
Abstract
The epithelial to mesenchymal transition (EMT) is a crucial event for renal fibrosis that can be elicited by TGF-β1/Smads signaling and its downstream mediator connective tissue growth factor (CTGF). As a distinct member of the TGF-β superfamily, Lefty A has been shown to be significantly downregulated in the kidneys of patients with severe ureteral obstruction, suggesting its role in renal fibrosis induced by obstructive nephropathy. In order to determine whether Lefty A prevents TGF-β1-induced EMT, human proximal tubule epithelial cells (HK-2) were stably transfected with Lefty A or control vectors and stimulated with 10 ng/ml TGF-β1 for 48 h. The results show that stimulation with TGF-β1 led to EMT including cell morphology changes, Smad2/3 signaling pathway activation, increased α-SMA, collagen type I, and CTGF expression, and decreased E-cadherin expression in mock-transfected HK-2 cells. Overexpression of Lefty A efficiently blocked p-Smad2/3 activation and attenuated all these EMT changes induced by TGF-β1. This finding suggests that Lefty A may serve as a potential new therapeutic target to inhibit or even reverse EMT during the process of renal fibrosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Obstructive nephropathy is an important cause of chronic kidney disease (CKD) that ultimately progresses to tubulointerstitial fibrosis and end-stage renal failure [1, 2]. In such cases, renal damage progresses even after the ureteral obstruction is relieved, and most patients with obstructive nephropathy are identified only when they have already reached end-stage renal failure [3]. To date, there is no effective treatment available to prevent tubulointerstitial fibrosis and the progressive loss of renal function. One of the main effector cells that contribute to the development of progressive renal fibrosis in obstructive nephropathy is the tubulointerstitial myofibroblast [4]. A large proportion of tubulointerstitial myofibroblasts is known to derive from tubular epithelial cells undergoing the epithelial to mesenchymal transition (EMT) process in the progression of tubulointerstitial fibrosis [2, 5]. Consequently, this process induces renal tubular destruction and the accumulation of myofibroblasts [2, 6]. Many studies have suggested that blockading the EMT process can relieve the severity of renal fibrosis followed by obstructive nephropathy [7–14]. Therefore, novel therapy which could potentially inhibit or reverse EMT in the kidneys may retard disease progression to obstructive nephropathy.
Lefty is a new member of the TGF-β protein superfamily, including lefty 1 and lefty 2 in mouse [15, 16] and their homologues Lefty A and Lefty B in human [17, 18]. Lefty is one of the key embryonic signals that drive the development of an asymmetric body plan [15, 19, 20]. Unlike other members of the TGF-β superfamily, Lefty does not appear to exist as a dimer. Therefore, it may act as an inhibitor of the TGF-β1 signaling pathway [17]. It has been shown that Lefty interrupts TGF-β1 signaling by inhibiting the phosphorylation of Smad2/3 following activation of the TGF-β receptor. It also inhibits the events downstream from R-Smad phosphorylation, including hetero-dimerization of R-Smads (Smad2/3) with Smad4 and nuclear translocation of the R-Smad-Smad4 complex [21]. Moreover, it was recently shown that Lefty A/Ebaf (endometrial bleeding associated factor) significantly reduces the amount of collagen deposited in tissues by inhibiting connective tissue growth factor (CTGF) and collagen type I mRNA synthesis and increasing collagenolysis and elastolysis [22]. Thus, Lefty A may serve as an antagonist for TGF-β1 to ameliorate TGF-β1-induced EMT.
Although both human Lefty isoforms show similar sequence identity and could therefore share similar function [15], our previous study has shown that only the expression of Lefty A mRNA was downregulated significantly (↓13.55 folds) in the kidneys of patients with severe ureteral obstruction using gene microarray analyses [23]. We further confirmed the downregulation of lefty 1 and lefty 2 mRNA and protein in the mice model of unilateral ureteral obstruction (UUO) [23]. In this study, we examined the in vitro effect of Lefty A on the prevention of TGF-β1-induced human proximal tubule cell (HK-2) EMT. In order to assess the progression of EMT and the potential attenuation by Lefty A, the signaling molecular p-Smad2/3, the epithelial marker E-cadherin, and the mesenchymal marker α-smooth muscle actin (α-SMA) were assayed by Western blotting; CTGF, the downstream mediator of TGF-β1/Smads, was assayed by quantitative real-time PCR.
