
Abstract
Gram-negative bacilli are one of the main causes for development of cholera, causing intestinal infections and acute watery diarrhea. Vaccination is one of the most important ways to prevent such infections. Some important immunogenic factors, such as toxin-co-regulated pilus A (TcpA), outer membrane protein W (OmpW), and cholera toxin B subunit (ctxB) as adjuvant that is non-toxic portion of cholera toxin can be used in process of vaccine design. For this purpose, in the present research, various types of bioinformatics software were used to study physiochemical properties of different refined structures, and validate them using bioinformatics tools as well as allergenic properties. One of the protein sequences with the highest antigenic index was the protein with sequence of Otc, which had a suitable tertiary structure according to the studies performed by the RaptorX server. Codon adaptation index (CAI) of this protein was increased to 0.98. The predicted structure showed stable condition according to thermodynamic analysis of mRNA. Structural analysis of chimeric vaccine-TLR-2 receptor protein–protein docking predicted efficient binding. Methodological approach and results obtained from studying candidate vaccine could be introduced to further experimental validations after efficacy of which was confirmed by experimental immunological assays in order to control and eliminate cholera in the future.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
During 5 years ago, more than 1.3 million deaths have occurred globally caused by diarrhoea as one of the major causes of mortality among children under 5 years of age. Although, responsibility is higher in some countries where people suffer from economic problems along with inadequate amount of safe water, sanitation, and urgent medical care, and diarrhoea infection is also a main cause of outpatient visits and hospital admissions in developed countries with high revenue and is one of important health problems among people all around the world (Troeger et al. 2017).
Three indicators of pathogenicity by microorganisms causing intestinal diseases are (1) attachment to intestinal epithelial cells, (2) attack to epithelial cells and epithelium, and (3) production of toxins that may be fatal to cells or lead to dehydration and influencing nervous system (Behrozie et al. 2018).
Pathogen of cholera was discovered by Filippo Pacini for the first time in 1854 in Italy. This organism was rediscovered by Robert Koch about 30 years later (Sanchez and Holmgren 2008).
Cholera toxin is an oligomeric protein subunit of 84 kDa produced in nature by gram-negative bacterium, V. cholerae, and is the main cause of secretory diarrhea in the upper intestine (Odumosu et al. 2010).
Given remarkable antigenic properties of cholera toxin, many studies have been performed aimed at using this toxin and recombinant subunits to create immunity against this fatal disease (Kundu et al. 2009 and Price et al. 2014).
Binding subunit of cholera toxin consists of five parts of 103 identical amino acids binding to GM1 ganglioside receptor of intestinal epithelial cells, in a ring-like arrangement. Cholera toxin B subunit (ctxB) is responsible for binding the toxin to host cells҆ cytoplasmic membrane receptors and is non-toxic (Sanchez and Holmgren 2008).
One of the main properties of ctxB is that it is able to stimulate regulatory responses and thereby, providing moderate autoimmune responses in some animal models. Lack of toxicity and easy expression of ctxB whether in the forms of linear peptide or protein has made it an easy adjuvant to handle (Stratmann et al. 2015).
Outer membrane protein W (OmpW) of V. cholerae is a molecule with 22 kDa of molecular weight (Jalajkumari and Manning, 1990) the gene of which is located in chromosome II of organism (Heidelberg et al. 2000). While, preponderance of b-structure in some of these proteins is consistent with their possible localization in outer membrane (OM) (Baldermann et al. 1998).
Enhancing intestinal colonization (i.e., motility) and the activity of those that are stringently required for colonization, TcpA (Taylor et al. 1987 and Herrington et al. 1988), ctxB, and toxin-co-regulated pilus (TCP) antigens are important virulence factors of V. cholerae. TCP antigens are one of the main responsible factors for colonization of V. cholerae on intestinal surface and type IV pilus antigens are composed of repeating subunits of the major pilin subunit TcpA (Taylor et al. 1987), which is absolutely required for colonization in mice and humans (Taylor et al. 1987and Herrington et al. 1988).
Today, the use of recombinant immunogens and subunit vaccines has received a great deal of attention to combat against microbial diseases due to risks of the first-generation vaccines. Recombinant immunogens and vaccines are safer and cause fewer non-specific reactions in immune system. On the other hand, the use of chimeric immunogens and multi-subunit vaccines instead of single subunits provides more protection against pathogens (Nazarian et al. 2012).
Despite immunological properties of cholera toxin, which are of great interest to researchers, toxicity has limited its use in the field of human vaccination (Behrozie et al. 2018).
