
Abstract
Exogenous insulin therapy improves endothelial function in insulin resistant patients, indirectly indicating that nitric oxide synthase activity and NO production may be impaired. Insulin stimulates production of NO by activating a signaling pathway including insulin receptor substrate-1, phosphatidylinositol-3-kinase and protein kinase B (PKB/Akt). Angiotensin II type I (AT1) receptor-evoked oxidative stress is implicated in the inactivation of NO, impairing endothelium-dependent vasodilatation. Blocking the actions of Angiotensin II with an AT1 receptor antagonist (Losartan), has beneficial effects in patients with insulin resistance or type 2 diabetes mellitus. This study investigated whether elevated Angiotensin II influences myocardial insulin resistance, insulin signaling and NO production in a rat model of diet-induced obesity (DIO) by antagonizing the actions of the AT1 receptor with Losartan. Isolated, perfused hearts, Western blotting and flow-cytometric methods were utilized to determine myocardial function, expression and phosphorylation of key proteins and NO production, respectively. Results showed that hearts from DIO rats are insulin resistant (higher serine phosphorylation of IRS-1, lower insulin-stimulated phosphorylation of PKB/Akt and eNOS, lower NO production) and had poorer functional recovery and larger infarct development after ischaemia/reperfusion. Losartan improved the impaired functional recovery, and NO production and enhanced eNOS expression and phosphorylation and reduced infarct size in hearts from the DIO animals. Data obtained from Losartan treatment also revealed that Angiotensin II signaling modulates myocardial PKB/Akt expression. We conclude that Angiotensin II signaling exacerbates inhibition of NO production in insulin resistance and that this can be improved by AT1 antagonism.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
According to the latest statistics supplied by the International Obesity Taskforce (www.web.winltd.com), there are currently 58 million overweight people in the USA alone with reported increases in both childhood obesity and the diagnosis of type 2 diabetes mellitus. Obesity is a well-recognized risk factor for the development of type 2 diabetes [1] and a good predictor of the metabolic syndrome and coronary artery disease [2]. Insulin resistance and diabetes-associated atherosclerosis are major clinical problems and are thought to be preceded by early changes or abnormalities in endothelial function [3]. There is accumulating evidence that increased production of reactive oxygen species (ROS) plays a major role in impaired endothelium-dependent vasodilatation [4, 5]. Increased superoxide reacts rapidly with nitric oxide (NO) to form peroxynitrite thereby decreasing NO bioavailability. A major source of superoxide is the NADPH oxidase, an enzyme that is stimulated, amongst others, by Angiotensin II (Ang II) [6, 7].
Physiological concentrations of NO play an important role in maintaining normal vascular function, have vasodilatory effects, inhibit platelet aggregation and adhesion to the vascular wall and control expression of proteins involved in atherogenesis. In addition, it reduces vascular permeability and decreases the rate of oxidation of LDL to its proatherogenic form [3]. It has been suggested that NO synthase (NOS) activity and the production of NO are chronically impaired in insulin resistance and type 2 diabetes [3]. This view is indirectly supported by the observation that exogenous insulin therapy appears to improve endothelial function in patients with type 2 diabetes [8].
Insulin stimulates production of NO in endothelial cells and cardiomyocytes by activating a signaling pathway that involves insulin receptor substrate-1 (IRS-1), phosphatidylinositol-3-kinase (PI3-K) and protein kinase B (PKB/Akt) [9, 10]. PKB/Akt activates endothelial NOS (eNOS) via phosphorylation on Ser1177 [11]. Overexpression of an inhibitory mutant of either PI3-K or PKB/Akt, inhibits insulin-stimulated NO production [9].
The renin-angiotensin system (RAS) is a central component of the cardiovascular system. Ang II, the biologically active peptide of the RAS, mediates physiological effects of vasoconstriction and blood pressure regulation. Recent studies have highlighted a pivotal role for Ang II in the development of atherosclerosis, myocardial infarction as well as vascular and myocardial remodeling [12, 13]. Signal transduction via the Ang II type 2 (AT2) receptor has been implicated in the regulation of expression of eNOS both in endothelial cells [6] as well as in murine cardiomyocytes [14]. Activation of both the mitogen-activated kinases Erk 1/2 and p38 MAP kinase may be involved in this process. Ang II type I (AT1) receptor-evoked oxidative stress has in turn been implicated in the inactivation of NO, leading to impaired endothelium-dependent vasodilatation [12, 13].