Materials and methods
Reagents
A line of human renal proximal tubular epithelial cells (HK-2) was purchased from American Type Culture Collection (VA, USA); Dulbecco’s modified eagle medium, nutrient mixture F-12 (DMEM/F12), fetal bovine serum (FBS), and trypsin/EDTA solution were purchased from Hyclone (UT, USA). The pcDNA3.1/Hygro (+) plasmid vector was a gift from Professor Siamak Tabibzadeh at Stony Brook University (Stony Brook, NY, USA). Full-length DNA sequences of human Lefty A were purchased from Wuhan Genesil Biotechnology Company (Hubei, China); recombinant human TGF-β1 was from Pepro Techec (London, UK); TRIzol and Lipofectamine™ 2000 were purchased from Invitrogen (CA, USA); the First-strand cDNA Synthesis Kit was from Fermentas (UAB); the quantitative real-time PCR kit was from Toyobo (Osaka, Japan); the BCA assay kit was from the Beyotime Institute of Biotechnology (Jiangsu, China); the monoclonal mouse antibody against E-cadherin was from Cell Signaling Technology (MA, USA); the monoclonal mouse antibody against Lefty A was from R&D Systems (MN, USA); the polyclonal antibody against α-SMA was from Abcam (Cambridge, UK); the polyclonal antibody against p-Smad2/3 and the polyclonal anti-Actin antibody were both purchased from Santa Cruz Biotechnology Inc (CA, USA).
Plasmid construction, cell culture, and stable transfection
The coding sequence (CDS) of human Lefty A (1.1 kb) was amplified from the full-length sequence of Lefty A. The primer sequences are as the follows: sense 5′-CCC AAGCTT GCC ACC ATG TGG CCC CTG TGG C-3′, antisense 5′-CGC GGATCC CTA TGG CTG GAG CCT CCT T-3′. PCR products were digested with the two enzymes and inserted into the Hind III and BamH I site of a pcDNA3.1/Hygro (+) vector. The recombinant plasmid was confirmed by DNA sequencing.
HK-2 cells were maintained in DMEM/F12 supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C under a humidified 5% CO2 atmosphere. Cells were seeded onto 6-well plates and transfected with 1 μg of either recombinant plasmid DNA or pcDNA3.1 empty vector using Lipofectamine™ 2000, according to the manufacturer’s instructions. Stable transfectants were generated from a HK-2 cell pool after selection with hygromycin (300 μg/ml). Overexpression of Lefty A in the stable transfected cell line was confirmed by Western blotting.
In order to examine the effects of Lefty A overexpression on the TGF-β1-induced EMT, the stably transfected cell line was cultured in serum-free medium for 24 h before TGF-β1 treatment. Cellular lysates were harvested 6, 12, 24, and 48 h after TGF-β1 treatment.
Western blotting
For Western blotting analysis, 2 × 106 cells were washed with ice-cold PBS and scraped into lysis buffer (1% NP40, 150 mM NaCl, 50 mM Tris/HCL, 1 mM PMSF, 1 mM EGTA, 10 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO5, 10 μg/ml leupeptin, and 20 μg/ml aprotinin). The protein concentrations were measured using a BCA assay kit according to the manufacturer’s instructions. After the addition of the protein loading buffer (10% glycerol, 50 mM Tris/HCl, 2% β-mercaptoethanol, 0.02% BPB, and 5% SDS, pH 6.8) and denaturation at 95°C for 5 min, 40 μg of total cellular protein from each sample was separated by 8% SDS-PAGE and transferred onto nitrocellulose membranes. After blocking in 5% skim milk for 1 h at 37°C, the membranes were incubated with the indicated primary antibody at 4°C overnight, followed by a horseradish peroxidase-conjugated second antibody for 1 h at room temperature. Signals were detected using the enhanced chemiluminescence (ECL) method.
RNA isolation and real-time RT-PCR
Total RNA was isolated with TRIzol Reagent, and first-strand cDNA was synthesized using the First-strand cDNA Synthesis Kit according to the manufacturer’s instructions. Quantitative real-time PCR was performed using the Bio-Rad iQ5 Real-Time PCR Detection System (CA, USA). SYBR green real-time PCR mix was used for the PCR reaction, with a primer concentration of 10 μM. The primers for real-time PCR are shown in Table 1. Reaction conditions were as follows: 95°C for 5 min; 40 cycles at 95°C for 20 s, 62°C for 30 s, and 72°C for 30 s; followed by a final extension at 72°C for 5 min. GAPDH was used as a reference gene. Relative quantification of gene expression was performed using the 2 − ΔΔCt method based on Ct values for both target and reference genes. In order to calculate the fold increase, the results of real-time PCR analysis are given as means ± SD.
