
Abstract
Altered microRNA expression is associated with tumor proliferation, metastasis, and tumorigenesis. In this study, we studied the role of miR-3117 in hepatocellular carcinoma (HCC) cell proliferation and found that miR-3117 was upregulated in HCC tissues and cells. MTT assay, soft agar growth assay, BrdU assay, and cell cycle assay revealed that miR-3117 overexpression promoted HCC HepG2 cell proliferation and that knockdown of miR-3117 suppressed HepG2 proliferation. Mechanism analysis suggested PH domain and leucine-rich repeat protein phosphatase-like (PHLPPL) as the target of miR-3117. Luciferase reporter assay suggested that miR-3117 directly binds to the 3′UTR of PHLPPL. Double knockdown of miR-3117 and PHLPPL copied the phenotypes caused by miR-3117 overexpression, suggesting that miR-3117 contributes to the proliferation of HepG2 by targeting PHLPPL. Our study provided a target for HCC therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
HCC is one of the most common malignancies and the second common cause of cancer-related death worldwide [1]. HBV and HCV infections, aflatoxin exposure, chronic alcohol consumption, and non-alcoholic fatty liver disease are the risk factors for HCC [2, 3]. Despite the development of surgery and chemotherapeutics such as Sorafenib [4, 5], the survival rate has not improved significantly.
microRNA constitutes non-coding RNA of about 22 nt long, and silences the target gene expression by base-pairing with the complementary sequences of 3′UTR of mRNAs, resulting in mRNA degradation or/and translation suppression [6]. Many microRNAs have been reported to regulate the proliferation, tumorigenesis, and metastasis of HCC. miR-183-96-182 cluster promotes HCC cell proliferation, invasion, and metastasis by targeting EFNA5, PDCD4, SOCS-6, FOXO1, FOXO3, and CEBPA [7]. miR-133a is a tumor suppressor of HCC and is negatively correlated with tumor differentiation, TNM stage, and lymph node metastasis. Its overexpression inhibits cell proliferation, migration, invasion, and tumorigenesis. Molecular mechanism analysis reveals that IGF-1R is the target of miR-133a, and inhibition of IGF-1R can inactivate Akt, and ERK signaling, leading to the suppression of HCC progression [8]. Neerincx et al. used RNA-seq to analyze miRNA expression profiles in paired non-cancerous primary and metastatic colorectal cancer tissues, and found five microRNAs including miR-3117 to have specific expression in advanced colorectal cancer tissues [9]. However, the role of miR-3117 in tumor progression has not been studied yet. In this study, we studied the expression of miR-3117 in HCC tissues and the role of miR-3117 in the proliferation of HCC HepG2 cell. We also studied the molecular mechanisms of miR-3117 in regulating HCC proliferation. We found that miR-3117 contributes to HepG2 proliferation by targeting PHLPPL.
Materials and methods
Cell culture
HCC cell lines Hep3B, MHCC97H, MHCC97L, Huh7, QGY-7703, HepG2, and HCCC-9810 were maintained in DMEM medium (Hyclone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (GIBCO, Carlsbad, CA) in a humidified atmosphere of 5% CO2 at 37 °C. The immortal hepatic cell lines LO2 and THLE3 were cultured in bronchial epithelial growth medium (Clonetics Corporation, Walkersville, MD) supplemented with 5 ng/mL epithelial growth factor (EGF), 70 ng/mL of phosphoethanolamine, and 10% of FBS.
Clinical specimens
Eight pairs of surgical HCC specimens (T) and matched adjacent normal liver tissues (ANT) were obtained from the Department of Hepatopathy, Liaocheng People’s Hospital. Ethical approval for this study was provided by the Institute Ethics Committee. All patients gave their informed consent in written format. The clinical specimens were snap-frozen in liquid nitrogen and were stored until use.
Oligonucleotides and transfection
miR-3117 mimic and inhibitor, mutational miR-3117 mimic, small interference RNAs (siRNAs) of PHLPPL and their negative control were synthesized by Guangzhou RiboBio Co., Ltd. Oligonucleotides were transfected into HepG2 cell using Lipofectamine 2000 (Life Technologies, Carlsbad, CA).