Price reported that both TcpA and ctxB are the antigens providing a secure and helpful subunit vaccine against cholera (Price et al. 2014). Kundu et al. showed immunogenicity of recombinant TcpA and ctxB proteins separately and in combination together against V. cholerae. Their results showed that the use of cholera toxin binding subunit as immunogen caused 45.70% of immunity and the use of TcpA antigen alone caused 1.41% of immunity; and simultaneous use of both antigens resulted in 100% of protection against V. cholerae in animal model (Kundu et al. 2009).
In the recent years, bioinformatics has been used in design of recombinant immunogens and vaccines, which includes studying new protein structures and designing algorithms for predicting epithelial B and T cell balls as well as checking stability of immunogens. This reduces time and cost required for laboratory tests. The use of bioinformatics tools helps to identify new immunogens (Soria-Guerra et al. 2015).
Immunogens and chimeric proteins include protein subunits, linkers, and sequences, such as ctxB having adjuvant properties that can enhance immunogenicity of recombinant proteins and stimulate appropriate immune responses. (Nazarian et al. 2012).
Considering advantages of using chimeric proteins and pathogenic mechanism of V. cholerae, the present study was conducted to design a recombinant chimeric immunogen against binding factors and V. cholerae toxin.
Materials and Methods
Protein Sequence Retrieva and Databases
Amino acid sequences of the three V. Cholera proteins including TcpA, OmpW, and ctxB with the profiles of Q60153, P17266, and P01556 were obtained completely using corresponding protein database of UniProt server (http://www.uniprot.org) in FASTA format. Also, length of TcpA, OmpW, and ctxB proteins was equal to 217, 244, and 124 amino acids, respectively. For analyzing the desired data, latest versions of online and offline software were used.
B Cell and T cell Epitope Prediction
The IEDB web server (https://tools.iedb.org/bcell/) (Peters and Sette 2005) was used to predict B cell epitopes, MHC Ι as well as was used to identify the second type of immune T cells (MHC ΙΙ) in the proteins selected for vaccine production in selected proteins for the design of the recombinant vaccine. This Web server predicts epitopes in proteins based on flexibility, accessibility, antigenicity, and hydrophobicity. (Emini et al. 1985; Karplus and Schulz 1985; Kolaskar and Tongaonkar 1990; Parker et al. 1986; Larsen et al. 2006).
Evaluation of Allergenicity and Antigenicity of Recombinant Vaccine
For ensuring that our recombinant vaccine was non-allergic, allergenicity of the designed vaccine was evaluated by two different servers of AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP) (Dimitrov et al. 2014) and AllergenFP1.0 (http://www.ddgpharmfac.net/AllergenFP/index.html) In the AllerTOP server, an alignment-free method was developed based on the major physicochemical characteristics of proteins. ANTIGENpro (http://www.scratch.proteomics.ics.uci.edu/) (Magnan et al. 2010) and VaxiJen v2.0 servers (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (Doytchinova et al. 2007) were employed for antigenicity assessment of multi-peptide vaccine. ANTIGENpro is a software that can predict protective likelihood of a protein or peptide and is pathogen-independent, sequence-based, and alignment-free (Magnan et al. 2010). In the current research, bacteria were selected as target organism, and threshold was set at 0.5. The VaxiJen v2.0 is a server, which predicts antigens based on auto cross-covariance (ACC) transformation of protein sequence (Doytchinova and Flower 2007).
Physiochemical Properties of Recombinant Vaccine
ProtParam online server (http://web.expasy.org/protparam/) was used for prediction of physicochemical properties of multi-epitope vaccine protein. SOLpro server is a software used for predicting solubility of recombinant fusion protein (http://scratch.proteomics.ics.uci.edu) (Magnan et al. 2009).
Predicting Structure of Recombinant Vaccine
For predicting secondary structure of the designed peptide vaccine, PSIPRED v3.3 workbench (http://bioinf.cs.ucl.ac.uk/psipred/) (Buchan et al. 2019) was used and for identifying the intrinsically disordered regions, DISOPRED 2 server (https://iupred2a.elte.hu/) (Ward et al. 2004) was applied. I-TASSER server, which is based on sequence-to-structure-to-function paradigm, was applied for modeling 3D structure.
Refinement of Tertiary Structure and Validation of recombinant vaccine
Galaxy Refine bio tool (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) that is based on CASP10 was used to test refinement technique. RAMPAGE, ProSA-web, and ERRAT servers are various types of software used for analysis of the refined tertiary structure. Stereochemical properties of protein structure were assessed by the RAMPAGE server (Ramachandran plot analysis) at (http://mordred.bioc.cam.ac.uk/rapper/rampage.php), to indicate the number of residues located in the favored, allowed, and outlier regions (Lovell et al. 2003). ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) (Wiederstein et al. 2007) is another server used for validation of tertiary structure, using which tertiary structure was validated. Also, ERRAT (http://services.mbi.ucla.edu/ERRAT/) server was applied in order to analyze statistics of non-bond interaction between different types of atoms (Chris Colovos 1993).
