-induced inflammation is thought to be a promising target to control the development of disease complications. In the present study, we used LPS-stimulated murine macrophage RAW 264.7 cells to explore the anti-inflammatory effect of a natural polyacetylene compound FAD.
Generally, in the field of natural products research, crude extracts are used to screen for their pharmacological properties. Such crude extracts are complicated mixtures containing various components differing in their characteristic features. At times, effective components are found too low in concentration or their effects are compromised by other interfering components. Hence, isolation of individual components offers a promising way to select a compound with valuable therapeutic potential. In our previous study, we isolated FAD from the rhizome of C. officinale Makino, which is reported to have an inhibitory effect on LPS-induced NO production in RAW 264.7 cells [16]. Similar results were also observed in the present study. The FAD-induced, decreased production of NO in LPS-challenged murine macrophages may be due to its inhibitory effect on the expression of the corresponding enzyme, iNOS. Further, existing literature has also shown that FAD reduced iNOS-mediated NO production in LPS-activated microglia cells, the resident macrophages in brain tissues. So, it can be concluded that FAD is a potent inhibitor of NO production in activated macrophages. In addition, the present study provides evidence that FAD has a significant, suppressive effect on LPS-induced expression of pro-inflammatory cytokines such as TNFα, IL-6, and IL-1β. These results suggest that FAD possesses a significant anti-inflammatory effect in LPS-activated murine macrophages.
Next, we explored the mechanisms of FAD on the inhibition of LPS-induced production of pro-inflammatory mediators. LPS induces an inflammatory response in macrophages by activating a number of signaling pathways. LPS initiates the inflammatory response in macrophages by first binding with membrane-bound Toll-like receptor 4 (TLR4). Then, this receptor activates its downstream signaling molecules, mainly MAPKs and NFκB, through induction by phosphorylation [27]. Activated MAPKs, such as phospho-p38, phospho-JNK and phospho-ERK, pass their signal to downstream molecules, which can translocate to the nucleus and induce the expression of inflammatory molecules [28]. FAD had a significant inhibitory effect on LPS-induced phosphorylation of JNK and ERK but not p38. Further, we found that both JNK inhibitors and FAD showed suppressive effects on the production of NO, TNFα, IL-6, and IL-1β, whereas an ERK inhibitor could suppress TNFα, IL-6, and IL-1β, but would promote NO production. Previous studies also clearly demonstrated the influences of MAPK inhibitors on the LPS-induced expression of inflammatory mediators in macrophages [29,30,31,32]. Unlike MAPKs, activated NFκB can directly enter into the nucleus and induce the production of NO, TNFα, IL-6, and IL-1β [33, 34]. Generally, NFκB is sequestered by its inhibitory protein IκB within the cytoplasm. LPS is reported to release NFκB by increasing phosphorylation and proteasome-mediated degradation of IκB [35]. Here, we found that FAD had no inhibitory effects on LPS-induced, increased phosphorylation of IκB and nuclear levels of NFκB molecules in RAW 264.7 cells. These mechanistic study results indicated that FAD may decrease the production of NO, TNFα, IL-6, and IL-1β by inhibiting the LPS-induced activation of their regulatory signaling proteins, JNK, and ERK, in RAW 264.7 murine macrophage cells.
In addition to MAPKs and NFκB, the JAK-STAT pathway has also been shown to be involved in LPS-induced, increased expression of inflammatory molecules, especially iNOS, IL-6, and IL-1β, in macrophage cells [36]. In this respect, we also found that AG490, an inhibitor of the JAK-STAT pathway, suppressed the LPS-induced production of iNOS, IL-6, and IL-1β in RAW 264.7 cells [36]. Upon activation of macrophages by LPS, membrane-bound JAK receptor proteins increase the phosphorylation of their substrate molecules, such as STAT proteins. Phosphorylated STAT proteins can translocate to the nucleus to regulate the expression of target genes encoding for inflammatory mediators [37]. Accumulating evidence demonstrates that STAT1 and STAT3 are the major downstream signaling proteins in the JAK-STAT pathway of RAW 264.7 murine macrophage cells [38, 39]. STAT1 is reported to regulate the transcription of the iNOS gene, and STAT3 is said to regulate transcription of both IL-6 and IL-1β genes [40, 41]. Pre-treatment of FAD showed a significant suppressive effect on LPS-induced activation of STAT1 and STAT3 signaling proteins in RAW 264.7 cells. While it is known that FAD inhibits JNK and ERK activation to regulate the LPS-induced expression of pro-inflammatory mediators iNOS, TNFα, IL-6, and IL-1β, we believe that its inhibitory effect on STAT1 and STAT3 activation may also be partly responsible for the decreased expression of iNOS, IL-6, and IL-1β in RAW 264.7 murine macrophages.