Statistical analysis
All the data examined are expressed as means ± SD. For Western blotting analysis, quantification was performed by scanning and analyzing the average volume density of the hybridization signals corrected for actin using GEL pro3.0. Statistical analysis of the data was performed with the Student’s t test using the SPSS 11.5 software package. A two-sided P < 0.05 was considered statistically significant.
Results
TGF-β1 treatment induced the EMT process in HK-2 cells
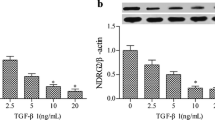
TGF-β1 is known to induce EMT in renal tubular cell lines isolated from humans, rats, mice, and pigs [24–27]. In order to determine whether Lefty A has the ability to inhibit EMT in renal tubular cells, we first developed an EMT cell model by treating HK-2 cells with TGF-β1. As shown in Fig. 1a, incubation of HK-2 cells with TGF-β1 was able to inhibit the expression of E-cadherin, an epithelial marker that is essential for the structural integrity of renal epithelium, and to induce the expression of α-SMA protein, a phenotypic marker for myofibroblast cells. The downregulation of E-cadherin and the induction of α-SMA occurred at a concentration as low as 2.5 ng/ml, suggesting that it is readily achievable in vivo under pathological conditions. The induction of α-SMA expression in tubular epithelial cells reached a plateau at TGF-β1 levels of 10 ng/ml, which was chosen for the following experiments. Next, we further investigated the time course of TGF-β1-induced expressions of epithelial and mesenchymal markers such as E-cadherin and α-SMA. As shown in Fig. 1b, HK-2 cells exhibited a time-dependent decrease in E-cadherin expression and a time-dependent increase in α-SMA expression over 48 h after TGF-β1 stimulation. The results revealed that the loss of E-cadherin expression was an early event which occurred as early as 6 h after TGF-β1 treatment, whereas induction of de novo expression of α-SMA was a delayed response requiring 24 h of incubation. Consistent with these changes, incubation of the cells with 10 ng/ml TGF-β1 for 48 h resulted in a significant loss of the typical cobblestone pattern of their epithelial monolayer. Instead, the cells displayed a fibroblast-like morphology identifiable by the presence of elongated lamellipodia and a spindle shape, as shown in Fig. 3c. The results suggested that HK-2 cells were converted to myofibroblasts by TGF-β1 treatment.

TGF-β1-induced EMT in HK-2 cells. a TGF-β1 downregulated the expression of E-cadherin and induced de novo expression of α-SMA in tubular epithelial cells in a dose-dependent style. HK-2 cells were incubated with TGF-β1 at various concentrations as indicated for 24 h. b Western blot analysis was performed to examine the time course of E-cadherin and α-SMA expression in HK-2 cells treated with 10 ng/ml TGF-β1 over 48 h. * P < 0.05, ** P < 0.01, ∆ P > 0.05 compared with the previous value (n = 3–5)
Overexpression of Lefty A blocked the TGF-β1-induced EMT
As illustrated in Fig. 2, the abundance of Lefty A protein in normal HK-2 cells was very low and progressively decreased to undetectable levels after 12 h treatment with TGF-β1. In order to examine whether overexpression of Lefty A would affect the progress of EMT, a recombinant Lefty A plasmid construct was stably transfected into HK-2 cells, and overexpression of Lefty A was confirmed by Western blotting (Fig. 3a). As indicated in Fig. 3a, overexpression of Lefty A did not affect the protein levels of E-cadherin or α-SMA in unstimulated HK-2 cells, however, overexpression of Lefty A partially blocked decrease of E-cadherin expression or increase of α-SMA expression in TGF-β1 stimulated HK-2 cells (Fig. 3b). Lefty A also inhibited the collagen type I mRNA expression induced by TGF-β1, as assessed by quantitative real-time PCR (Fig. 4c). Moreover, overexpression of Lefty A attenuated the morphology changes toward a myofibroblast phenotype induced by TGF-β1 (Fig. 3c). The data demonstrated that Lefty A partially inhibited TGF-β1-induced EMT in HK-2 cells.