Codon Optimization and In-Silico Cloning of the Designed Multi-Peptide Vaccine
Java Codon Adaptation tool (http://www.jcat.de/) (Grote et al. 2005) was used for cloning of the designed construct, and codon optimization of vaccine. SnapGene (GSL Biotech, California, USA) tool was used for cloning of the model developed for this construct through inserting the optimized coding sequence into a plasmid vector. Sites of NdeI and EcoR1 restriction enzymes were added to N and C-terminals of the sequence, respectively.
Molecular Docking of Multi-Epitope Vaccine with TLR-4
Toll-like receptor 4 (TLR-4) that is responsible for immune response against 17D vaccine has been described as one of TL receptors and stimulates a mixed Th2 and Th1 cell profile (Pulendran, 2009). TLR-4 structure was retrieved from the RCSB PDB database (PDB ID: 3fxi). Molecular docking of construct vaccine was done by TLR-4 using with the Cluspro 2.0 server (available at http://cluspro.bu.edu/login.php) in order to evaluate the affinity of binding of profiling ligand and TLR4 (Vajda et al. 2017). Finally, the result of docking was visualized using PyMOL software (DeLano 2002).
Molecular Dynamics Simulation
One of the methods to determine the stability of vaccine structure is designed through molecular dynamic simulation which is done from the package GROMACS 2016.1 (Abraham et al. 2015). Full system MD simulation was run with the CHARMM54a7 force field. The modeled structure of these epitopes was used as an input file for 30 ns (ns) MD at 310˚ K and at 1 bar pressure. We used a Cubic box with periodic boundary conditions and SPC/E water molecules (Narayanan and Johnston 2007) for MD simulation of epitopes. The steepest descent algorithm is used to reduce the energy of system as well as due to loosening solvent molecule.
A LINear Constraint Solver (LINCS) (Berendsen 1995) and SETTLE (Schmid 2011) Algorithms were used to eliminate chemical bonds among atomic epitopes also to eliminate chemical bonds among solvent molecules, respectively. Particle Mesh Ewald (PME) summation method (Narayanan and Johnston, 2007) was used to calculate the total electrostatic energy in the periodic box. Using L-J model with 10 Å cutoff distances, other non-bonded properties and changes were performed. The addition of Berendsen weakcoupling algorithm (Schmid 2011) 1.0 picosecond (Ps) was used to adjust the pressure and temperature of each system during simulation and connection time period. The interactions that occur in each peptide structure after equilibrium in molecular dynamics simulation can make a difference in RMSD values. To determine the dynamic behavior of each amino acid in the structure of vaccine designed from RMSF value, which was defined by peak elevation in all simulations used to determine the stability of vaccine designed against the time of the radius of gyration (RG) and RMSD of the complex of designed vaccine and TLR-4 receptor were plotted versus time (Ps) during 30 ns MD simulations.
Calculation of Binding Free Energy
The entropic component of binding free energy was disregarded. The average binding energy and its standard deviation were calculated by MmPbSaStat.py python script (http://rashmikumari.github.io/g_mmpbsa/). To estimate the contribution of each residue to the total binding free energy, MmPbSaDecomp.py python script was used (Kollman et al. 2000; Kumari et al. 2014).
Immune Simulation
C-ImmSim server (http://150.146.2.1/C-IMMSIM/index.php) (Rueckert et al. 2012) was used for identification of immunogenicity and immune response profile of chimeric peptide, in in-silico immune simulations. C-ImmSim is a software that uses an agent-based model to forecast immune interactions using a position-specific scoring matrix (PSSM) for prediction of immune epitope and machine learning techniques. It “simultaneously simulates three compartments representing three separate anatomical regions found in mammals including bone marrow,, thymus,and a tertiary lymphatic organ, such as a lymph node”[124]. All the simulation parameters were set at default with time steps set at 1, 84, and 168 (each time step consists of 8 h and time step 1 includes injection at time 0).
Results
Identification of different epitopes of B and T cells Selection of Antigenic Section and Designing of Multi-Epitope Vaccine
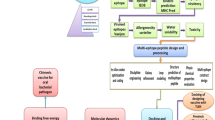
After identifying different epitopes of ompW and tcpA proteins, these short amino acid sequences were likned by a flexible GDGDG linker. At the end of this sequence, ctxB protein was added to this structure using EAAAK hard linker for better binding of the receptor to this structure (Fig. 1). Using IEDB software, epitopes were selected proteins for B immune cells based on antigenicity, accessibility of surface, flexibility, hydrophobicity, and predictions of linear epitope. Epitopes were also identified for MHC Ι and MHC ΙΙ. The predicted epitopes are presented in Table 1. Nine non-overlapping epitopes of each protein with the highest score were selected and linker using appropriate linkers. After binding the adjuvant to the end of this sequence, the final recombinant vaccine contained 416 amino acids (Fig. 2), which was evaluated for subsequent analysis.