Quantitative reverse transcription-PCR (qRT-PCR)
miRNA was isolated from cells or tissues using mirVana miRNA Isolation Kit (Ambion, Austin, TX). cDNA was synthesized using TaqMan miRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and miR-3117 specific primers (Applied Biosystems). miR-3117 expression was detected using specific primers and TaqMan Universal PCR Master Mix (Applied Biosystems) on a Bio-Rad IQ5 real-time PCR system (Bio-Rad). PCR for each sample was run in triplicate. miR-3117 expression was calculated based on threshold cycle (Ct) value and \(2^{{ - \Delta \Delta C_{t} }}\) method. U6 was used as an endogenous control for miR-3117 data normalization.
Western blot
Western blot was performed according to standard methods [8, 10]. Antibodies for PHLPPL (A300-661A, Bethyl Laboratories, Montgomery, TX), p21 (sc-271532, Santa Cruz Biotech, Santa Cruz, CA, USA), and p27 (sc-776, Santa Cruz Biotech) were used, and α-Tubulin (sc-8035, Santa Cruz Biotech) was used as an internal control.
BrdU cell proliferation assay
Cells were seeded on cover slips (Fisher, Temecula, CA) and incubated with bromodeoxyuridine (BrdU) for 1 h, and then stained with anti-BrdU antibody (Upstate, Pittsburgh, PA). DAPI (Sigma, MO, USA) was used to stain nucleus. Images were acquired using a fluorescence microscope (Olympus IX81). The experiments were performed in triplicates.
Soft agar assay
Cells were trypsinized into single cell and resuspended in a complete medium with 0.3% agar. The agar–cell mixture was plated on top of a bottom layer consisting of 1% agar in complete medium, and the cells were grown for 2 weeks and colonies were stained with 1% crystal violet. The number of colonies with diameter of >0.1 mm was counted using a microscope. The experiments were performed in triplicates.
Cell cycle assay
Cells were harvested and fixed in 70% ethanol at 4 °C for overnight. The cells were incubated with 10 ug/ml RNase A and 20 ug/ml propidium iodide (Sigma) in the dark for 30 min at 37 °C. DNA content analysis was carried out by flow cytometry (FACSCalibur, BD Biosciences, Franklin Lakes, NJ). The experiments were performed in triplicates.
Luciferase reporter assay
The sequence of 3′UTR of PHLPPL was amplified and cloned into psiCHECK2 vector (Promega, WI, USA). For the luciferase reporter assay, cells were cotransfected with the constructed vector and miR-3117 mimic, inhibitor, mutant miR-3117 mimic, or negative control using Lipofectamine 2000 (Life Technologies). 48 h after transfection, the luciferase and renilla signals were determined using the Dual Luciferase Reporter Assay Kit (Promega) according to the instructions of manufacturer. Renilla luciferase was used for normalization. The experiments were performed in triplicates.
Statistical analysis
The data were presented as mean ± standard deviation (SD), and comparisons between the groups were analyzed using two-tailed Student’s t tests. Differences between means were considered significant at p < 0.05. SPSS 19.0 was used for statistical analyses.
Results
miR-3117 is upregulated in HCC tissues and cells
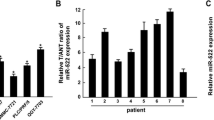
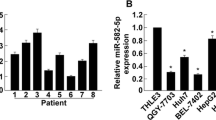
We first investigated the miR-3117 levels in HCC tissues and cells. TCGA dataset was used to analyze the miR-3117 levels in normal liver tissues and malignant liver tissues. Statistical analysis revealed that miR-3117 was significantly upregulated in malignant liver tissues compared to normal liver tissues (Fig. 1a). miR-3117 levels were also significantly higher in HCC cells (Fig. 1b). We also confirmed this result using clinical tissues collected by ourselves and found that miR-3117 was upregulated in HCC tissues compared to the adjacent non-cancerous tissues (Fig. 1c). Altogether, these findings suggested that miR-3117 is upregulated in HCC cells and tissues.