TGF-β1 suppressed Lefty A expression in HK-2 cells. A decrease of Lefty A expression was found 6 h after 10 ng/ml TGF-β1 treatment, and Lefty A was undetectable 12 h after treatment. ** P < 0.01 compared with the previous value (n = 3–5)

Overexpression of Lefty A inhibited TGF-β1-induced EMT. Panel a Western blotting images showing that the expression changes of Lefty A, E-cadherin, and α-SMA in untransfected cells and Lefty A stably transfected cells. Panel b HK-2 cells stably transfected with either Lefty A or a pcDNA3.1 empty vector were subsequently treated with 10 ng/ml of TGF-β1 for 48 h. C control untransfected cells, T untransfected cells treated with TGF-β1, E pcDNA3.1 empty vector stably transfected cells treated with TGF-β1, L HK-2 cells stably transfected with Lefty A treated with TGF-β1. ∆ P > 0.05 compared to TGF-β1-treated untransfected group; * P < 0.05 compared to both the TGF-β1-treated untransfected group and the TGF-β1-treated empty vector-transfected group (n = 3–5). Panel c Lefty A attenuated TGF-β1-induced morphology changes in HK-2 cells. a unstimulated cells, b mock-transfected cells stimulated with 10 ng/ml TGF-β1 48 h. The epithelial morphology of HK-2 cells changed dramatically to a fibroblast-like state, c Lefty A transfected cells stimulated with 10 ng/ml TGF-β1 48 h. Overexpression of Lefty A partially blocked the TGF-β1-induced morphology changes toward the myofibroblast phenotype

Overexpression of Lefty A blocked TGF-β1/Smads signaling pathway as well as the upregulation of collagen type I and CTGF mRNA induced by TGF-β1 in HK-2 cells. Panel a TGF-β1 activated p-Smad2/3 in tubular epithelial cells. HK-2 cells were incubated with 10 ng/ml of TGF-β1 for 12 h, and the expression of p-Smad2/3 was detected by Western blotting. * P < 0.05, ** P < 0.01 compared with the previous value. Panel b Overexpression of Lefty A blocked TGF-β1/Smads signaling pathway in tubular epithelial cells. HK-2 cells stably transfected with Lefty A (L) or not (T) were subsequently treated with 10 ng/ml of TGF-β1 for 1 h, and the expression of p-Smad2/3 was detected by Western blotting. ** P < 0.01 compared with TGF-β1-treated untransfected group. Panel c HK-2 cells stably transfected with either Lefty A or pcDNA3.1 empty vector were treated with 10 ng/ml of TGF-β1 for 48 h. The expression levels of collagen type I and CTGF mRNA were detected using real-time PCR. C Control untransfected cells, T untransfected cells treated with TGF-β1, E pcDNA3.1 empty vector stably transfected cells treated with TGF-β1, L HK-2 cells stably transfected with Lefty A treated with TGF-β1. ∆ P > 0.05 compared to TGF-β1-treated untransfected group; * P < 0.05 compared to both the TGF-β1-treated untransfected group and the TGF-β1-treated empty vector-transfected group (n = 3–5)
Lefty A inhibited TGF-β1/Smads signaling pathway and the expression of CTGF mRNA
Our study demonstrated that Lefty A partially inhibited TGF-β1-induced EMT in HK-2 cells, but the mechanisms remained unknown. As it has been reported that in stem cells Lefty A inhibits TGF-β1/Smads signaling pathway which plays a significant role in the process of EMT in renal fibrosis [2], we first investigated whether Lefty A could block TGF-β1 induced Smad2/3 activation in HK-2 cells. Western blot analyses showed that there was no expression of p-Smad2/3 in normal HK-2 cells, and TGF-β1 induced a rapid activation of p-Smad2/3 in a time-dependent manner with a peak at 1 h (Fig. 4a). Overexpression of Lefty A by transfection blocked activation of Smad2/3 by TGF-β1 in HK-2 cells (Fig. 4b), indicating that Lefty A could inhibit TGF-β1/Smads signaling pathway in the process of EMT.