Amino acid sequence of the recombinant vaccine, which are connected by a suitable linker

3D model of multiepitope vaccine that was obtained after prediction of final multi-epitope by I-TASSER server
Prediction of Antigenicity and Allergenicity of Vaccine Candidate
Three different types of software were used for prediction of antigenicity and allergenicity including ANTIGENpro v2.0, AllerTOP, and AllergenFP1.0 and the results indicated that chimeric construct was non-allergenic. ANTIGENpro showed that antigenicity of chimeric protein was equal to 0.88% and overall prediction for protective antigen was estimated as 1.1023 by VaxiJen (default threshold). Therefore, it can be said that our construct is antigen with a high probability. The results indicated that the generated sequences were antigenic in nature.
Prediction of Physicochemical and Biological Properties
Output of the ProtParam server regarding analysis of the final multi-epitope vaccine with 595 amino acids showed a molecular weight of 42,678.45 D and its theoretical isoelectric point (PI) was calculated by 4.16. The total number of atoms was equal to 5836 with an atomic composition including Carbon (1841), Hydrogen (2836), Nitrogen (508), Oxygen (640), and Sulfur (11). The total number of the negatively charged residues was equal to 59 and the total number of the positively charged residues was equal to 24. Half-life of the predicted structure was 1 h in mammalian reticulocytes in-vitro, and it was 2 min in yeast and E. coli in-vivo, respectively.
Prediction of Secondary Structure
PSIPRED is one of the best software for prediction of secondary structure according to which secondary structure contained 20% of alpha helix, 26% of beta strand, and 52% of coil. In addition, regarding solvent accessibility of amino acid residues, 45% of secondary structure was predicted to be exposed, 20% of it was medium-exposed, and 33% of it was predicted to be buried. RaptorX Property server predicted that a total of 31 residues (7%) were located in the disordered domains. (Fig. 3).

Schematic representation of secondary structure prediction of the multi-epitope vaccine. It shows fractions of α-helix, β-strands and coils secondary structures by RaptorX server
Refinement and Validation of Vaccines҆ Three-Dimensional Structure
Totally, five tertiary structure models of the designed multi-epitope vaccine, such as 1ltrA, 6vs7A, 3chbD, 4l6t, and 6ozdA were the best based on ten threading templates predicted by I-TASSER server. All the ten chosen templates showed good alignment according to their Z-score values (ranging from 1.19 to 7.52). C-score indicated confidence of the model predicted by I-TASSER server. The five predicted models had C-score values ranging from −1.69 to −2.68. The highest C-score of −1.69 was selected as the best model for further refinement representing a model with higher stability and confidence that is close to the score recommended by I-TASSER server (−1.5) for accuracy, and has the highest frequency in the top cluster with respect to size. This model had an estimated TM-score of 0.51 ± 0.15 with an estimated RMSD of 10.9 ± 4.6 Å. For prediction of 3D structure modeling (Fig. 2), three different servers, such as SWISS-MODEL, Galaxy web, and I-TASSER were used. The results of these servers were validated by ProSA-web, Ramachandran plot analysis (RAMPAGE), and ERRAT. Validation results illustrated that model 1 was the best model based on SWISS-MODEL server. ProSA-web data indicated that Z-score of the model was equal to −2.47 (Fig. 4). The chosen model had an overall quality factor of 95% after refinement according to ERRAT server (Fig. 5). Ramachandran plot analysis showed that 59.6, 30.8, and 3% of the residues were located in the favored, allowed, and outlier regions, respectively (Fig. 6).

ProSA-web Z-score plot for 3D structure of fusion construct. The Z-score of the best model is -3.36 (shown in a large black spot), which is in range of native protein conformations. Z-score plot includes the Z-scores of all experimentally protein chains in PDB determined by NMR spectroscopy (dark blue) and X-ray crystallography (light blue) by ProSA-web server

ERRAT plot. The overall quality factor of the final model is 99. Values around 95% or higher usually show high resolution of structures by SAVES server

Ramachandran plot formation to validate the 3D modeled structure exhibiting 55.9% of residues in the favored region by SAVES server
Codon Adaptation and In-Silico Cloning
In this study, Java Codon Adaptation Tool (JCat) was used in order to optimize codon usage of vaccine construct in E. coli (strain K12) for maximal protein expression. Length of the optimized codon sequence was equal to 1260 nucleotides. The results of JCat showed that CAI and average GC content of the adapted sequence were equal to 0.98 and 53%, respectively. So that, there was a possibility of good expression of vaccine candidate in E. coli as a host. GC content of the final vaccine was equal to 0.53. Cloning of the optimized gene sequence in the final vaccine construct was done by SnapGene tool in E. coli pET-28a ( +) vector, NdeI was introduced to N-terminals and EcoRI restriction sites were introduced to C-terminals of the sequence. Finally, the optimized sequence (with restriction sites) was inserted into pET-28a ( +) vector to ensure vaccine expression (Fig. 7).