miR-3117 is overexpressed in HCC. (a) Analysis of miR-3117 expression in normal liver tissues (N) and HCC tissues (T) using TCGA dataset. (b) Analysis of miR-3117 expression in normal liver cells and HCC cells by qRT-PCR. (c) Analysis of miR-3117 expression in HCC tissues (T) and the adjacent non-cancerous tissues (ANT) by qRT-PCR. Values normalized to U6. Error bars represent the mean ± SD. *p < 0.05
miR-3117 promotes HCC cell proliferation
We overexpressed miR-3117 in HCC HepG2 cell to examine the role of miR-3117 in HCC cell proliferation, and qRT-PCR analysis suggested that miR-3117 was significantly upregulated in HepG2 transfected with miR-3117 mimic (Fig. 2a). MTT assay suggested that miR-3117 overexpression promoted the proliferation of HepG2 (Fig. 2b). Soft agar growth assay suggested that miR-3117 overexpression increased the colony formation ability in soft agar (Fig. 2c). BrdU assay found that miR-3117 overexpression significantly increased the number of BrdU-positive cells compared to the negative control (NC) (Fig. 2d). Cell cycle assay revealed that miR-3117 overexpression increased the percentage of cells in S phase and reduced the percentage of cells in G1/G0 phase (Fig. 2e). These findings suggested that miR-3117 overexpression promoted HepG2 proliferation. In a reciprocal approach, we downregulated miR-3117 in HepG2 cell (Fig. 3a). MTT assay suggested that miR-3117 knockdown inhibited the proliferation of HepG2 (Fig. 3b). Soft agar growth assay suggested that miR-3117 knockdown reduced the colony formation ability in soft agar (Fig. 3c). BrdU assay found that miR-3117 knockdown significantly reduced the number of BrdU-positive cells compared to the negative control (NC) (Fig. 3d). Cell cycle assay revealed that miR-3117 knockdown reduced the percentage of cells in S phase and increased the percentage of cells in G1/G0 phase (Fig. 3e). Altogether, miR-3117 contributed to the proliferation of HCC cell HepG2.

miR-3117 overexpression promotes HCC HepG2 cell proliferation. (a) Analysis of miR-3117 expression in HepG2 cell transfected with miR-3117 mimic and negative control (NC). Values normalized to U6. (b) Proliferation curves of HepG2 cells transfected with miR-3117 mimic and NC. (c) Soft agar growth assay of HepG2 cells transfected with miR-3117 mimic and NC. (d) BrdU staining of HepG2 cells transfected with miR-3117 mimic and NC. (e) Cell cycle assay of HepG2 cells transfected with miR-3117 mimic and NC. Error bars represent the mean ± SD. *p < 0.05

miR-3117 knockdown inhibits HCC HepG2 cell proliferation. (a) Analysis of miR-3117 expression in HepG2 cell transfected with miR-3117 inhibitor (miR-3117-in) and negative control (NC). Values normalized to U6. (b) Proliferation curves of HepG2 cells transfected with miR-3117 inhibitor and NC. (c) Soft agar growth assay of HepG2 cells transfected with miR-3117 inhibitor and NC. (d) BrdU staining of HepG2 cells transfected with miR-3117 inhibitor and NC. (e) Cell cycle assay of HepG2 cells transfected with miR-3117 inhibitor and NC. Error bars represent the mean ± SD. *p < 0.05
miR-3117 promotes HCC cell proliferation through targeting PHLPPL
To screen the target of miR-3117, we utilized the prediction software TargetScan and Pictar to predict the target of miR-3117. PHLPPL (also named as PHLPP2) was its target (Fig. 4a). PHLPP which is a Ser/Thr protein phosphatase regulates many pathways, such as Ras, Akt, and PKC [11, 12]. miR-3117 overexpression suppressed PHLPPL expression, while miR-3117 knockdown increased PHLPPL expression (Fig. 4b). Meanwhile, we cloned the sequence of 3′UTR of PHLPPL, including the conserved predicted binding sites of miR-3117 into luciferase reporter vector, and transfected vector into HepG2. Wild-type miR-3117 mimic transfection resulted in a significant reduction of luciferase activity in a dose-dependent manner, whereas miR-3117 inhibitor transfection resulted in a significant increase of luciferase activity in a dose-dependent manner, but the mutant miR-3117 mimic transfection did not result in any change in the luciferase activity, suggesting that miR-3117 bound to the 3′UTR of PHLPPL (Fig. 4c). We have found that miR-3117 accelerated cell cycle progression, and hence, we also determined the levels of the two well-known cell cycle inhibitors p21 and p27 after miR-3117 overexpression or knockdown. qRT-PCR and western blot analysis suggested that miR-3117 overexpression significantly suppressed their expression both in mRNA and protein levels, and miR-3117 knockdown significantly promoted their expression both in mRNA and protein levels (Fig. 4d, e). These suggest that miR-3117 indeed promoted cell proliferation.

