
Abstract
Chronic fatigue syndrome (CFS) is a relatively common disorder defined as a status of severe persistent disabling fatigue and subjective unwellness. While the biological basis of the pathology of this disease has recently been confirmed, its pathophysiology remains to be elucidated. Moreover, since the causes of CFS have not been identified, treatment programs are directed at symptom relief, with the ultimate goal of the patient regaining some level of pre-existing function and well-being. Several studies have examined whether CFS is associated with: (i) a range of infectious agents and or immune disturbance; (ii) specific changes of activity in the central or peripheral nervous systems; and (iii) elevated stress periods, which may be associated with the pathology via genetic mechanisms. The role of oxidative stress in CFS is an emerging focus of research due to evidence of its association with some pathological features of this syndrome. New data collectively support the presence of specific critical points in the muscle that are affected by free radicals and in view of these considerations, the possible role of skeletal muscle oxidative imbalance in the genesis of CFS is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The definition of chronic fatigue syndrome.
A status of severe persistent disabling fatigue and subjective unwellness is termed chronic fatigue syndrome (CFS). CFS is a relatively common disorder that affects more than 500 women and 250 men from all age groups (including children) per 100,000 individuals (Jason et al. 1999). CFS affects individuals differently, as for all diseases, and not everybody reaches the severe end of the spectrum.
Clinical CFS is characterized by incapacitating fatigue of at least 6 months duration, usually with an abrupt onset accompanied by an infectious-like illness. CFS can affect virtually every major system in the body, as evident from reports on related neurological, immunological, hormonal, gastro-intestinal, musculoskeletal, and psychological problems (Wyller 2007).
Many patients with CFS are unable to work, and drastically reduce their social activities. Symptoms are often severely debilitating, and may last for several months or years. Individuals with CFS perform a lower level of activity than they were originally capable of before the onset of the illness. Importantly, any active medical condition that explains the presence of these symptoms may preclude the diagnosis of CFS (Fukuda et al. 1994; Reeves et al. 2003). Today, the pathogenesis of CFS remains to be conclusively delineated, and no specific diagnostic tests with adequate sensitivity and specificity are currently available Even if recent studies indicate that Vis-NIR spectroscopy for sera combined with chemometrics analysis may provide a promising tool for the objective diagnosis of CFS (Sakudo et al. 2006)
Therefore, this syndrome can currently be diagnosed only with the most widely used criteria established in 1994 by the Center for Disease Control and Prevention (Fukuda et al. 1994) and revised in 2003 by the International chronic fatigue syndrome study group (Reeves et al. 2003) (Fig. 1) while there are no comprehensive reports on mortality among patients with chronic fatigue and CFS, several sources suggest that CFS is associated with an elevated risk of heart failure and suicide (Smith et al. 2006).

Clinical definition of the chronic fatigue syndrome by the international CFS study group
CFS is a disease of physical origin
While some researchers consider CFS a psychological rather than physical illness (Sakudo et al. 2006), several studies suggest an organic origin of the disease. Elevated bioactive transforming growth factor-beta levels are observed in sera from patients with chronic fatigue (Bennett et al. 1997), and many CFS patients have atypical circulating lymphocytes and immune complexes (Bates et al. 1995), and increased concentrations of C-reactive protein and β-2 microglobulin (Buchwald et al. 1997). The presence of other immunologically significant factors, (Linde et al. 1992; Hanson et al. 2001) strongly implies significant immune impairment in CFS patients. Moreover, specific neurological changes are evident, including white matter lesions in the CNS (Lange et al. 1999), cerebral hypoperfusion, and significant reduction in global gray matter volume, compared to matched controls (de Lange et al. 2005). Other findings supporting CNS involvement include vestibular dysfunction and gait abnormalities (Saggini et al. 1998).