There is increasing evidence that Ang II interferes with insulin signaling by affecting insulin-induced tyrosine phosphorylation of IRS-1, impeding its interaction with PI3-K [15, 16]. It has also been shown that blocking the actions of Ang II with an AT1 receptor antagonist like Losartan, has beneficial effects in animal models [17], as well as patients with insulin resistance or type 2 diabetes mellitus [1, 18].
In view of the above, we investigated whether elevated Ang II influenced myocardial insulin resistance, insulin signaling and NO production in a rat model of insulin resistance caused by diet-induced obesity (DIO). Our results demonstrated impaired NO production in hearts from the DIO animals. This could be improved by treatment with Losartan, an AT1 receptor antagonist. In addition, we showed that Losartan treatment of DIO rats resulted in higher myocardial eNOS and PKB/Akt expression levels. Losartan treatment furthermore protected the hearts of DIO animals against ischaemia/reperfusion injury.
Materials and methods
Animal model
Male Wistar rats (180–200 g) were randomly divided into control and diet-induced obese (DIO) animals. DIO animals were placed on a diet supplemented with sucrose and condensed milk for a period of 16 weeks to induce obesity by hyperphagia [19]. Animals had free access to food and water and were kept on a 12 h day/night cycle in an AAALAC accredited facility. After 15 weeks on the respective diets, half of each group of animals was treated with 10 mg/kg/day Losartan supplied in their drinking water. After 1 week treatment, rats were anaesthetized by intraperitoneal injection of sodium pentobarbital (160 mg/kg) before killing. The study conformed to the Guide for the Care and Use of Laboratory Animals of the NIH (Publication No. 85-23, revised 1996).
Materials
Collagenase type II were from Worthington; BSA fatty acid free from Boehringer Mannheim; all antibodies were from Cell Signaling and a 1:1,000 dilution of primary antibody was used throughout. Losartan was kindly supplied by Merck research laboratories. DHR-123, DHE and DAF were bought from Molecular Probes (Oregon, USA). Ang II levels were determined using a Euro-Diagnostica kit (Malmö, Sweden) while AmprepTM Phenyl PH Minicolumns (Amersham) were used to enrich Ang II levels. All other chemicals were of the highest grade available.
Methods
Perfusion of isolated hearts
Hearts were excised from anaesthetized animals and mounted on a perfusion rig. Two different perfusion protocols were used in the study.
-
1.
Hearts were perfused in the Langendorff mode using Krebs–Henseleit (KH) bicarbonate buffer containing in mM: NaCl 119; NaHCO3 24.9; KCL 4.74; KH2PO4 1.19; MgSO4 0.6; Na2SO4 0.6; CaCl2 1.25; glucose 10. The buffer was gassed with 95% O2/5% CO2. Hearts from control and DIO animals were randomly perfused for either 30 min with KH or 15 min with KH followed by 15 min stimulation with 0.3 mIU/ml insulin. Afterwards, hearts were snap-frozen and stored in liquid nitrogen. This tissue was used for determination of proteins via western blotting.
-
2.
Hearts were perfused in the working mode with KH at 15 cm H2O preload, 100 cm H2O afterload and a constant temperature of 37°C. The latter was monitored via a temperature probe inserted into the coronary sinus. After a 40 min stabilization period, the left anterior descending artery was occluded by tying it off with a silk suture to induce regional ischaemia for a period of 35 min. This was followed by 60 min reperfusion consisting of 10 min retrograde, 20 min work, 30 min retrograde perfusion. Infarct size was determined as described previously and expressed as a percentage of the area at risk [20]. Aortic output was measured during the stabilization period and again after the ischaemic period. The post-ischaemic aortic output was calculated as a percentage of the pre-ischaemic values to determine recovery.
Preparation of lysates and western blotting
Frozen tissues were pulverized with a liquid nitrogen pre-cooled mortar and pestle and then extracted in lysis buffer containing in mM: Tris–HCl 20 (pH 7.5), EGTA 1, EDTA 1, NaCl 150, Na2VO3 1, beta-glycerophosphate 1, sodium-pyrophosphate 2.5, PMSF 0.3, Triton X-100 1% (v/v) plus 10 μg/ml leupeptin and aprotinin, respectively, using a Polytron PT10 homogenizer, 2 × 4 s, setting 4. Lysates were cleared from particulate matter by centrifuging for 15 min at 14,000 rpm in a microfuge. Equal amounts of cytosolic proteins were separated on an SDS poly-acrylamide gel and electro-transferred to ImmobilonTM-P membranes. Transfer and equal loading of proteins were visualized and monitored with Ponceau Red reversible stain. The membranes were blocked for 2 h in Tris-buffered saline (TBS) containing 0.1% Tween-20 and 5% non-fat milk powder. Membranes were probed with primary antibodies directed against: total eNOS protein and Ser1177 phosphorylated eNOS, total PKB/Akt protein and Ser473 phosphorylated PKB/Akt or total IRS-1 and Ser612 phosphorylated IRS-1 protein followed by a horse-radish peroxidase coupled anti-rabbit secondary antibody. Bands were visualized using the ECL detection system and quantified by laser scanning densitometry and suitable software (Silk-science).