PHLPPL is the target of miR-3117. (a) Localization and site-specific mutagenesis of the predicted binding site of miR-3117 within the 3′UTR of PHLPPL. (b) Western blot analysis of PHLPPL expression in HepG2 cell transfected with miR-3117 mimic, inhibitor, and negative control. α-Tubulin was used as the loading control. (c) Analysis of luciferase activity of HepG2 cells cotransfected with luciferase reporter vector and miR-3117 mimic, inhibitor, mutant miR-3117 mimic, or negative control. (d) Analysis of p27 and p21 expressions in HepG2 cell transfected with miR-3117 mimic, inhibitor, or negative control by qRT-PCR. Values normalized to GAPDH. (e) Analysis of p27 and p21 expressions in HepG2 cell transfected with miR-3117 mimic, inhibitor, or negative control by western blot. α-Tubulin was used as the loading control. Error bars represent the mean ± SD. *p < 0.05
To assess whether miR-3117 contributes to HCC cell proliferation by targeting PHLPPL, we double knocked-down miR-3117 and PHLPPL in HepG2. Western blot analysis suggested PHLPPL siRNAs inhibit PHLPPL expression (Fig. 5a). Soft agar growth assay suggested that knockdown of miR-3117 and PHLPPL significantly increased the colony formation ability in soft agar (Fig. 5b). BrdU assay revealed that the number of BrdU-positive cells was significantly increased when miR-3117 and PHLPPL were knocked-down simultaneously (Fig. 5c). These results suggested the phenotype caused by the double knockdown of miR-3117 and PHLPPL was the same as caused by miR-3117 overexpression, suggesting that miR-3117 regulated HCC cell proliferation by targeting PHLPPL.

miR-3117 promotes HepG2 proliferation by targeting PHLPPL. (a) Analysis of PHLPPL expression in HepG2 cell transfected with PHLPPL siRNAs and miR-3117 inhibitor by western blot. α-Tubulin was used as the loading control. (b) Soft agar growth assay of HepG2 cells transfected with PHLPPL siRNAs and miR-3117 inhibitor. (c) BrdU staining of HepG2 cells transfected with PHLPPL siRNAs and miR-3117 inhibitor. Error bars represent the mean ± SD. *p < 0.05
Discussion
In this study, we found that miR-3117 was upregulated in HCC cells and tissues. Overexpression of miR-3117 promotes cell proliferation and accelerates cell cycle progression, while knockdown of miR-3117 inhibits cell proliferation and arrested cell cycle progression. A Ser/Thr protein phosphatase PHLPPL was the target of miR-3117 that is directly bound to the 3′UTR of PHLPPL. Double knockdown of miR-3117 and PHLPPL promoted HCC cell proliferation, confirming miR-3117 promoted HCC cell proliferation by targeting PHLPPL.