Central activation is diminished in CFS patients, presenting a possible cause of physiologically defined changes, such as variations in corticospinal excitability and neurotransmitter concentrations (Schillings et al. 2004). Consistent with this finding, the brain activity of CFS patients is altered during voluntary motor actions (isometric handgrip contractions at 50% maximal voluntary contraction level), compared to healthy individuals, particularly when motor activities induce fatigue (Siemionov et al. 2004). The most striking symptoms include debilitating fatigue, muscle pain, and muscle weakness, indicative of neuromuscular dysfunction. Serum acylcarnitine deficiency in CFS patients (Kuratsune et al. 1994) may induce a decrease in oxidative metabolism (Wong et al. 1992) and higher levels of plasma lactate. Moreover, serum creatine kinase levels are sometimes mildly increased (Archard et al. 1988). These metabolic defects may contribute to the reduced physical endurance of CFS patients. During dynamic exercise, individuals with CFS display similar skeletal muscle metabolic patterns to control subjects, but reach exhaustion (in the presence of reduced sarcoplasmic ATP concentrations) much more rapidly. The data imply defects in oxidative metabolism, with resultant acceleration of glycolysis in working skeletal muscles. McCully and colleagues (McCully et al. 1996) drew a similar conclusion in a 31P magnetic resonance spectroscopy study aimed at determining whether CSF is characterized by abnormalities in oxidative muscle metabolism. However, the decrease in the oxidative capacity of muscle fibers of CFS subjects is possibly the consequence, at least in part, of reduced oxygen delivery. This theory is supported by the abnormal autonomic control of blood flow in patients. In fact, reaching the age-predicted target heart rate is a limiting factor in achieving maximal effort, possibly due to autonomic disturbances (De Becker et al. 2000). CFS muscles exhibit non-specific histological abnormalities (Edwards et al. 1993), and occasionally, alterations in muscle fiber type proportions and sizes (Lanea et al. 1998).
In conclusion, while it is difficult to distinguish between the initial factors that trigger CFS and other pathogenic factors maintaining it, there is significant evidence that the disease is correlated with several modifications in different tissues and organs.
The pathophysiology of CFS remains to be clarified, and the current lack of a biologically plausible mechanism of pathology mediation impedes the development of effective medical strategies. Several etiopathogenic hypotheses have been proposed, beginning from 1988 when Holmes et al. (1988) initially described this syndrome. CFS is unlikely to be due to persisting infection, although a number of infectious agents are associated with the condition. In fact, CFS is also referred to as Chronic Fatigue and Immune Dysfunction Syndrome (CFIDS) (Gerrity et al. 2004) or Chronic Epstein-Barr Virus (CEBV) (Katz 2002). Coxsackie, rubella and varicella viruses are sporadically implicated in some well-documented cases of CFS (Koelle et al. 2002). However, no one specific virus appears to be associated with this syndrome, and therefore, it is possible that CFS is triggered by a number of distinct viruses. In other cases, bacterial or parasitic infections appear to trigger the fatigue response, leading to the overall theory of post-infectious fatigue syndrome (Nicolson et al. 2003). Enteroviruses are well-known causes of acute respiratory and gastrointestinal infections with tropism for the central nervous system, muscles, and heart. In vitro experiments and animal models clearly establish a state of chronic persistence through the formation of double-stranded RNA, analogous to findings from muscle biopsies of patients with CFS. This evidence supports a plausible role for enteroviruses in the etiology of pathology (Chia 2005). An immune disturbance of some type mediated through the central, autonomic and/or peripheral nervous systems is suggested, in keeping with reports of excessive cytokine release (Quan and Herkenham 2002). Additionally, there is an indication of reduced perforin levels within the cytotoxic T cells of CFS subjects, suggesting a T cell-associated cytotoxic deficit in CFS. Since perforin is involved in homeostasis of the immune system, its deficiency is an important factor in CFS pathogenesis (Maher et al. 2005). However, the present data indicate that individuals with CFS display immune response changes that fall outside the normative ranges. There is currently no definitive evidence on whether these immune abnormalities are a cause or result of the illness.