Preparation of ventricular cardiomyocytes
The myocyte isolation technique was based on a method described previously [21]. Hearts were retrogradely perfused with a nominally calcium-free buffer, solution A (containing in mM: KCl 6, Na2HPO4 1, NaH2PO4 0.2, MgSO4 1.4, NaCl 128, HEPES 10, d-glucose 5.5 and pyruvic acid 2), to rinse blood from the coronary vessels. After 5 min, perfusion was switched to a digestion buffer, solution B (solution B = solution A containing 0.7% fatty acid free BSA + 0.1% collagenase + 15 mM 2,3-butanedione monoxime (BDM)). Calcium (200 μM in total) was added at 20 and 25 min, respectively, of total perfusion time. After 30–35 min, the heart was removed from the perfusion apparatus, the atria removed and the ventricles gently torn apart. The ventricular tissue was then placed in a post-perfusion digestion buffer, solution C (solution C = 1 part solution A + 1 part solution B + 200 μM calcium + 1% fatty acid free BSA), followed by incubation in a shaking water bath (37°C) for 15 min, and a step-wise calcium re-administration period to render calcium-tolerant cells (200 μM/min for 4 min and 250 μM during the final minute; total calcium: 1.25 mM). After filtering and gentle centrifugation (100 rpm × 3 min) the supernatant was removed and the pellet resuspended in an incubation solution, solution D (solution D = solution A + 1.25 mM CaCl2 + 2% fatty acid free BSA), allowed to settle through solution D for 5 min and the pellet resuspended in solution D. Cells were stabilized for 1 h by slow rotation at room temperature. The average digested heart yielded 3–5 × 106 ventricular myocytes. Investigations were repeated on myocyte preparations from different hearts, with sample sizes varying from n = 6 to 8. The overwhelming majority of cells isolated in this manner (>70%) demonstrated typical rod-shaped morphology with clear striations, as assessed by light microscopy.
Flow-cytometric determinations
Oxygenated control conditions were simulated by incubating isolated cardiomyocytes in solution D (500,000 cells/35 mm petri dish) in a standard tissue culture incubator under an O2 atmosphere (21% O2, 5% CO2, 40–60% humidity, 37°C) for the full duration of the experiments (180 min).
Protocols for the detection of NO, peroxynitrite (ONOO−) and superoxide were employed as previously described for endothelial cells and isolated cardiomyocytes, with minor modifications [7, 22, 23]. The specificity of all probes has previously been confirmed. At the beginning of experiments (t = 0 min), samples (500,000 cells/sample) were loaded with non-limiting concentrations of the cell-permeable fluorescent probes diaminofluorescein-diacetate (DAF-2/DA, 10 μM in 1 ml solution D), di-hydrorhodamine-123 (DHR-123, 2 μM in 1 ml solution D) or di-hydro-ethidium (DHE, 20 μM in 1 ml solution D) for detection of intracellular production of NO, ONOO− and superoxide, respectively. These probes were present throughout the experiment. At t = 180 min, probes were washed out from all samples and cells resuspended in probe-free solution D followed by immediate FACS analysis. Exposure to light was avoided as far as possible in all samples due to the light sensitivity of the fluorescent probes. All 3 probes were always used comparatively by dividing the same myocyte preparation into 3 fractions. Reported n values indicate myocyte preparations from n different hearts.
Angiotensin II levels
Blood was collected at the time of killing, allowed to clot on ice, centrifuged (14,000 rpm for 15 min) and stored at −20°C. Ang II was enriched in samples by passing the serum through an Amprep cartridge according to the manufacturer’s specifications. The final eluates were freeze dried and stored until assayed. Ang II levels in these extracts were determined using a RIA kit. Each assay included a standard curve from 0 to 150 pmol/l.