In-silico cloning of optimized and reversed codon sequences into pET28a ( +) vector. showing the region of choice in blue color surrounded between Nde1 and EcoR1 while the vector has shown in black lines by SnapGene tool
Molecular Docking of Multi-Epitope Vaccine with TLR-4
Molecular docking of multi-epitope vaccine construct was done by TLR-4 using the Cluspro 2.0 server to investigate binding affinity of TLR-4 and a total of 117 models were constructed based on biophysical features of ligand (multi-epitope vaccine) and receptor (TLR-4). Among them, just one model was chosen, which was definitely connected to receptors and having the lowest energy score in this regard, the model number 4.29 was found to fulfill the desired criteria explaining the reason for its selection as the best-docked complex. Energy score obtained for the cluster 4.29 of chimeric vaccine and TLR-4 was found to be1.8. PDB format of the docked structure was visualized using PyMOL software (Fig. 8). The docked complexes were preferred for molecular dynamic analysis.

Docking model of TLR-4 molecule and fusion protein obtained by the Cluspro 2.0 server. TLR-4 is shown in green and ligand and adjuvant are shown in blue and red respectively. The interacting residues between TLR4 molecule and CTXB are colored in yellow. Pymol software was used for visualization of Docked model. (Color Fig online)
Molecular Dynamics Simulation
The stability of reaction between TLR4 receptor and the epitopes at the microscopic level was evaluated using GROMACS 2016.1 package; finally, the stability of selected epitopes was examined. The interactions between ligand and receptor which indicate the stability of designed vaccine structure were examined by mean square root deviation (RMSD) of ligand (multi-epitope vaccine) and receptor (TLR-4).
The interaction between Rg and RMSD was evaluated in 30 ns. Rg diagram shows the stability of simulation until the end of period (Fig. 9a). Also, in a period of 30 ns, RMSD chart shows fluctuations from 0.4 to 1.7 nm (Fig. 9b). In this case, the mean RMSD difference for the epitope was about 0.2 Å.

Molecular dynamics simulation of the ligand-receptor complex (vaccine and human TLR-4). a Rg of vaccine receptor complex during 100 ns MD simulations. b RMSD—Root Mean Square Deviation of the ligand-receptor complex demonstrates no significative deviation, reflecting a stable microscopic interaction between the two molecules and c RMSF-Root Mean Square Fluctuation plot of the ligand-receptor complex reflects the flexibility of side chain of the docked protein complex
Besides, due to RMSF diagram, the sequence of epitopes with residues does not cause instability. As shown in Fig. 9c, the minimum and maximum difference is approximately 1.3 nm, based on which it can be concluded that the proposed vaccine is stable in the human body.
Binding Free Energy Estimates
Results are related to free-binding energy in 0–3000 PS using MM/PBSA method presented in Table 2. Regarding the analysis, it was found that the proposed vaccine will be stable in the cloning stage due to the negative binding energy of epitopes to the protein. The number of hydrogen bonds between peptide and receptor showed significant variation during the simulation (Fig. 10). The highest average of Hydrogen bonds was −248.168 (kJ/mol), and −162.964(kJ/mol) was related to electrostatic interaction, and −657.647 (kJ/mol) was due to van der Waals. Amino acids contribution to epitopes binding free energies of amino acid residue in all peptides was shown in (see Additional file). For GLU-324, ASP-12, and ASP 24 as the highest amino acid with negative charge and LYS190, ARG-1, with negatively charged had more relevant effect on vaccine interaction with TLR4 receptor. In general, it was shown that the proposed recombinant vaccine is a stable protein with a rigid interaction with the TLR4 receptor.

Estimated binding free energy for the vaccine construction. Calculated with the MM/PBSA method on the 100–200 ns period of one of the simulation replicates
Immune Simulation
Results of the C-ImmSim server immune simulation showed that high levels of IgM was one of the characteristics of primary response and increases in the B-cell populations and levels of immunoglobulin G 1(IgG1) + immunoglobulin G 2(IgG2), immunoglobulin M (IgM), and IgG + IgM antibodies were properties of secondary and tertiary responses, which were correspondingly decreased in the antigen concentration. So, these properties show development of immune memory and consequently, the increased clearance of antigen upon subsequent exposures. Similarly, a high response was observed in the Th (helper) and Tc (cytotoxic) cell populations corresponding with memory development (Fig. 11).