More recently, a genetic component in the development of CFS was proposed. A number of studies demonstrate a possible association between human leukocyte antigen (HLA) class II and chronic fatigue immune dysfunction. Smith et al. (2005) reported that CFS is possibly associated with the HLA-DQA1 gene, although a role for other class II alleles in linkage immune disequilibrium cannot be excluded. To identify the gene markers of CFS-associated post-exertional fatigue, Whistler and colleagues measured the peripheral blood gene expression profiles of women with CFS and matched controls before and after exercise challenge. The expression of exercise-responsive genes differed between CFS patients and controls, particularly those implicated in chromatin and nucleosome assembly, cytoplasmic vesicles, membrane transport, ion transport, ion channel activity, and G protein-coupled receptor ontology (Whistler et al. 2005). Notably, last year, the U.S. Centers for Disease Control and Prevention in Atlanta, Georgia announced in an editorial in Science (Kaiser 2006), that “CFS is a pathology having a biological and, probably, genetic basis. These claims have attracted widespread media attention. However, like most aspects of CFS, the study and its findings remain controversial. Some scientists believe that the agency is overstating the case for a link between the syndrome and genetic mutations. While the expression patterns of several genes in the peripheral blood of patients with CFS have been established using polymerase chain reaction (PCR), the precise genes and metabolic pathways involved remain to be determined. It is important to ensure that a gene identified is specific for CFS, and does not occur in other diseases and infections”. Details of these studies are available in Pharmacogenomics (Vol 7 2006).
Clearly, CFS cannot be understood on the basis of single measurements of genetic, immune, endocrine, cardiovascular, or autonomic nervous system dysfunction (Prins et al. 2006). It remains a debilitating disease of uncertain etiology that is characterized by unexplained severe fatigue associated with a number of typical symptoms. A novel multifactorial model for the etiology of the disease, which includes aspects other than those analyzed, is required.
The role of oxidative stress in CFS
Oxidative stress is an emerging focus of research, in view of recent findings that it contributes to the pathology and clinical symptoms of CFS (Fulle et al. 2000; Steinberg et al. 2005; Fulle et al. 2003; Pall and Satterlee 2001).
Theoretically, oxidative stress is caused by an increase in the generation of reactive oxygen species (ROS), of which mitochondrial dysfunction is believed to be a main source. On the other hand, the condition may be induced by a decline in the efficiency of antioxidant enzyme systems. In most situations, both of these circumstances are involved (Sen 2001). Various studies have examined both possibilities (mainly the first) by attempting to identify markers of oxidative stress and protective antioxidant systems in vitro [36, 41] and in vivo (Steinberg et al. 2005; Vecchiet et al. 1996). In particular, experiments designed to measure malondialdehyde, methemoglobin, mean erythrocyte volume, and 2,3-diphosphoglycerate in sera of 33 patients diagnosed with CFS revealed an increase in all these parameters, relative to 27 age and sex-matched controls. Interesting data were obtained from the analysis of lipid peroxidation status in the plasma of CFS patients, an appropriate target to establish the damage induced by ROS accumulation. Keenoy and colleagues showed that patients with this pathology have increased LDL and VLDL susceptibility to copper-induced peroxidation, which is related to both lower levels of serum transferrin and other unidentified pro-oxidizing effects of CFS (Manuel y Keenoy et al. 2001). Recently, Kennedy et al. (2005) published results obtained from a large number of CFS patients divided into two groups those previously identified with cardiovascular risk factors and those that were not). Both groups displayed significantly increased levels of isoprostanes and oxidized low-density lipoproteins, indicative of lipid peroxidation induced by ROS accumulation. Moreover, CFS symptoms correlated with isoprostane levels in patients with low cardiovascular risk. This is the first report on elevated levels of the gold-standard measure of in vivo oxidative stress and its association with CFS symptoms.
Protein carbonyl levels, a measure of protein oxidation, were significantly enhanced in the sera of CFS patients, compared to controls (Smirnova and Pall 2003). In conclusion, the elevated protein carbonyl levels, alteration of lipidic peroxidation status in sera of CFS subjects, and the general absence of conflicting data support earlier reports that oxidative stress is associated with this syndrome.