Statistical analyses
Data are presented as mean ± SEM. Comparisons between multiple groups were performed by either one-way or two-way ANOVA followed by Bonferroni post-hoc test (Graph-Pad Prism 5). A value of P < 0.05 was considered statistically significant.
Results
Animal model
We have utilized a previously described [19] model of hyperphagia-induced obesity in rats, generating animals that suffer from insulin resistance and the metabolic syndrome [24].
As shown in Table 1, after 16 weeks on diet (DIO), animals gained significantly more weight than their age-matched controls on normal rat chow, their intraperitoneal fat depots (IP fat) were significantly more and their fasting blood glucose- and insulin-levels were significantly elevated. One week of a low dose of Losartan treatment did not change the blood glucose levels.
Ang II levels were also significantly elevated in the DIO animals. Losartan treatment resulted in serum Ang II levels of such magnitude that values of both control treated and DIO treated animals were outside the sensitivity range of the assay, therefore indicated as >150 pmol/l.
Demonstration of insulin resistance
Stimulating hearts from control as well as DIO, untreated animals ex vivo with 0.3 mIU/ml insulin resulted in significant elevation of phosphorylation of PKB/Akt on Ser473 above levels in unstimulated hearts (Fig. 1a). However, the response in hearts from DIO animals was very low in comparison to control hearts.

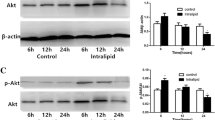
Western blotting was performed as described in “Materials and methods” section using equal amounts of protein per lane. In a and b lysates prepared from freeze-clamped ventricular tissue were used and the graphs represent insulin-stimulated phosphorylation of Ser473 on PKB/Akt and Ser1177 on eNOS with and without Losartan treatment (* P < 0.05, ** P < 0.01 and *** P < 0.001 as determined by a 2-way ANOVA, n = 5–8 individual hearts). c represents basal Ser612 phosphorylation on IRS-1 determined by FACS analysis on isolated cardiomyocytes. * P < 0.05 control vs. DIO, n = 4 preparations, and d shows the total IRS-1 protein in the same preparations analysed in tandem
Similarly, insulin stimulation of hearts from control, untreated animals resulted in significantly higher levels of phospho-eNOS (Ser1177) detected than in hearts from control animals with no elevation in hearts from DIO animals (Fig. 1b).
Furthermore, under basal conditions, significantly higher levels of Ser612 phosphorylation of IRS-1 were detected in cardiomyocytes prepared from DIO animals (Fig. 1c) while expression of IRS-1 protein was lower (Fig. 1d).
Together, these results are a clear indication of insulin resistance being present in hearts from the DIO animals, inferring down regulation of the PI-3-K, PKB/Akt, eNOS signaling pathway.
Losartan treatment (10 mg/kg/day for 1 week) per se did not stimulate phosphorylation of PKB/Akt nor did it enhance insulin-stimulated phosphorylation of this kinase (Fig. 1a) in either experimental group. However, both basal and insulin-stimulated phosphorylation of eNOS were enhanced in hearts from the DIO animals (Fig. 1b). The representative blots are shown in Fig. 2a with a Ponceau red stain to show equal loading in Fig. 2c.

Representative western blots including beta-tubulin to demonstrate equal expression of a different protein of total and phosphorylated PKB/Akt and eNOS. Lysates from frozen hearts with or without treatment were subjected to PAGE as described in “Methods” section: 30 μg protein per lane, 10% PAGE for PKB/Akt and 80 μg protein per lane, 7.5% PAGE for eNOS. a depicts phosphorylation stimulated by insulin in comparison to control protein levels. b shows the total protein levels with and without Losartan treatment while c is a Ponceau Red stain to show equal loading of protein
Functional performance of hearts
Hearts from control animals and DIO animals with or without Losartan treatment were perfused as described in “Methods” section. Baseline parameters of aortic output and total work performed were not different in the groups of animals (Table 2). However, on reperfusion after 35 min regional ischaemia, hearts of DIO recovered significantly worse than hearts from control animals and produced less total work (Fig. 3a, b). This poor aortic output recovery as well as total work performed of the DIO animals, were significantly improved by Losartan treatment (Table 2; Fig. 3). In addition, the infarct size that developed, expressed as a percentage of the area at risk, was significantly larger in hearts of the DIO animals versus controls (Fig. 4). The area at risk did not differ between control and DIO hearts. The protective effects of Losartan were further evidenced by the significantly smaller infarcts found in control and DIO animals after Losartan treatment (Fig. 4).