Since it has been reported that Lefty A degrades the ECM protein through inhibiting CTGF transcription which is an important downstream mediator of TGF-β1/Smads signaling pathway, we further studied the effects of Lefty A overexpression on the expression of CTGF mRNA in the TGF-β1-induced EMT process in HK-2 cells by quantitative real-time PCR. As shown in Fig. 4c, the expression of CTGF mRNA increased dramatically in HK-2 cells after TGF-β1 treatment for 48 h, however, this increase was abolished by overexpression of Lefty A, indicating that CTGF is one of the major downstream targets for Lefty A. It is possible that Lefty A inhibits the TGF-β1-induced EMT process via inhibition of TGF-β1/Smads signaling pathway and expression of its downstream mediator CTGF.
Discussion
Although specific therapies that inhibit the progression of chronic renal disease are currently unavailable, preventing or inhibiting EMT has been shown to be a promising therapeutic option through which renal fibrosis and subsequent obstructive nephropathy could potentially be inhibited. In the present study, we demonstrated that overexpression of Lefty A was able to attenuate TGF-β1-induced EMT in HK-2 cells. This process is likely mediated via the inhibition of p-Smad2/3 activation and CTGF expression induced by TGF-β1 [21, 22, 28]. Therefore, in addition to its role in the development of an asymmetric body plan and tissue remodeling of the human endometrium, our findings suggest that Lefty A may be applied to inhibit progression toward renal fibrosis, at least in part by attenuating the TGF-β1-induced EMT.
Tubulointerstitial fibrosis is a common final step leading to end-stage renal disease, irrespective of the nature of initial renal injury. Myofibroblasts have been shown to play a critical role in the process, and the major source of myofibroblasts is presumed to be tubular epithelial cells undergoing EMT process [6, 7, 29, 30]. Since TGF-β1, as a sole factor, is thought to initiate and complete the entire course of EMT [6, 31], we used it as an inducer of EMT to stimulate HK-2 cells. Our results showed that TGF-β1 stimulated HK-2 cells underwent the EMT process as assessed by the presence of de novo expression of α-SMA and downregulation of E-cadherin as well as the upregulation of collagen type I mRNA and the loss of epithelial morphology. Of note, the loss of E-cadherin expression was an early event which occurred as early as 6 h after TGF-β1 treatment. In contrast, induction of de novo expression of α-SMA was a delayed response requiring 12 h of incubation. As E-cadherin plays an important role in cell adherence, these data suggest that the dissociation of structural integrity of renal epithelia is an early event during EMT induced by TGF-β1. The downregulation of E-cadherin precedes de novo α-SMA expression, as reported previously [6].
Because of the important role of EMT in the process of obstructive nephropathy-induced tubulointerstitial fibrosis, any treatment or intervention that inhibits or even reverses EMT may serve as a new therapeutic option for tubulointerstitial fibrosis. Previously, our group found that the gene and protein expression levels of Lefty A were significantly downregulated in the kidneys of patients with severe ureteral obstruction and in a rat model of UUO, respectively [19]. Given its role in embryonic development via inhibition of TGF-β1/Smads signaling [17, 21], we postulate that Lefty A could have renoprotective ability against renal fibrosis induced by UUO. Even though the protein expression level of Lefty A was low in human proximal tubular cells in the present study, we demonstrated for the first time that it was downregulated in a time-dependent manner after TGF-β1 stimulation. Considering Lefty A’s role in inhibiting TGF-β1/Smads signaling, these results suggest that there may be mutual antagonism between Lefty A and TGF-β1/Smads signaling in the process of EMT. In order to validate our hypothesis, we further established a cell line stably expressing Lefty A and found that the overexpression of Lefty A partially blocked TGF-β1-induced EMT by preserving E-cadherin suppression and preventing α-SMA expression. These findings suggest that Lefty A is likely to be effective in attenuating EMT in proximal tubular cells.