Oxidative damage in muscle
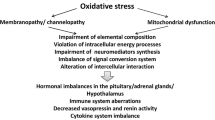
Muscle weakness and pain are the primary symptoms of CFS, suggesting that muscle is the major target of eventual oxidative stress status (Fig. 2). In fact, the sarcolemmal and sarcoplasmic membranes clearly present signs of oxidative damage caused by an increase in the generation of ROS without the possibility that the antioxidant enzyme system counteracts this event (Fulle et al. 2000; Steinberg et al. 2005; Fulle et al. 2003). The mitochondrial respiratory chain is the major site of ROS production in muscle cells. Electrons produced by the metabolism of glucose and fatty acids flow through the electron transport chain, leading to ATP generation. Mitochondrial muscle dysfunction may be implicated in the genesis of CFS pathology. In the early 1990s, Kuratsune et al. (Kuratsune et al. 1994) showed that the low acylcarnitine content in the serum of CFS patients is related to reduced energy production by muscle mitochondria. Pall (2000), followed by several other authors (Nijs et al. 2005), proposed that elevated peroxynitrite in CFS muscles induces mitochondrial dysfunction directly and/or positive feedback derived from increased cytokine levels.

Sites of cell damage induced by ROS as a consequence of oxidative stress. The dashed lines indicate the presence of cell damage in CFS muscles
Augmented cytokine levels trigger the production of new nitric oxide (NO), which combines with the anion superoxide, forming the more potent oxidant, peroxynitrite, which induces mitochondrial dysfunction in CFS muscles. In this case, an inverse correlation is possible between increased NO levels in the sarcoplasm derived from ex novo NO formation by nitric oxide synthase (NOS) stimulation and work produced in muscle of CFS patients. In vitro experiments performed in the presence of 95% pO2 indicate that NOS inhibition and the consequent decrease in NO intracellular levels positively regulates muscle contractility (Eu et al. 2003). However, this last observation is inconsistent with the general opinion that neoformation of NO is induced during skeletal muscle activity. This finding may indicates a positive trend in the force generation mechanism in both slow- and fast-twitch mammalian skeletal muscles (Murrant and Barclay 1995). The results are consistent with the positive action of other reactive oxygen intermediates on excitation-contraction (E-C) coupling in unfatigued skeletal muscle. Interestingly, when amphibian skeletal muscles were stimulated in vitro in the presence of NO donors and unsaturated pO2, which slow frequency rate (2 Hz), praecox onset of fatigue occurred, but only after a transitory increase in the first twitch tensions, due to an increase in Ca2+ release from the sarcoplasmic reticulum (Belia et al. 1998). This result was confirmed by Eu et al. (2003). The group demonstrated significant enhancement of muscle performance at low physiological pO2 (1%) and inhibition at higher physiological pO2 (20%), which directly influenced nNOS activity.
A number of investigators have focused on whether CFS is associated with defective oxidative metabolism of the skeletal muscle. It is difficult to establish this issue because of conflicting results and accounts of differences in methods and patient populations. A number of reports show lowering of maximal oxygen consumption and/or delivery (McCully et al. 1996), and/or abnormality in the muscle metabolism (Belia et al. 1998; McCully et al. 2004), whereas others do not (Gibson et al. 1993). An additional possibility is that abnormal function of the autonomic nervous system leads to reduced blood flow to active muscles, resulting in decreased O2 delivery and/or utilization by muscle, which, in turn suppresses mitochondrial capacity and exercise performance (Sargent et al 2002).
In conclusion, despite numerous doubts regarding the presence of ultrastructural mitochondrial abnormalities in CFS patients (Plioplys and Plioplys 1995), several lines of evidence suggest that aspects of mitochondrial alterations determined in these muscles (Vecchiet et al. 1996) are not related to a deficit in energy production. However in skeletal muscle, a contractile deficit or modification on fatigue onset may derive from energetic deficit due to mitochondrial activity, in addition to several other factors, some of which are present in the same muscle.
Our group previously demonstrated specific oxidative alterations in the vastus lateralis muscle of CFS patients, both as an increased value of oxidative damage markers (8OH-dG, MDA) and membrane fluidity and an imbalance in the oxidant–antioxidant system (Fulle et al. 2003). These findings are in agreement with the hypothesis that impairment of mitochondrial activity underlies an increase in the production of ROS that leads to muscle fatigue, similar to normal ageing (Mecocci et al. 1999). This theory is supported by CFS-associated muscle magnesium deficiency, a possible further cause of oxidative stress (Manuel y Keenoy et al. 2000).