a Functional recovery of aortic output after 35 min regional ischaemia of perfused hearts from control, DIO and Losartan treated animals (n = 6–9). b Recovery of the calculated total work performed in hearts after 35 min regional ischaemia of perfused hearts with significance as indicated (n = 6–9)

Infarct development expressed as a percentage of the area at risk of infarction after 35 min regional ischaemia of the same hearts as in Fig. 2, with significance as indicated (n = 6–9)
FACS analysis
Figure 5a shows a typical FACS image of cardiomyocytes indicating forward scatter on the x-axis and side scatter on the y-axis with the window indicating the gating that was used consistently. Figure 5b, c are representative fluorescent scans of DAF fluorescence of isolated cardiomyocytes prepared from control (Fig. 5b) and DIO (Fig. 5c) animals, with or without Losartan treatment.

a A typical scatter plot of a cardiomyocyte preparation as shown by FACS scanning. The area normally gated is encircled. b The shift in fluorescence in cells from control animals with or without Losartan treatment; and c the same analysis on cells from DIO animals with or without Losartan treatment
NO production by cardiomyocytes
DAF fluorescence, as a direct measure of the ability of cardiomyocytes to produce NO, showed that cells from DIO animals produced significantly less NO than cells from control animals (Fig. 6a). Treating DIO rats with Losartan restored their ability to produce NO. Surprisingly, treating control animals with Losartan resulted in lower NO production measured in the cardiomyocytes.

FACS analysis and probes specific for the different ROS species were used to determine their intracellular levels in cardiomyocytes prepared from control or DIO animals, with or without Losartan treatment. Data represents 7–11 individual preparations and P values (* P < 0.05) were determined by 2-way ANOVA
Peroxynitrite and superoxide production by cardiomyocytes
The 2 probes used for measurement of ONOO− and superoxide production showed no change in the production of these free radicals either in myocytes taken from DIO animals or from animals treated with Losartan versus the respective controls (Fig. 6b, c).
Expression of key proteins
PKB/Akt
While hearts from DIO animals were more insulin resistant (Fig. 1), the condition per se resulted in significantly elevated levels of PKB/Akt expression (Fig. 7a). After 1 week treatment with Losartan, hearts from control animals presented with significantly attenuated expression of PKB/Akt, showing hitherto unrecognized regulation of the expression of this key protein via Ang II signaling. Furthermore, in hearts from DIO animals, this regulation was absent and Losartan treatment resulted in an additional elevation in the expression of PKB/Akt, underscoring regulation of this pathway via Ang II signaling. A representative blot is shown in Fig. 1b.

eNOS
In order to further clarify the observations made by direct measurement of NO production by cardiomyocytes, the expression levels of eNOS protein was measured. Basal levels of expression of eNOS were not affected by DIO (Fig. 7b). However, Losartan treatment again unmasked regulation of the expression of eNOS protein via Ang II evoked signaling. In hearts from control animals, expression of eNOS was significantly attenuated after Losartan treatment while expression of this protein was significantly elevated after treatment in hearts from the DIO animals, underscoring results obtained by FACS analysis of NO production. A representative blot is shown in Fig. 2b.
Discussion
It is currently recognized worldwide that the ever-increasing incidence of obesity contributes largely to the escalation in cardiovascular disease. Changes in arterial patency because of early changes in endothelial function and development of atherosclerosis have partly been blamed for this [3, 25, 26]. These changes have been linked to insulin resistance via indirect evidence obtained in patients [2, 27]. In addition, ROS production through activation of the RAS system has been implicated in early endothelial dysfunction [12, 13] and this is supported by the observation that an AT1 receptor antagonist like Losartan, has beneficial effects in patients with insulin resistance or type 2 diabetes mellitus [1, 17, 18].
In the current study, we have addressed these issues by using a rat model of obesity induced cardiovascular dysfunction with elevated levels of Ang II and insulin resistance. In addition, this model is known to develop endothelial dysfunction [28]. We measured expression and activation of eNOS and PKB/Akt as key proteins in the insulin signaling pathway leading to NO production and correlated this with direct measurement of NO, peroxynitrite and superoxide production by cardiomyocytes from these animals with or without Losartan treatment. We furthermore subjected hearts from the different groups to ex vivo perfusion and ischaemia followed by reperfusion, to determine whether Losartan treatment afforded myocardial protection.