PHLPPL is the target of many microRNAs, for example, miR-224 promotes colorectal cancer and esophageal squamous cell carcinoma proliferation by repressing PHLPPL1 and PHLPPL [13, 14]. miR-141 promotes non-small cell lung cancer (NSCLC) cell proliferation by targeting PHLPPL1 and PHLPPL [15]. miR-205 contributes to NSCLC cell proliferation and blood vessel formation by targeting PTEN and PHLPPL, and in turn activates AKT/FOXO3a and AKT/mTOR pathway [16]. miR-135a promotes bladder cancer proliferation by targeting PHLPPL and FOXO1 [17]. miR-372 promotes glioma cell proliferation and invasion by targeting PHLPPL [18]. These results suggest that PHLPPL is a bona fide tumor suppressor. Our study suggested that PHLPPL is the target of miR-3117 and inhibited HCC cell proliferation. PHLPPL inactivates many pathways which promote HCC development and progression. It can interact with IKKβ and inactivates NF-κB pathway by inhibiting its phosphorylation. In addition to this, IKKβ activation requires Bcl10-MALT1 ubiquitin ligase complex which results in NEMO/IKKγ non-degradative ubiquitination and IKKβ activation. PHLPPL also forms complex with Bcl10 and competitively displaces MALT1, leading to NF-κB inactivation [19]. PHLPPL also inhibits Akt pathway by dephosphorylation of Akt1 and Akt3, leading to the arrest of cell cycle progression and induction of apoptosis [12, 20]. Both NF-κB [21, 22] and Akt [23, 24] pathways promote HCC progression; therefore, we thought miR-3117 contributes to HCC proliferation by targeting PHLPPL which inactivates NF-κB and Akt pathway.
In summary, we found that miR-3117 contributes to HCC cell proliferation by targeting PHLPPL, and might provide a target for HCC therapy.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65(2):87–108. doi:10.3322/caac.21262
Testino G, Leone S, Borro P (2014) Alcohol and hepatocellular carcinoma: a review and a point of view. World J Gastroenterol 20(43):15943–15954. doi:10.3748/wjg.v20.i43.15943
Pezzuto F, Izzo F, Buonaguro L, Annunziata C, Tatangelo F, Botti G, Buonaguro FM, Tornesello ML (2016) Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget. doi:10.18632/oncotarget.9801
Cheng S, Yang J, Shen F, Zhou W, Wang Y, Cong W, Yang GS, Cheng H, Hu H, Gao C, Guo J, Li A, Meng Y, Jiang X, Yang Y, Qian G, Luo M, Hu B, Man X, Zhang B, Su C, Zhou F, Li N, Shi J, Wang M, Zheng Y, Guo W, Sun J, Wang H, Lau WY, Wu MC (2016) Multidisciplinary management of hepatocellular carcinoma with portal vein tumor thrombus—Eastern Hepatobiliary Surgical Hospital consensus statement. Oncotarget. doi:10.18632/oncotarget.8386
Harding JJ, Abou-Alfa GK (2014) Treating advanced hepatocellular carcinoma: how to get out of first gear. Cancer 120(20):3122–3130. doi:10.1002/cncr.28850
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Ma Y, Liang AJ, Fan YP, Huang YR, Zhao XM, Sun Y, Chen XF (2016) Dysregulation and functional roles of miR-183-96-182 cluster in cancer cell proliferation, invasion and metastasis. Oncotarget. doi:10.18632/oncotarget.8715
Zhang W, Liu K, Liu S, Ji B, Wang Y, Liu Y (2015) MicroRNA-133a functions as a tumor suppressor by targeting IGF-1R in hepatocellular carcinoma. Tumour Biol 36(12):9779–9788. doi:10.1007/s13277-015-3749-8
Neerincx M, Sie DL, van de Wiel MA, van Grieken NC, Burggraaf JD, Dekker H, Eijk PP, Ylstra B, Verhoef C, Meijer GA, Buffart TE, Verheul HM (2015) MiR expression profiles of paired primary colorectal cancer and metastases by next-generation sequencing. Oncogenesis 4:e170. doi:10.1038/oncsis.2015.29
Wang YQ, Ren YF, Song YJ, Xue YF, Zhang XJ, Cao ST, Deng ZJ, Wu J, Chen L, Li G, Shi KQ, Chen YP, Ren H, Huang AL, Tang KF (2014) MicroRNA-581 promotes hepatitis B virus surface antigen expression by targeting Dicer and EDEM1. Carcinogenesis 35(9):2127–2133. doi:10.1093/carcin/bgu128
Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, Stein J, Stein GS, Iglehart JD, Shi Q, Pardee AB (2010) Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol Cell 38(4):512–523. doi:10.1016/j.molcel.2010.03.017
Brognard J, Niederst M, Reyes G, Warfel N, Newton AC (2009) Common polymorphism in the phosphatase PHLPP2 results in reduced regulation of Akt and protein kinase C. J Biol Chem 284(22):15215–15223. doi:10.1074/jbc.M901468200
Liao WT, Li TT, Wang ZG, Wang SY, He MR, Ye YP, Qi L, Cui YM, Wu P, Jiao HL, Zhang C, Xie YJ, Wang JX, Ding YQ (2013) microRNA-224 promotes cell proliferation and tumor growth in human colorectal cancer by repressing PHLPP1 and PHLPP2. Clin Cancer Res 19(17):4662–4672. doi:10.1158/1078-0432.CCR-13-0244
He X, Zhang Z, Li M, Li S, Ren L, Zhu H, Xiao B, Shi R (2015) Expression and role of oncogenic miRNA-224 in esophageal squamous cell carcinoma. BMC Cancer 15:575. doi:10.1186/s12885-015-1581-6
Mei Z, He Y, Feng J, Shi J, Du Y, Qian L, Huang Q, Jie Z (2014) MicroRNA-141 promotes the proliferation of non-small cell lung cancer cells by regulating expression of PHLPP1 and PHLPP2. FEBS Lett 588(17):3055–3061. doi:10.1016/j.febslet.2014.06.020
Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y, Zhu X, Chen B, Wu J, Li M (2013) miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res 73(17):5402–5415. doi:10.1158/0008-5472.CAN-13-0297
Mao XP, Zhang LS, Huang B, Zhou SY, Liao J, Chen LW, Qiu SP, Chen JX (2015) Mir-135a enhances cellular proliferation through post-transcriptionally regulating PHLPP2 and FOXO1 in human bladder cancer. J Transl Med 13:86. doi:10.1186/s12967-015-0438-8
Chen X, Hao B, Han G, Liu Y, Dai D, Li Y, Wu X, Zhou X, Yue Z, Wang L, Cao Y, Liu J (2015) miR-372 regulates glioma cell proliferation and invasion by directly targeting PHLPP2. J Cell Biochem 116(2):225–232. doi:10.1002/jcb.24949
Agarwal NK, Zhu X, Gagea M, White CL 3rd, Cote G, Georgescu MM (2014) PHLPP2 suppresses the NF-kappaB pathway by inactivating IKKbeta kinase. Oncotarget 5(3):815–823. doi:10.18632/oncotarget.1774
Brognard J, Sierecki E, Gao T, Newton AC (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell 25(6):917–931. doi:10.1016/j.molcel.2007.02.017
Luedde T, Schwabe RF (2011) NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nature Rev Gastroenterol Hepatol 8(2):108–118. doi:10.1038/nrgastro.2010.213
Bao C, Li Y, Huan L, Zhang Y, Zhao F, Wang Q, Liang L, Ding J, Liu L, Chen T, Li J, Yao M, Huang S, He X (2015) NF-kappaB signaling relieves negative regulation by miR-194 in hepatocellular carcinoma by suppressing the transcription factor HNF-1alpha. Sci Signal 8:387. doi:10.1126/scisignal.aaa8441
Zhang PF, Li KS, Shen YH, Gao PT, Dong ZR, Cai JB, Zhang C, Huang XY, Tian MX, Hu ZQ, Gao DM, Fan J, Ke AW, Shi GM (2016) Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3 K/AKT signaling. Cell Death Dis 7:e2201. doi:10.1038/cddis.2015.324
Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, Guzman G, Hay N (2016) Spontaneous hepatocellular carcinoma after the combined deletion of akt isoforms. Cancer Cell 29(4):523–535. doi:10.1016/j.ccell.2016.02.008
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Additional information
Xia Cui and Qingyan Li contributed equally to this work.
Rights and permissions
About this article

Cite this article
Cui, X., Li, Q. & He, Y. miR-3117 regulates hepatocellular carcinoma cell proliferation by targeting PHLPPL. Mol Cell Biochem 424, 195–201 (2017). https://doi.org/10.1007/s11010-016-2855-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2855-2