Perspectives
The available data imply an imbalance in the redox status in some CFS muscle species. If this interferes with the contractile mechanism of skeletal muscle, at least three different levels of structure/function modifications may be related to the persistent presence of ROS:
-
E-C coupling (Sarcoplasmic reticulum)
-
Force production (Fibers and filaments)
-
Repair mechanism (Satellite cells).
The release of Ca2+ from sarcoplasmic reticulum cisternae mediated by ryanodine receptor (RyR) Ca2+ release channels is essential for muscle contraction. RyR channels in skeletal muscle are regulated by membrane potential, and do not require Ca2+ entry. Instead, Ca2+ release is necessary to trigger cardiac muscle contraction. Endogenous redox molecules formed by several mechanisms that induce S-nitrosylation or S-glutathionylation of several RyR cysteines can alter receptor function (Hidalgo et al. 2005). The modifications may include both activation and inhibition, and are dependent on the concentration of sulfhydryl-modifying compounds present in the muscle, period of exposure to these agents, and the nature of the chemical reaction with sulfhydryl groups (Pessah et al. 2002). In particular, in skeletal muscle, the oxidation status of RyR1 channel thiols (the RyR isoform mainly active in this tissue) is directly correlated to the functional status of the channel. In fact, oxidation of ca. 10 of these thiols had little effect on channel activity, whereas more extensive oxidation (resulting in 13 free thiols remaining per subunit) irreversibly inactivated the channel, reducing the opening status (Sun et al. 2001). In vitro studies performed with CFS muscle sarcoplasmic membranes indicate a diminished capacity of RyR-channels derived from pathological muscles to be maintained in the open status. This finding was confirmed in experiments in which caffeine (an alkaloid that stimulates the opening of SR Ca2+ channels) was unable to activate full load Ca2+ release from RyR (Fulle et al. 2003).
Compared to healthy sedentary people, CFS patients display significantly decreased exercise capacity, similar to that recorded in an aged population (CFS = old muscle in young body). Ageing is a complex process usually associated with a decrease in mass, strength, and velocity of contraction (sarcopenia) in muscle. This process is the result of several cellular changes, such as reduction in the number of motor units, modification of the fiber type composition, decreased synthesis of myofibrillar components, atrophy due to disuse, and accumulation of connective tissue. Sarcopenia is possibly triggered by ROS that have accumulated throughout an individual’s lifetime.
Initial in vivo data on the contractile properties of muscles of CFS patients revealed no consistent correlations between symptoms and changes in fiber type prevalence, fiber size, degenerative or regenerative features. Moreover, the contractile properties of quadriceps (maximal isometric force, force generation pathway, and relaxation rate) examined using symptom-limited incremental exercise tests, were not significantly altered, relative to normal controls (Edwards et al. 1993). Data from other experiments performed on quadriceps needle muscle biopsies in a larger CFS patient population (n = 105) are partially inconsistent with this conclusion (Lane et al. 1998). The authors cross-examined the proportions of type 1 and type 2 muscle fibers and the degree of muscle fiber atrophy in patients with chronic fatigue syndrome to determine to the extent of abnormalities due to inactivity (inactivity is expected to result in a shift to type 2 fiber predominance and fiber atrophy). No changes were evident, even when patients with abnormal lactate responses to exercise had a significantly lower proportion of mitochondria-rich type 1 muscle fibers. Unfortunately, no results clarifying the mechanisms of force production from muscle biopsies of CFS patients are available. Due to the lack of direct mechanical measurements in single fiber experiments, damage of filaments involved in the contraction mechanism induced by free radical accumulation is yet to be established.