Animals were kept on an obesity inducing diet (DIO) for a period of 16 weeks and treated with 10 mg/kg/day Losartan for the last week of this period. According to Navarro-Cid et al. [29], even 1 mg/kg/day Losartan could prevent a rise in blood pressure and could blunt Ang II induced vasoconstriction in rats fed a high fructose diet. Body weight, IP fat weight, fasting blood glucose, insulin- and Ang II levels were all significantly elevated in the DIO animals (Table 1). In accordance with the observations of Van den Meiracker et al. [30] and Shinozaki et al. [17], Losartan treatment resulted in elevation of plasma Ang II levels, showing that this low dose of Losartan could interrupt the negative feedback mechanism on renin release [30].
Our results showed that hearts from the DIO animals were insulin resistant as evidenced by a severely reduced ability of insulin to stimulate PKB/Akt and eNOS phosphorylation (Fig. 1a, b). In conjunction with the latter, IRS-1 expression was significantly attenuated and highly serine phosphorylated (Fig. 1c, d). Several researchers [16, 31] demonstrated that Ang II signaling can increase serine phosphorylation of IRS-1 as well as serine phosphorylation of the p85 subunit of PI3-Kinase. Both of these will inhibit the interaction of PI-3-Kinase with IRS-1 and will result in reduced signaling through the pathway and lower phosphorylation of PKB/Akt and eNOS. We speculate that the treatment period we used was too short for Losartan to effectively lower blood glucose levels as Hotta et al. [32] found lower levels after 2 weeks of treatment with 3 mg/kg Losartan. That insulin resistance per se was not changed by the Losartan treatment is underscored also by the finding that we could not detect a higher level of phosphorylation of PKB/Akt protein via insulin stimulation after treatment (Fig. 1a).
Despite this, Losartan treatment translated into improved myocardial function in the DIO animals. As shown in Figs. 3 and 4, hearts from DIO animals presented with larger infarct development after regional ischaemia as well as poorer functional ability. Both of these parameters were significantly improved after Losartan treatment. In a recent article, Hotta et al. [32] described cardioprotective effects of Valsartan treatment with restoration of JAK2-PI3-K/PKB/Akt signaling and suppression of calcineurin activity. They postulated that this was the main route of protection via AT1 receptor antagonism together with alleviation of ER stress. However, in the present study, we could not demonstrate restoration of PKB/Akt signaling despite eliciting protection.
It has been proposed that Ang II signaling causes not only insulin resistance but also lowers NO production via activation of NADPH oxidase and ROS production [16, 33, 34]. Most of these effects of Ang II are mediated by signaling pathways activated via the AT1 receptor, therefore antagonism of this signaling with blockers like Losartan, should not only alleviate insulin resistance, as shown by Henriksen [34] and Hotta et al. [32] but also increase NO production, thereby improving endothelial function. By directly measuring NO production in freshly isolated adult ventricular myocytes taken from control and DIO animals, we clearly demonstrated this. NO production in myocytes from DIO animals was significantly attenuated under basal conditions (Fig. 6a) and treatment with Losartan significantly improved this. It may be argued that the enhanced NO bio-availability after Losartan treatment is as result of reduced ROS production via the NADPH system, therefore less scavenging by superoxide to form peroxynitrite. However, using established probes to measure both peroxynitrite and superoxide [22, 23], in the same cell preparation where we measured NO levels, we could not confirm a role for ROS in our observations (Fig. 6b, c). The lower basal production of NO by cardiomyocytes from the DIO animals with equal expression and phosphorylation of eNOS may reflect the contribution of the diverse other cell types present in whole heart tissue in comparison to isolated cardiomyocytes.
Concurrently, the expression of eNOS protein in DIO hearts was enhanced by Losartan treatment (Fig. 7b). Enhanced expression of eNOS after Losartan treatment corroborates the findings of Li et al. [6] in endothelial cells while Brede et al. [14] found lower eNOS expression in cardiomyocytes in an AT2 receptor deficient mouse model. The latter observation would indicate that either signaling via the AT1 receptor or absence of signaling via the AT2 receptor or both, as the effects elicited by signaling through the AT2 receptors have been reported to functionally antagonize effects of the AT1 receptors [35, 36], will lower eNOS expression. In the presence of AT1 receptor blockade, the very high levels of Ang II may enhance signaling via the AT2 receptor leading to eNOS and PKB/Akt expression as seen in the DIO animals. However, the opposite effect is seen in control animals, negating a conclusion as to the influence of this. In addition, we documented enhanced phosphorylation of eNOS in DIO hearts after Losartan treatment (Fig. 1b). This may be ascribed to inhibition of calcineurin, as described by Hotta et al. [32].