The mechanism by which Lefty A inhibits TGF-β1-induced EMT remains unknown. It is more likely that multiple pathways are involved in the inhibitory effect of Lefty A, among which Smad2/3 activation and CTGF expression may be the two major events during the process. Since Lefty A has been reported to inhibit the process of TGF-β1/Smads signaling by interfering with both the hetero-dimerization of R-Smads (Smad2/3) with Smad4 and the nuclear translocation of the R-Smad-Smad4 complex in stem cells during embryonic development [21], we investigated whether it could also block TGF-β1-induced Smad2/3 activation in HK-2 cells. Our results showed that Lefty A inhibited the p-Smad2/3 activation induced by TGF-β1 in HK-2 cells, as well as in stem cells. The results confirmed that there was a mutual antagonism between Lefty A and TGF-β1/Smads signaling in the process of EMT in the renal fibrosis, which has never been shown previously. We also further studied the effects of Lefty A on CTGF expression, which was the important downstream mediator of TGF-β1/Smads signaling pathway [32]. Our results showed that overexpression of Lefty A in HK-2 cells attenuated TGF-β1-induced upregulation of CTGF transcription as well as TGF-β1-induced ECM, in line with previous reports that inhibition of CTGF expression attenuated the EMT process in vitro [33, 34]. The present study demonstrated that Lefty A could block TGF-β1-induced EMT probably by inhibition of p-Smad2/3 activation and CTGF expression in HK-2 cells. However, although it is reported that Lefty can be regulated by Wnt/β-catenin signaling in stem cells [35, 36], there is no evidence showing such regulation exists in renal fibrosis, since the fact that Wnt/β-catenin signaling can promote the process of renal fibrosis [37–40]. Thus, the regulation mechanism of Lefty A during renal fibrosis remained unclear, and further studies are required to investigate the mechanisms by which Left A inhibits the EMT process.
Our data present, for the first time, the important information that Lefty A, as a novel growth factor in embryonic development and endometrium remodeling, inhibits TGF-β1-induced EMT in HK-2 cells. These findings may pave the road toward an effective therapy for tubulointerstitial fibrosis.
References
Walsh PC (2002) Campbell’s urology. W.B. Saunders Company, Philadelphia
Liu Y (2004) Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 15:1–12
Ito K, Chen J, El Chaar M, Stern JM, Seshan SV, Khodadadian JJ, Richardson I, Hyman MJ, Vaughan ED Jr, Poppas DP, Felsen D (2004) Renal damage progresses despite improvement of renal function after relief of unilateral ureteral obstruction in adult rats. Am J Physiol Renal Physiol 287:F1283–F1293
Simonson MS (2007) Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int 71:846–854
Strutz F, Zeisberg M (2006) Renal fibroblasts and myofibroblasts in chronic kidney disease. J Am Soc Nephrol 17:2992–2998
Yang J, Liu Y (2001) Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol 159:1465–1475
Yang J, Liu Y (2002) Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol 13:96–107
Park SH, Choi MJ, Song IK, Choi SY, Nam JO, Kim CD, Lee BH, Park RW, Park KM, Kim YJ, Kim IS, Kwon TH, Kim YL (2007) Erythropoietin decreases renal fibrosis in mice with ureteral obstruction: role of inhibiting TGF-beta-induced epithelial-to-mesenchymal transition. J Am Soc Nephrol 18:1497–1507
Tumbarello DA, Turner CE (2007) Hic-5 contributes to epithelial-mesenchymal transformation through a RhoA/ROCK-dependent pathway. J Cell Physiol 211:736–747
Tan X, Li Y, Liu Y (2007) Therapeutic role and potential mechanisms of active Vitamin D in renal interstitial fibrosis. J Steroid Biochem Mol Biol 103:491–496
Tan X, Li Y, Liu Y (2006) Paricalcitol attenuates renal interstitial fibrosis in obstructive nephropathy. J Am Soc Nephrol 17:3382–3393
Wu MJ, Wen MC, Chiu YT, Chiou YY, Shu KH, Tang MJ (2006) Rapamycin attenuates unilateral ureteral obstruction-induced renal fibrosis. Kidney Int 69:2029–2036
Copeland JW, Beaumont BW, Merrilees MJ, Pilmore HL (2007) Epithelial-to-mesenchymal transition of human proximal tubular epithelial cells: effects of rapamycin, mycophenolate, cyclosporin, azathioprine, and methylprednisolone. Transplantation 83:809–814
Lange-Sperandio B, Trautmann A, Eickelberg O, Jayachandran A, Oberle S, Schmidutz F, Rodenbeck B, Hömme M, Horuk R, Schaefer F (2007) Leukocytes induce epithelial to mesenchymal transition after unilateral ureteral obstruction in neonatal mice. Am J Pathol 171:861–871
Meno C, Ito Y, Saijoh Y, Matsuda Y, Tashiro K, Kuhara S, Hamada H (1997) Two closely-related left-right asymmetrically expressed genes, lefty-1 and lefty-2: their distinct expression domains, chromosomal linkage and direct neuralizing activity in Xenopus embryos. Genes Cells 2:513–524