C-ImmSim presentation of an in-silico immune simulation with the chimeric peptide. a Immunoglobulin production in response to antigen injections (black vertical lines); specific subclasses are indicated as coloured peaks. b The evolution of B-cell populations after the three injections. c The evolution of T-helper, and d T-cytotoxic cell populations per state after the injections. The resting state represents cells not presented with the antigen while the anergic state represents tolerance of the T-cells to the antigen due to repeated exposures by C-ImmSim server
Discussion
According to global statistics and prior studies that have highlighted importance of diarrhea, this infection is the second cause of mortality among children in the world so that, in 2012, it caused the death of 1600 children per day and more than 580,000 children have died due to this fatal disease in the recent years. Intestinal pathogens, such as V. cholerae are common causes of diarrhea and a vaccination program for controlling this bacterium can help to reduce disease mortality (Troeger et al. 2017).
For this purpose, possible surface proteins are identified by a reverse method starting with genome instead of microorganism using computational techniques and pattern recognition. Therefore, in addition to identifying all the antigens that can be studied by conventional methods, this method is also able to identify new antigens playing an important role in immunogenicity of new vaccines (Davies and Flower 2007). Therefore, the present study was conducted to develop a cholera vaccine and reduce mortality in the world and the obtained results would corroborate findings of the previous works in this field.
In the field of research on subunit and recombinant vaccines, a new approach regarding the use of multicomponent antigens can be used with greater safety, specific response, and less adverse reactions in designing of vaccine candidates. In multi-capacity chimer analysis and analysis of bioinformatics data, valuable tools and new strategies can provide different structural and safety features combined with epitopes, resulting in cost and time savings in design process (Nuccitelli et al. 2011 and Rostami et al. 2016).
Given the above-mentioned reasons, retrieving sequences of ctxB adjuvant, TcpA, and OmpW proteins of V. cholerae from Uniport was considered the first step and before designing tertiary structure of epitopes, appropriate epitopes with the highest score were selected by software according to I-Tasser servers for analysis in the next steps. Finally, after predicting the three-dimensional structure of epitopes, five epitopes were selected based on the results of docking. The selected epitopes showed good ability to bind to TLR-4 cavity.
After confirming stability of peptide TLR-4, epitopes were linked together. Linkers are used in designing of chimeric proteins. The use of a suitable linker enables immune system to respond and produce antibodies against linear epitopes, especially structural epitopes of each protein subunit. In this study, a hard linker consisting of EAAAK amino acids containing glutamic acid, alanine, and lysine was used. One of important factors of EAAAK linker is the ability to separate proteins and reduce interaction of vaccines as well as increasing thermal stability of chimeric protein (Chen v 2017; Saadi et al. 2017).
In this study, due to immunogenicity of B-Cell and T-Cell epitopes for TcpA and OmpW proteins and antigenic and adjuvant properties of proteins containing all the three subunits were designed. Bioinformatics studies have shown that this protein has the ability to stimulate immune system.
Prior studies have shown importance of CT in development of profuse watery diarrhea, and TCP (composed of repeating copies of the major pilin TcpA) has been found to be required for intestinal colonization by V. cholerae. TcpA protein, as a large subunit of pili and due to its location on bacterial surface, is a good candidate for developing anti-bacterial colonization immunity (Price and Holmes 2014).
On the other hand, ctxB protein has a hemopentameric and non-toxic structure located on large chromosome of the V. cholerae. In addition to producing antibodies and protecting against cholera toxin, ctxB protein is an important bioadjuvant that can elicit immune responses including mucosal immunity.
Findings of the current study are consistent with those in the studies by Heidelberg (2000) and Schoolnik and Yildiz (2000) who found involvement of the OmpW factor in development of cholera. (Heidelberg v 2000; Schoolnik and Yildiz 2000).
Price and Holmes (2014) clearly showed that in infant mouse model, TcpA and ctxB combination could be used as an immunogenic subtype of cholera. However, further studies are needed to understand efficacy of TcpA and ctxB combination and effective vaccines against cholera (Price and Holmes 2014).
One of the reasons leds to the increased use of ctxB is expression of this protein in different organisms. Another factor that makes ctxB a cost-effective adjuvant is the capacity to reduce antigen level up to 100 times for immunogenicity as well as attachment of agent to antigen-presenting cells (APCs) and epithelium and epithelial surface. The ctxB is currently used in vaccines, such as Dukoral, an anti-V. cholerae vaccine consisting of the killed V. cholerae and recombinant ctxB (Stratmann 2015).