A novel finding of this study is that PKB/Akt protein expression in the heart is modulated by AT1 or AT2 signaling similar to what we found for eNOS expression (Fig. 7a). In addition, we documented higher levels of PKB/Akt in hearts from the DIO animals, possibly coupled to the development of myocardial hypertrophy in this model. Losartan treatment of DIO animals further elevated PKB/Akt levels underscoring the results of Kawahara et al. [37] in aortic tissue of spontaneously hypertensive rats.
Contrary to expectation, Losartan treatment of control animals resulted in lower NO production measured in cardiomyocytes. This is reflected by the absence of improved functional recovery after ischaemia seen in control hearts despite smaller infarct size development (Figs. 3, 4). This discrepancy between infarct size development and functional recovery is well-documented in different species and is explained by differences in the levels of stunning in the heart after different treatment [38]. Underscoring the result obtained, eNOS expression was attenuated in hearts from control animals treated with Losartan accompanied by no improvement in the levels of phospho-eNOS (Figs. 1b, 7b). In addition, Losartan treatment lowered PKB/Akt protein levels (Fig. 1a), emphasizing the conclusion that signaling modulates PKB/Akt expression similar to eNOS expression. The differential response of control and DIO tissue in the regulation of the expression both PKB/Akt and eNOS and NO production is interesting but currently not well understood and needs further investigation to clarify.
From the results of our study, we conclude that AT1 receptor antagonism for a short period with a low dose of Losartan, could restore the suppressed NO production in cardiomyocytes from DIO animals by increasing eNOS and PKB/Akt protein expression and enhancing phosphorylation of eNOS. Changes in the expression level of these 2 key proteins are therefore occurring before alleviation of whole-body insulin resistance, plasma insulin- or glucose levels could be detected and may therefore represent the first changes affected by AT1 antagonism leading to protection of the cardiovascular system.
References
Sharma AM, Engeli S (2006) The role of rennin-angiotensin system blockade in the management of hypertension associated with the cardiometabolic syndrome. J Cardiometab Syndr 1:29–35
Ruige JB, Assendelft WJ, Dekker JM, Kostense PJ, Heine RJ, Bouter LM (1998) Insulin and risk of cardiovascular disease: a meta-analysis. Circulation 97:996–1001
Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M (2002) Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest 108:1341–1348
Cai H, Harrison DG (2002) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Landmesser U, Dikalov S, Price SR et al (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111:1201–1209
Li J, Zhao X, Li X, Lerea KM, Olson SC (2007) Angiotensin II type 2 receptor-dependent increases in nitric oxide synthase expression in the pulmonary endothelium is mediated via a G alpha i3.Ras/Raf.MAPK pathway. Am J Physiol Cell Physiol 292:C2185–C2196
Wei Y, Sowers JR, Nistala R et al (2006) Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem 281:35137–35146
Vehkavaara S, Mäkimattila S, Schlenzka A, Vakkilainen J, Westerbacka J, Yki-Järvinen H (2006) Insulin therapy improves endothelial function in type 2 diabetes. Arterioscler Thromb Vasc Biol 20:545–550
Zeng G, Nystrom FH, Ravichandran LV et al (2000) Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 101:1539–1545
Sowers JR (2004) Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol 286:H1597–H1602
Montagnani M, Chen H, Barr VA, Quon MJ (2001) Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt and Ser1179. J Biol Chem 276:30392–30398
Metha PD, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292:C82–C97
Werner N, Nickenig G (2003) AT1 receptors in atherosclerosis: biological effects including growth, angiogenesis and apoptosis. Eur Heart J Supp 5:9–13
Brede M, Roell WS, Ritter O et al (2003) Cardiac hypertrophy is associated with decreased eNOS expression in angiotensin AT2 receptor-deficient mice. Hypertension 42:1177–1182
Velloso LA, Folli F, Sun XJ, White MF, Saad MJA, Kahn CR (1996) Cross-talk between the inhsulin and angiotensin signaling systems. Proc Natl Acad Sci USA 93:12490–12495
Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G (2004) Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res 94:1211–1218