Conclusion
Previous studies focused primarily on the inhibitory effect of FAD on NO production in activated macrophages. Our present findings demonstrate the inhibitory effect of FAD on the inflammatory cytokines TNFα, and on IL-6 and IL-1β production in LPS-challenged murine macrophages. FAD also significantly reduced the respective mRNA levels of these inflammatory cytokines as well as iNOS, which is a corresponding enzyme responsible for NO production, in murine RAW 264.7 macrophage cells. Further, the inhibitory effect of FAD on JNK, ERK, STAT1, and STAT3 activation revealed that FAD may exhibit its anti-inflammatory effects via inhibition of the MAPK and JAK-STAT signaling pathways.
Abbreviations
- BCA:
-
Bicinchoninic acid
- BSA:
-
Bovine serum albumin
- cDNA:
-
Complementary deoxyribonucleic acid
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- E. coli :
-
Escherichia coli
- ERK:
-
Extracellular signal–regulated kinase
- FAD:
-
Falcarindiol
- FBS:
-
Fetal bovine serum
- IL-1β:
-
Interleukin-1β
- HBSS:
-
Hank’s balanced salt solution
- HEPES:
-
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- HRP:
-
Horseradish peroxidase
- IκB:
-
Inhibitory kappa B protein
- IL-6:
-
Interleukin-6
- iNOS:
-
Inducible nitric oxide synthase
- JAK-STAT:
-
Janus kinase-signal transducers and activators of transcription
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- MAPKs:
-
Mitogen-activated protein kinases
- mRNA:
-
Messenger RNA
- MTT:
-
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
- NaCl:
-
Sodium chloride
- NFκB:
-
Nuclear transcription factor kappa B
- NO:
-
Nitric oxide
- P38:
-
P38 Mitogen-activated protein kinase
- PBS:
-
Phosphate-buffered saline
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- RIPA:
-
Radioimmunoprecipitation assay
- RNA:
-
Ribonucleic acid
- SDS:
-
Sodium dodecyl sulfate
- STAT1:
-
Signal transducers and activators of transcription factor 1
- STAT3:
-
Signal transducers and activators of transcription factor 3
- TLR4:
-
Toll-like receptor 4
- TNF-α:
-
Tumor necrosis factor α
References
Ramalingam M, Yong-Ki P (2010) Free radical scavenging activities of Cnidium officinale Makino and Ligusticum chuanxiong Hort. methanolic extracts. Pharmacogn Mag 6:323–330
Lee SH, Lee JH, Oh EY, Kim GY, Choi BT, Kim C, Choi YH (2013) Ethanol extract of Cnidium officinale exhibits anti-inflammatory effects in BV2 microglial cells by suppressing NF-κB nuclear translocation and the activation of the PI3K/Akt signaling pathway. Int J Mol Med 32:876–882
Kim JM, Son D, Lee P, Lee KJ, Kim H, Kim SY (2003) Ethyl acetate soluble fraction of Cnidium officinale MAKINO inhibits neuronal cell death by reduction of excessive nitric oxide production in lipopolysaccharide-treated rat hippocampal slice cultures and microglia cells. J Pharmacol Sci 92:74–78
Kim JJ, Cho HW, Park HR, Jung U, Jo SK, Yee ST (2013) Preventative effect of an herbal preparation (HemoHIM) on development of airway inflammation in mice via modulation of Th1/2 cells differentiation. PLoS ONE 8:e68552
Kim JJ, Choi J, Lee MK, Kang KY, Paik MJ, Jo SK, Jung U, Park HR, Yee ST (2014) Immunomodulatory and antidiabetic effects of a new herbal preparation (HemoHIM) on streptozotocin-induced diabetic mice. Evid Based Complement Alternat Med 2014:461685
Jeong SI, Kwak DH, Lee S, Choo YK, Woo WH, Keum KS, Choi BK, Jung KY (2005) Inhibitory effects of Cnidium officinale Makino and Tabanus fulvus Meigan on the high glucose-induced proliferation of glomerular mesangial cells. Phytomedicine 12:648–655
de la Cruz J, Kim DH, Hwang SG (2014) Anti cancer effects of Cnidium officinale Makino extract mediated through apoptosis and cell cycle arrest in the HT-29 human colorectal cancer cell line. Asian Pac J Cancer Prev 15:5117–5121