Meno C, Saijoh Y, Fujii H, Ikeda M, Yokoyama T, Yokoyama M, Toyoda Y, Hamada H (1996) Left-right asymmetric expression of the TGF beta-family member lefty in mouse embryos. Nature 381:151–155
Kothapalli R, Buyuksal I, Wu SQ, Chegini N, Tabibzadeh S (1997) Detection of ebaf, a novel human gene of the transforming growth factor beta superfamily association of gene expression with endometrial bleeding. J Clin Invest 99:2342–2350
Kosaki K, Bassi MT, Kosaki R, Lewin M, Belmont J, Schauer G, Casey B (1999) Characterization and mutation analysis of human LEFTY A and LEFTY B, homologues of murine genes implicated in left-right axis development. Am J Hum Genet 64:712–721
Hamada H, Meno C, Watanabe D, Saijoh Y (2002) Establishment of vertebrate left-right asymmetry. Nat Rev Genet 3:103–113
Dvash T, Mayshar Y, Darr H, McElhaney M, Barker D, Yanuka O, Kotkow KJ, Rubin LL, Benvenisty N, Eiges R (2004) Temporal gene expression during differentiation of human embryonic stem cells and embryoid bodies. Hum Reprod 19:2875–2883
Ulloa L, Tabibzadeh S (2001) Lefty inhibits receptor-regulated Smad phosphorylation induced by the activated transforming growth factor-beta receptor. J Biol Chem 276:21397–21404
Mason JM, Xu HP, Rao SK, Leask A, Barcia M, Shan J, Stephenson R, Tabibzadeh S (2002) Lefty contributes to the remodeling of extracellular matrix by inhibition of connective tissue growth factor and collagen mRNA expression and increased proteolytic activity in a fibrosarcoma model. J Biol Chem 277:407–415
Yao Y, Zhang J (2008) Lefty: a new member of TGF-beta family with anti-fibrosis function. Int J Urol Nephrol (Chinese) 28:237–240
Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, Thomas MC, Cooper ME, Kantharidis P (2006) Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol 17:2484–2494
Masszi A, Di Ciano C, Sirokmány G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A (2003) Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol 284:F911–F924
Li Y, Yang J, Dai C, Wu C, Liu Y (2003) Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis [erratum appears in J Clin Invest. 2004 Feb;113(3):491]. J Clin Invest 112:503–516
Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Müller GA, Neilson EG (2002) Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 61:1714–1728
Abdel Wahab N, Mason RM (2004) Connective tissue growth factor and renal diseases: some answers, more questions. Curr Opin Nephrol Hypertens 13:53–58
Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG (1995) Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130:393–405
Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG (2002) Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110:341–350
Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY (1999) Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 56:1455–1467
Qi W, Chen X, Poronnik P, Pollock CA (2008) Transforming growth factor-beta/connective tissue growth factor axis in the kidney. Int J Biochem Cell Biol 40:9–13
Chen L, Liu BC, Zhang XL, Zhang JD, Liu H, Li MX (2006) Influence of connective tissue growth factor antisense oligonucleotide on angiotensin II-induced epithelial mesenchymal transition in HK2 cells. Acta Pharmacol Sin 27:1029–1036
Liu BC, Zhang JD, Zhang XL, Wu GQ, Li MX (2006) Role of connective tissue growth factor (CTGF) module 4 in regulating epithelial mesenchymal transition (EMT) in HK-2 cells. Clin Chim Acta 373:144–150
Tabibzadeh S, Hemmati-Brivanlou A (2006) Lefty at the crossroads of “stemness” and differentiative events. Stem Cells 24:1998–2006
Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of WNT signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10:55–63
Hermens JS, Thelen P, Ringert RH, Seseke F (2007) Alterations of selected genes of the Wnt signal chain in rat kidneys with spontaneous congenital obstructive uropathy. J Pediatr Urol 3:86–95
Surendran K, McCaul SP, Simon TC (2002) A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol 282:431–441
He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y (2009) Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20:765–776
Surendran K, Schiavi S, Hruska KA (2005) Wnt-dependent beta-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J Am Soc Nephrol 16:2373–2384
Acknowledgments
This work was supported by Ph.D. Programs Foundation of the Ministry of Education of China (20060486054).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Y., Zhang, J., Fang, L. et al. Lefty A attenuates the TGF-β1-induced epithelial to mesenchymal transition of human renal proximal epithelial tubular cells. Mol Cell Biochem 339, 263–270 (2010). https://doi.org/10.1007/s11010-010-0389-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-010-0389-6