During adult life, skeletal muscle fibers undergo drastic structural and functional modifications, and as a consequence, skeletal muscles activate a repair mechanism, based on the recruitment of satellite cells when required. These cells are usually committed to myogenic phenotype, and are in a quiescent state between the basal lamina and plasma membrane of myofibers. Satellite cells responsible for pre- and postnatal muscle growth are capable of both proliferation and differentiation, in order to repair skeletal muscle fibers following injury or other such stimuli (Zammit et al. 2002). Our recent observations show that the damage induced by ROS generation and accumulation during muscle life also affects quiescent satellite cells, which spend their life in close anatomic and functional contact with adult fibers. This status, which is derived from a decrease in the antioxidative capacity of these cells during ageing, may negatively affect the ability of ageing satellite cells to effect muscle repair (Fulle et al. 2005). Apart from an already cited paper published more than 10 years ago (Edwards et al. 1993) that discloses no alterations in the muscle regenerative ability in CFS patients, no data are available, and overall, no experiments have been performed on satellite cells derived from CFS muscle. In our opinion, there is a significant possibility that oxidative stress affects satellite cells, and that this status increases the functional deficit of these patients (Fig. 3).

Schematic representation of possible cellular targets of ROS accumulation in skeletal muscles of CFS patients
Conclusions
In conclusion, real progress in establishing that the etiopathogenesis of CFS is a ROS-dependent process will be possible only with definitive evidence that an excess of free radicals in CFS muscles (not balanced by an adequate increase in endogenous scavenger system activity) is directly correlated with modifications in critical factors, such as E-C coupling, force generation, and the satellite cell repair system.
References
Archard LC, Bowles NE, Behan PO et al (1988) Postviral fatigue syndrome: persistence of enterovirus RNA and elevated creatine kinase. J Roy Soc Med 81:326–329
Bates DW, Buchwald D, Lee J et al (1995) Clinical laboratory test findings in patients with chronic fatigue syndrome. Arch Intern Med 155:97–103
Belia S, Pietrangelo T, Fulle S et al (1998) Sodium nitroprusside, a NO donor, modifies Ca2+ transport and mechanical properties in frog skeletal muscle. J Muscle Res Cell Motil 19:865–876
Bennett AL, Chao CC, Hu S et al (1997) Elevation of bioactive transforming growth factor-beta in serum from patients with chronic fatigue syndrome. J Clin Immunol 17:160–166
Buchwald D, Wener MH, Pearlman T et al (1997) Markers of inflammation and immune activation in chronic fatigue and chronic fatigue syndrome. J Rheumatol 24:372–376
Chia JK (2005) The role of enterovirus in chronic fatigue syndrome. J Clin Pathol 58:1126–1132
De Becker P, Roeykens J, Reynders M et al (2000) Exercise capacity in chronic fatigue syndrome. Arch Intern Med 160:3270–3277
de Lange FP, Kalkman JS, Bleijenberg G et al (2005) Gray matter volume reduction in the chronic fatigue syndrome. Neuroimage 26:777–781
Edwards R, Gibson H, Clague J et al (1993) Muscle physiology and histopathology in chronic fatigue syndrome. In: Kleinman A, Straus S (eds) Chronic fatigue syndrome. Wiley & Sons, Chichester, pp 101–131
Eu JP, Hare JM, Hess DT et al (2003) Concerted regulation of skeletal muscle contractility by oxygen tension and endogenous nitric oxide. Proc Natl Acad Sci USA 100:15229–15234
Fukuda K, Straus SE, Hickie I et al (1994) The Chronic Fatigue Syndrome, a comprehensive approach to its definition and study. Ann Intern Med 121:953–959
Fulle S, Belia S, Vecchiet J et al (2003) Modification of the functional capacity of sarcoplasmic reticulum membranes in patients suffering from chronic fatigue syndrome. Neuromuscul Disord 13:479–484