Kundu v in an experiment on elimination of cholera by nasal vaccination evaluated the ability of ctxB and TcpA antigens of V. cholerae to create immunity against bacteria and toxins. As a result, they found that action of these antigens together can protect the rabbit against a homologous challenge, while each antigen alone can only protect the rabbit to some extent (Kundu et al. 2009).
Another important finding about immune simulation was that consistent with typical immune responses, there was a general increase in the generated immune responses following repeated exposure to antigen. In V. cholerae, IgG1, immunoglobulin G 3(IgG3), and nIgE responses to bacterial antigens are implicated in protection against disease. Development of memory B-cells and T- cells was evident, with production of memory B-cells lasting for several days.
In this study, codon optimization was performed using E. coli (strain K12) to achieve high expression of recombinant vaccine protein. As a result, some factors, such as codon compatibility index and GC content were found to be desirable (0.98 and 49.74%, respectively). According to the molecular dynamical analysis and the RMSD design results, the designed vaccine has shown stability and stability.Besides, the results of studying molecular behavior, RMSD, RMSF, Rg, SASA, hydrogen bond, and free binding energy were obtained from trajectory to investigate show that hydrogen bonds hydrophobic interactions play an important role in the formation and stabilization of epitopes and receptor interactions.
Moreover, due to the analyses performed and the results obtained from molecular dynamics and docking, it was found that this proposed vaccine has a stable structure; hence, we expect this structure to be stable in the body environment.
It is interesting to note that epitopes were predicted based on various parameters, such as hydrophilicity, flexibility, accessibility, and surface exposure using sequence and structure-based methods. The results showed that B-cell epitopes were scattered throughout all three parts of chimeric structure and could potentially produce a protective immune response. Studies have shown that chimeric structure is not allergenic and is one of the most important immunogenic candidates for designing of vaccines.
Conclusion
It can be concluded that bioinformatics methods have a significant effect on analysis of immunogens҆ design. Evaluation of in-silico chimeric vaccine showed that this gene construct was effectively expressed in E. coli and induced immune responses against binding factors and bacterial toxins.
Abbreviations
- ACC:
-
Auto cross-covariance
- APCs:
-
Antigen-presenting cells
- CAI:
-
Codon adaptation index
- ctxB:
-
Cholera toxin B subunit
- GM1:
-
Monosialotetrahexosylganglioside
- IgG2:
-
Immunoglobulin G 2
- IgM:
-
Immunoglobulin M
- I-TASSER:
-
Iterative Threading ASS Embly Refinement
- JCat:
-
Java Codon Adaptation Tool
- LINCS:
-
Linear constraint solver
- MD:
-
Molecular dynamics
- PDB:
-
Protein data bank
- PI:
-
Protrusion index
- PSSM:
-
Position-specific scoring matrix
- RMSD:
-
Root Mean Square Deviation
- RMSF:
-
Root Mean Square Fluctuation
- SASA:
-
Solvent accessible surface area
- TCP:
-
Toxin-co-regulated pilus
- TcpA:
-
Toxin-co-regulated pilus A
- TLR-4:
-
Toll-like receptor 4
References
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Baldermann C, Lupas A, Lubieniecki J, Engelhardt H (1998) The regulated outer membrane protein Omp21 from Comamonas acidovorans is identified as a member of a new family of eight-stranded β-sheet proteins by its sequence and properties. J Bacteriol 180(15):3741–3749
Berendsen HJ, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91(1–3):43–56
Behrozie, M., Nazarian, S., & Aghaie, S. M. (2018). Bioinformatic Design and Analysis of Chimeric Immunogen Against Adherence and Toxicity of Vibrio Cholera.
Buchan DW, Jones DT (2019) The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res 47(W1):W402–W407
Chen H, Chen Z, Wu B, Ullah J, Zhang T, Jia J, Tan T (2017) Influences of various peptide linkers on the Thermotoga maritima MSB8 nitrilase displayed on the spore surface of Bacillus subtilis. J Mol Microbiol Biotechnol 27(1):64–71
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2(9):1511–1519
Davies MN, Flower DR (2007) Harnessing bioinformatics to discover new vaccines. Drug Discovery Today 12(9–10):389–395
DeLano WL (2002) The PyMOL molecular graphics system. http://www.pymol.org.