Jeong JB, Park JH, Lee HK, Ju SY, Hong SC, Lee JR, Chung GY, Lim JH, Jeong HJ (2009) Protective effect of the extracts from Cnidium officinale against oxidative damage induced by hydrogen peroxide via antioxidant effect. Food Chem Toxicol 47:525–529
Park HR, Jo SK, Choi NH, Jung U (2012) HemoHIM ameliorates the persistent down-regulation of Th1-like immune responses in fractionated γ-irradiated mice by modulating the IL-12p70-STAT4 signaling pathway. Radiat Res 177:676–684
Kwak DH, Kim JK, Kim JY, Jeong HY, Keum KS, Han SH, Rho YI, Woo WH, Jung KY, Choi BK, Choo YK (2002) Anti-angiogenic activities of Cnidium officinale Makino and Tabanus bovinus. J Ethnopharmacol 81:373–379
Lee YM, Lee YR, Kim CS, Jo K, Sohn E, Kim JS, Kim J (2016) Cnidium officinale extract and butylidenephthalide inhibits retinal neovascularization in vitro and in vivo. BMC Complement Altern Med 16:231
Ku SK, Kim H, Kim JW, Kang KS, Lee HJ (2017) Ameliorating effects of herbal formula hemomine on experimental subacute hemorrhagic anemia in rats. J Ethnopharmacol 198:205–213
Lee WS, Shin JS, Jang DS, Lee KT (2016) Cnidilide, an alkylphthalide isolated from the roots of Cnidium officinale, suppresses LPS-induced NO, PGE2, IL-1β, IL-6 and TNF-α production by AP-1 and NF-κB inactivation in RAW 264.7 macrophages. Int Immunopharmacol 40:146–155
Tomoda M, Ohara N, Shimizu N, Gonda R (1994) Characterization of a novel heteroglucan from the rhizome of Cnidium officinale exhibiting high reticuloendothelial system-potentiating and anti-complementary activities. Biol Pharm Bull 17:973–976
Ozaki Y, Sekita S, Harada M (1989) Centrally acting muscle relaxant effect of phthalides (ligustilide, cnidilide and senkyunolide) obtained from Cnidium officinale Makino. Yakugaku Zasshi 109:402–406
Bae KE, Choi YW, Kim ST, Kim YK (2011) Components of rhizome extract of Cnidium officinale Makino and their in vitro biological effects. Molecules 16:8833–8847
Kim JM, Lee P, Son D, Kim H, Kim SY (2003) Falcarindiol inhibits nitric oxide-mediated neuronal death in lipopolysaccharide-treated organotypic hippocampal cultures. Neuroreport 14:1941–1944
Ma Y, Pope RM (2005) The role of macrophages in rheumatoid arthritis. Curr Pharm Des 11:569–580
Dickhout JG, Basseri S, Austin RC (2008) Macrophage function and its impact on atherosclerotic lesion composition, progression, and stability: the good, the bad, and the ugly. Arterioscler Thromb Vasc Biol 28:1413–1415
Patel SK, Janjic JM (2015) Macrophage targeted theranostics as personalized nanomedicine strategies for inflammatory diseases. Theranostics 5:150–172
Venkatesan T, Park EJ, Choi YW, Lee J, Kim YK (2017) Anti-inflammatory activity of Ternstroemia gymnanthera stem bark extracts in bacterial lipopolysaccharide-stimulated RAW264.7 murine macrophage cells. Pharm Biol 55:837–846
Cekici A, Kantarci A, Hasturk H, Van Dyke TE (2014) Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000 64:57–80
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210
Fujiwara N, Kobayashi K (2005) Macrophages in inflammation. Curr Drug Targets Inflamm Allergy 4:281–286
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737
Jou IM, Lin CF, Tsai KJ, Wei SJ (2013) Macrophage-mediated inflammatory disorders. Mediators Inflamm 2013:316482
Kim SH, Lee TH, Lee SM, Park JH, Park KH, Jung M, Jung H, Mohamed MA, Baek NI, Chung IS, Kim J (2015) Cynandione A attenuates lipopolysaccharide-induced production of inflammatory mediators via MAPK inhibition and NF-κB inactivation in RAW264.7 macrophages and protects mice against endotoxin shock. Exp Biol Med 240:946–954
Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, Unkeless JC, Mayer L, Xiong H (2009) AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem 284:24006–24016
Lee YS, Lan Tran HT, Van Ta Q (2009) Regulation of expression of matrix metalloproteinase-9 by JNK in Raw 264.7 cells: presence of inhibitory factor(s) suppressing MMP-9 induction in serum and conditioned media. Exp Mol Med 41:259–268
Lahti A, Jalonen U, Kankaanranta H, Moilanen E (2003) c-Jun NH2-terminal kinase inhibitor anthra(1,9-cd)pyrazol-6(2H)-one reduces inducible nitric-oxide synthase expression by destabilizing mRNA in activated macrophages. Mol Pharmacol 64:308–315
Koide N, Ito H, Mu MM, Sugiyama T, Hassan F, Islam S, Mori I, Yoshida T, Yokochi T (2005) Inhibition of extracellular signal-regulated kinase 1/2 augments nitric oxide production in lipopolysaccharide-stimulated RAW264.7 macrophage cells. FEMS Immunol Med Microbiol 45:213–219
Arkusz J, Stepnik M, Lewińska D, Stańczyk M, Palus J, Dziubałtowska E (2007) Interleukin-1beta expression in murine J774A.1 macrophages exposed to platinum compounds: the role of p38 and ERK 1/2 mitogen-activated protein kinases. Toxicol In Vitro 21:371–379
Kim JB, Han AR, Park EY, Kim JY, Cho W, Lee J, Seo EK, Lee KT (2007) Inhibition of LPS-induced iNOS, COX-2 and cytokines expression by poncirin through the NF-kappaB inactivation in RAW 264.7 macrophage cells. Biol Pharm Bull 30:2345–2351
Shin KM, Shen L, Park SJ, Jeong JH, Lee KT (2009) Bis-(3-hydroxyphenyl) diselenide inhibits LPS-stimulated iNOS and COX-2 expression in RAW 264.7 macrophage cells through the NF-kappaB inactivation. J Pharm Pharmacol 61:479–486
Yun KJ, Koh DJ, Kim SH, Park SJ, Ryu JH, Kim DG, Lee JY, Lee KT (2008) Anti-inflammatory effects of sinapic acid through the suppression of inducible nitric oxide synthase, cyclooxygase-2, and proinflammatory cytokines expressions via nuclear factor-kappaB inactivation. J Agric Food Chem 56:10265–10272
Guo D, Li JR, Wang Y, Lei LS, Yu CL, Chen NN (2014) Cyclovirobuxinum D suppresses lipopolysaccharide-induced inflammatory responses in murine macrophages in vitro by blocking JAK-STAT signaling pathway. Acta Pharmacol Sin 35:770–778
Qi Z, Yin F, Lu L, Shen L, Qi S, Lan L, Luo L, Yin Z (2013) Baicalein reduces lipopolysaccharide-induced inflammation via suppressing JAK/STATs activation and ROS production. Inflamm Res 62:845–855
Kou X, Qi S, Dai W, Luo L, Yin Z (2011) Arctigenin inhibits lipopolysaccharide-induced iNOS expression in RAW264.7 cells through suppressing JAK-STAT signal pathway. Int Immunopharmacol 11:1095–1102
Zhu ZG, Jin H, Yu PJ, Tian YX, Zhang JJ, Wu SG (2013) Mollugin inhibits the inflammatory response in lipopolysaccharide-stimulated RAW264.7 macrophages by blocking the Janus kinase-signal transducers and activators of transcription signaling pathway. Biol Pharm Bull 36:399–406
Ohmori Y, Hamilton TA (2001) Requirement for STAT1 in LPS-induced gene expression in macrophages. J Leukoc Biol 69:598–604
Samavati L, Rastogi R, Du W, Hüttemann M, Fite A, Franchi L (2009) STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol Immunol 46:1867–1877
Funding
The funding was provided by Forest Science & Technology Projects, Forest Service, Republic of Korea (Grant No. S211316L010110), Kookmin University (KR) research grant (Grant No. 2016)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Venkatesan, T., Choi, YW., Lee, J. et al. Falcarindiol inhibits LPS-induced inflammation via attenuating MAPK and JAK-STAT signaling pathways in murine macrophage RAW 264.7 cells. Mol Cell Biochem 445, 169–178 (2018). https://doi.org/10.1007/s11010-017-3262-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3262-z