Fulle S, Di Donna S, Puglielli C et al (2005) Age-dependent imbalance of the antioxidative system in human satellite cells. Exp Gerontol 40:189–197
Fulle S, Mecocci P, Fano G et al (2000) Specific oxidative alterations in vastus lateralis muscle of patients with the diagnosis of chronic fatigue syndrome. Free Rad Biol Med 29:1252–1259
Gerrity TR, Papanicolaou DA, Amsterdam JD et al (2004) CFIDS Association of America. Immunologic aspects of chronic fatigue syndrome. Report on a Research Symposium convened by The CFIDS Association of America and co-sponsored by the US Centers for Disease Control and Prevention and the National Institutes of Health. Neuroimmunomodulation 11:351–357
Gibson H, Carroll N, Clague JE et al (1993) Exercise performance and fatigability in patients with chronic fatigue syndrome. J Neurol Neurosurg Psychiatry 56:993–998
Hanson SJ, Gause W, Natelson B (2001) Detection of immunologically significant factors for chronic fatigue syndrome using neural-network classifiers. Clin Diagn Lab Immunol 8:658–662
Hidalgo C, Donoso P, Carrascom MA (2005) The ryanodine receptors Ca2+ release channels: cellular redox sensors?. IUBMB Life 57:315–322
Holmes GP, Kaplan JE, Gantz NM et al (1988) Chronic fatigue syndrome: a working case definition. Ann Intern Med 108:387–389
Jason LA, Richman JA, Rademaker AW et al (1999) A community-based study of chronic fatigue syndrome. Arch Intern Med 159:2129–2137
Kaiser J (2006) BIOMEDICINE: Genes and Chronic Fatigue How Strong Is the Evidence?. Science 312:669–671
Katz BZ (2002) Update on chronic fatigue syndrome and Epstein-Barr virus. Pediatr Ann 31:741–744
Kennedy G, Spence VA, McLaren M et al (2005) Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med 39:584–589
Koelle DM, Barcy S, Huang ML et al (2002) Markers of viral infection in monozygotic twins discordant for chronic fatigue syndrome. Clin Infect Dis 35:518–525
Kuratsune H, Yamaguti K, Takahashi M et al (1994) Acylcarnitine deficiency in chronic fatigue syndrome. Clin Infect Dis 18:S62–S67
Lane RJ, Barrett MC, Woodrow D et al (1998) Muscle fibre characteristics and lactate responses to exercise in chronic fatigue syndrome. J Neurol Neurosurg Psychiatry 64:362–367
Lanea RJM, Barrett MC, Taylor DJ et al (1998) Heterogeneity in chronic fatigue syndrome: evidence from magnetic resonance spectroscopy of muscle. Neuromuscul Disord 8:204–209
Lange G, DeLuca J, Maldjian JA et al (1999) Brain MRI abnormalities exist in a subset of patients with chronic fatigue syndrome. J Neurol Sci 171:3–7
Linde A, Andersson B, Svenson SB et al (1992) Serum levels of lymphokines and soluble cellular receptors in primary Epstein-Barr virus infection and in patients with chronic fatigue syndrome. J Infect Dis 165:994–1000
Maher KJ, Klimas NG, Fletcher MA (2005) Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin Exp Immunol 142:505–511
Manuel y Keenoy B, Moorkens G, Vertommen J et al (2000) Magnesium status and parameters of the oxidant-antioxidant balance in patients with chronic fatigue: effects of supplementation with magnesium. J Am Coll Nutr 19:374–382
Manuel y Keenoy B, Moorkens G, Vertommen J et al (2001) Antioxidant status and lipoprotein peroxidation in chronic fatigue syndrome. Life Sci 68:2037–2049
McCully KK, Natelson BH, Iotti S et al (1996) Reduced oxidative muscle metabolism in chronic fatigue syndrome. Muscle Nerve 19:621–625
McCully KK, Smith S, Rajaei S et al (2004) Muscle metabolism with blood flow restriction in chronic fatigue syndrome. J Appl Physiol 96:871–878
Mecocci P, Fanò G, Fulle S et al (1999) Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic Biol Med 26:303–308
Murrant CL, Barclay JK (1995) Endothelial cell products alter mammalian skeletal muscle function in vitro. Can J Physiol Pharmacol 73:736–741
Nicolson GL, Gan R, Haier J (2003) Multiple co-infections (Mycoplasma, Chlamydia, human herpes virus-6) in blood of chronic fatigue syndrome patients: association with signs and symptoms. APMIS 111:557–566