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP v. 2—a server for in silico prediction of allergens. J Mol Model 20(6):1–6
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8(1):4
Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33(Suppl_2):W526–W531
Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Gill SR (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406(6795):477–483
Emini EA, Hughes JV, Perlow D, Boger J (1985) Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol 55(3):836–839
Herrington DA, Hall RH, Losonsky GENEVIEVE, Mekalanos JJ, Taylor RK, Levine MM (1988) Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168(4):1487–1492
Jalajakumari MB, Manning PA (1990) Nucleotide sequence of the gene, ompW, encoding a 22kDa immunogenic outer membrane protein of Vibrio cholerae. Nucleic Acids Res 18(8):2180
Karplus PA, Schulz GE (1985) Prediction of chain flexibility in proteins. NW Naturwissenschaften 72(4):212–213
Kolaskar AS, Tongaonkar PC (1990) A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 276(1–2):172–174
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Cheatham TE (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Kundu J, Mazumder R, Srivastava R, Srivastava BS (2009) Intranasal immunization with recombinant toxin-coregulated pilus and cholera toxin B subunit protects rabbits against Vibrio cholerae O1 challenge. FEMS Immunol Med Microbiol 56(2):179–184
Kumari R, Kumar R, Open Source Drug Discovery Consortium, Lynn A (2014) g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962
Larsen JEP, Lund O, Nielsen M (2006) Improved method for predicting linear B-cell epitopes. Immun Res 2(1):1–7
Lovell SC, Davis IW, Arendall WB III, De Bakker PI, Word JM, Prisant MG, Richardson DC (2003) Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Struct Funct Bioinform 50(3):437–450
Magnan CN, Randall A, Baldi P (2009) SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 25(17):2200–2207
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, Baldi P (2010) High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 26(23):2936–2943
Nazarian S, Gargari SLM, Rasooli I, Amani J, Bagheri S, Alerasool M (2012) An in silico chimeric multi subunit vaccine targeting virulence factors of enterotoxigenic Escherichia coli (ETEC) with its bacterial inbuilt adjuvant. J Microbiol Methods 90(1):36–45
Narayanan R, Johnston D (2007) Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron 56(6):1061–1075
Nuccitelli A, Cozzi R, Gourlay LJ, Donnarumma D, Necchi F, Norais N, Grandi G (2011) Structure-based approach to rationally design a chimeric protein for an effective vaccine against Group B Streptococcus infections. Proc Natl Acad Sci 108(25):10278–10283
Odumosu O, Nicholas D, Yano H, Langridge W (2010) AB toxins: a paradigm switch from deadly to desirable. Toxins 2(7):1612–1645
Parker JMR, Guo D, Hodges RS (1986) New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 25(19):5425–5432
Peters B, Sette A (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6(1):1–9
Price GA, Holmes RK (2014) Immunizing adult female mice with a TcpA-A2-CTB chimera provides a high level of protection for their pups in the infant mouse model of cholera. PLoS Negl Trop Dis 8(12):e3356
Pulendran B (2009) Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat Rev Immunol 9(10):741–747
Rostami A, Goshadrou F, Langroudi RP, Bathaie SZ, Riazi A, Amani J, Ahmadian G (2016) Design and expression of a chimeric vaccine candidate for avian necrotic enteritis. Protein Eng Des Sel 30(1):39–45
Rueckert C, Guzmán CA (2012) Vaccines: from empirical development to rational design. PLoS Pathog 8(11):e1003001
Vajda S, Yueh C, Beglov D, Bohnuud T, Mottarella SE, Xia B, Kozakov D (2017) New additions to the C lus P ro server motivated by CAPRI. Proteins: Struct Funct Bioinform 85(3):435–444
Saadi M, Karkhah A, Nouri HR (2017) Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect Genet Evol 51:227–234
Sanchez J, Holmgren J (2008) Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci 65(9):1347–1360
Schoolnik GK, Yildiz FH (2000) The complete genome sequence of Vibrio cholerae: a tale of two chromosomes and of two lifestyles. Genome Biol 1(3):1–3
Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J 40(7):843–856
Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S (2015) An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform 53:405–414
Stratmann T (2015) Cholera toxin subunit B as adjuvant––an accelerator in protective immunity and a break in autoimmunity. Vaccines 3(3):579–596
Taylor RK, Miller VL, Furlong DB, Mekalanos JJ (1987) Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci 84(9):2833–2837
Troeger C, Forouzanfar M, Rao PC, Khalil I, Brown A, Reiner RC Jr, Alemayohu MA (2017) Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis 17(9):909–948
Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337(3):635–645
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(suppl_2):W407–W410
Acknowledgements
We acknowledge the collaboration and assistance of all team members.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Research Involving Human and Animal Rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Raheem, S.G., Salh, K.K., Ibrahim, K.S. et al. In-Silico Designing a Multi-Peptide Vaccine: Against Vibrio Cholera. Int J Pept Res Ther 27, 1541–1553 (2021). https://doi.org/10.1007/s10989-021-10190-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-021-10190-3