Shinozaki K, Ayajiki K, Nishio Y, Sungaya T, Kashiwagi A, Okamura T (2004) Evidence for a causal role of the renin-angiotensin system in vascular dysfunction associated with insulin resistance. Hypertension 43:255–262
Dahlöf B, Devereux R, Kjeldsen SE et al (2002) LIFE Study Group. Losartan Intervention for Endpoint reduction in hypertension. Lancet 359:995–1003
Pickavance LC, Tadayyon M, Widdowson PS, Buckinham RE, Wilding JPH (1999) Therapeutic index for rosilitazone in dietary obese rats: separation of efficacy and haemodilution. Br J Pharmacol 128:1570–1576
Marais E, Genade S, Salie R, Huisamen B, Maritz S, Moolman JA, Lochner A (2005) The temporal relationship between p38 MAPK and HSP27 activation in ischaemic and pharmacological preconditioning. Basic Res Cardiol 100:35–47
Huisamen B, Donthi RV, Lochner A (2001) Insulin in combination with vanadate stimulates glucose transport in isolated cardiomyocytes from obese Zucker rats. Cardiovasc Drugs Ther 15:445–452
Strijdom H, Jacobs S, Hattingh S, Page C, Lochner A (2006) Nitric oxide production is higher in rat cardiac microvessel endothelial cells than ventricular cardiomyocytes in baseline and hypoxic conditions: a comparative study. FASEB J 20:314–316
Strijdom H, Muller C, Lochner A (2004) Direct intracellular nitric oxide detection in isolated adult cardiomyocytes: flow cytometric analysis using the fluorescent probe, diaminofluorescein. J Mol Cell Cardiol 37:897–902
Du Toit EF, Nabben M, Lochner A (2005) A potential role for angiotensin II in obesity induced cardiac hypertrophy and ischaemic/reperfusion injury. Basic Res Cardiol 100:346–354
Landmesser U, Hornig B, Drexler H (2004) Endothelial function: a critical determinant in atherosclerosis? Circulation 109:27–33
Galili O, Versari D, Sattler KJ et al (2006) Early experimental obesity is associated with coronary endothelial dysfunction and oxidative stress. Am J Physiol Heart Circ Physiol 292:H904–H911
Jaap AJ, Shore AC, Tooke JE (1997) Relationship of insulin resistance to microvascular dysfunction in subjects with fasting hyperglycaemia. Diabetologia 40:238–243
Naderali EK, Pickavance LC, Wilding John PH, Williams G (2001) Diet-induced endothelial dysfunction in the rat is independent of the degree of increase in total body weight. Clin Sci 100:635–641
Navarro-Cid J, Maeso R, Perez-Vizcaino F et al (1995) Effects of Losartan on blood pressure, metabolic alterations and vascular reactivity in the fructose-induced hypertensive rat. Hypertension 26:1074–1078
Van den Meiracker AH, Admiraal PJJ, Janssen JA et al (1995) Hemodynamic and biochemical effects of the AT1 receptor antagonist Irbesartan in hypertension. Hypertension 25:22–29
Folli F, Kahn CR, Hansen H, Bouchie JL, Feener EP (1997) Angiotensin II inhibits insulin signalling in aortic smooth muscle cells at multiple levels. A potential role of serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest 100:2158–2169
Hotta H, Miura T, Miki T et al (2010) Angiotensin II type 1 receptor mediated upregulation of calcineurin activity underlies impairment of cardioprotective signaling in diabetic hearts. Circ Res 106:129–132
Seshiah PN, Weber DS, Tocic P, Valppu L, Taniyama Y, Griendling KK (2002) Angiotensin II stimulation of NAD(P)H oxidase activity—upstream mediators. Circ Res 91:406–413
Henriksen EJ (2007) Improvement of insulin sensitivity by antagonism of the rennin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 293:R974–R980
Unger T, Chung O, Csikos T et al (1996) Angiotensin receptors. J Hypertens 14:3–9
Griendling KK, Lasseque B, Alexander RW (1996) Angiotensin receptors and their therapeutic implications. Annu Rev Pharmacol Toxicol 36:281–306
Kawahara S, Umemoto S, Tanaka M, Umeji K, Matsuda S, Kubo M, Matsuzaki M (2005) Up-regulation of Akt and eNOS induces vascular smooth muscle cell differentiation in hypertension in vivo. J Cardiovasc Pharmacol 45:367–374
Lochner A, Genade S, Moolman JA (2003) Ischemic preconditioning: infarct size is a more reliable endpoint than functional recovery. Basic Res Cardiol 98:337–346
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huisamen, B., Pêrel, S.J.C., Friedrich, S.O. et al. ANG II type I receptor antagonism improved nitric oxide production and enhanced eNOS and PKB/Akt expression in hearts from a rat model of insulin resistance. Mol Cell Biochem 349, 21–31 (2011). https://doi.org/10.1007/s11010-010-0656-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-010-0656-6