Nijs J, Van de Velde B, De Meirleir K (2005) Pain in patients with chronic fatigue syndrome: Does nitric oxide trigger central sensitization? Med Hypotheses 64:558–562
Pall ML (2000) Elevated, sustained peroxynitrite levels as the cause of chronic fatigue syndrome. Med Hypotheses 54:115–125
Pall ML, Satterlee JD (2001) Elevated nitric oxide/peroxynitrite mechanism for the common etiology of multiple chemical sensitivity, chronic fatigue syndrome, and posttraumatic stress disorder. Ann N Y Acad Sci 933:323–329
Pessah IN, Kim KH, Feng W (2002) Redox sensing properties of the ryanodine receptor complex. Front Biosci 7:a72–79
Plioplys AV, Plioplys S (1995) Electron-microscopic investigation of muscle mitochondria in chronic fatigue syndrome. Neuropsychobiology 32:175–181
Prins JB, Van Der Meer JW, Bleijenberg G (2006) Chronic fatigue Syndrome. Lancet 367:346–55
Quan N, Herkenham M (2002) Connecting cytokines and brain: a review of current issues. Histol Histopathol 17:273–288
Reeves WC, Lloyd A, Vernon SD et al. (2003) International Chronic Fatigue Syndrome Study Group. Identification of ambiguities in the 1994 chronic fatigue syndrome research case definition and recommendations for resolution. BMC Health Serv Res 3:25
Saggini R, Pizzigallo E, Vecchiet J et al (1998) Alteration of spatial-temporal parameters of gait in chronic fatigue syndrome patients. J Neurol Sci 154:18–25
Sakudo A, Kuratsune H, Kobayashi T et al (2006) Spectroscopic diagnosis of chronic fatigue syndrome by visible and near-infrared spectroscopy in serum samples. Biochem Biophys Res Commun 345(4):1513–1516
Sargent C, Scroop G, Nemeth P et al (2002) Maximal oxygen uptake and lactate metabolism are normal in chronic fatigue syndrome. Med Sci Sports Exerc 34:51–56
Schillings ML, Kalkman JS, van der Werf SP et al (2004) Diminished central activation during maximal voluntary contraction in chronic fatigue syndrome. Clin Neurophysiol 115:2518–2524
Sen CK (2001) Antioxidant and redox regulation of cellular signaling: introduction. Med Sci Sports Exerc 33:368–370
Siemionow V, Fang Y, Calabrese L et al (2004) Altered central nervous system signal during motor performance in chronic fatigue syndrome. Clin Neurophysiol 115:2372–2381
Smirnova IV, Pall ML (2003) Elevated levels of protein carbonyls in sera of chronic fatigue syndrome patients. Mol Cell Biochem 248:93–95
Smith WR, Noonan C, Buchwald D (2006) Mortality in a cohort of chronically fatigued patients. Psychol Med 36(9):1301–1306
Smith J, Fritz EL, Kerr JR et al (2005) Association of chronic fatigue syndrome with human leucocyte antigen class II alleles. J Clin Pathol 58:860–863
Steinberg JG, Mambrini O, Bregeon F et al (2005) Chronic fatigue syndrome: assessment of increased oxidative stress and altered muscle excitability in response to incremental exercise. J Intern Med 257:299–310
Sun J, Xu L, Eu JP et al (2001) Classes of thiols that influence the activity of the skeletal muscle calcium release channel. J Biol Chem 276:15625–15630
Vecchiet L, Montanari G, Pizzigallo E et al (1996) Sensory characterization of somatic parietal tissues in humans with chronic fatigue syndrome. Neurosci Lett 208:117–120
Whistler T, Jones JF, Unger ER et al (2005) Exercise responsive genes measured in peripheral blood of women with chronic fatigue syndrome and matched control subjects. BMC Physiol 5:5
Wong R, Lopaschuk G, Zhu G et al (1992) Skeletal muscle metabolism in the chronic fatigue syndrome. In vivo assessment by 31P nuclear magnetic resonance spectroscopy. Chest 102:1716–1722
Wyller VB (2007) The chronic fatigue syndrome- an update. Acta Neurol Scand Supp 187:7–14
Zammit PS, Heslop L, Hudon V et al (2002) Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp Cell Res 281:39–49
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fulle, S., Pietrangelo, T., Mancinelli, R. et al. Specific correlations between muscle oxidative stress and chronic fatigue syndrome: a working hypothesis. J Muscle Res Cell Motil 28, 355–362 (2007). https://doi.org/10.1007/s10974-008-9128-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-008-9128-y