
Abstract
This study explores the impact of different levels of halloysite nanotubes (HNT) and d-isomer on the non-isothermal crystallization behavior of the polylactic acid (PLA). The processing conditions, thermal flow, molecular orientation and the presence of nanofillers can affect the crystallinity and properties of PLA. In this work, PLA-based nanocomposite filaments were produced and their crystallization kinetics was studied. The data obtained through experiments, using differential scanning calorimetry at various cooling rates, were analyzed employing the Jeziorny and Mo models. And the activation energy of crystallization was determined by the Friedman method. The results showed that the growth of PLA crystals is three-dimensional, the HNT can accelerate the crystallization process, and crystallization occurs at higher temperatures in the presence of flow. The activation energy increased with addiction of more HNT in the nanocomposite. The study also found that the d-isomer has a more significant influence on the crystallization of PLA than HNT, which can inhibit crystal formation in some cases. These findings provide insights into the factors that can influence the properties of PLA nanocomposites and how they can be optimized.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The polylactic acid (PLA) is an aliphatic, non-toxic, transparent, biodegradable and biocompatible polyester. Because it has a chiral carbon (optical isomerism), this polymer can be found as poly(L-lactic acid) (PLLA), poly(d-lactic acid) (PLDA) or even poly(DL-lactic acid) (PDLLA), depending on the enantiomers present during synthesis. Its crystallization kinetics is generally slow and depends on the presence and content of its stereoisomers. The addition of even a small percentage of the d-isomer in PLLA makes the crystallization even slower and causes a reduction in the degree of crystallinity. This slow crystallization kinetic exhibits a crystallization half-time (t1/2) spanning from 17 to 40 min for the pure PLA, which is impractical for industrial-scale manufacturing (combined with poor heat and impact resistance of the PLA), particularly for processes like injection molding, where cycle times typically last 60 to 90 s [1,2,3]. Despite these limitations, PLA has shown considerable potential as a material in various applications such as packaging, nanocomposites, biomedical and three-dimensional (3D) printing [4,5,6].
In general, the use of nucleating agents is an effective strategy to increase the crystallization temperature and crystallinity of a material, resulting in improved properties and processability. For the PLA, the nucleating agents zinc phenylphosphonate [7]; N,N′,N″-tricyclohexyl-1,3,5-benzenetricarboxylamide (TMC) [8]; N,N′-bis(2-hydroxyethyl)-terephthalamide (BHET) [9]; Kraft and Organosolv lignin [10],ultrafine talc [10], dilithium hexahydrophthalate [11] and cadmium phenylmalonate (PMACd) [12] were studied. All of them reported increase in the degree of crystallinity and faster crystallization. Wu et al. [13] and Liu et al. [14] researched PLA/HNT nanocomposites and concluded that HNT worked as a nucleating agent, increased the mechanical properties, and was well dispersed in the PLA matrix.
Halloysite nanotubes (HNT) are clay minerals that can act as mechanical reinforcing nanoparticles in polymeric matrices. Additionally, HNT can be utilized as a material for hydrogen storage due to its hydrogen adsorption capabilities [15]. It can also function as a sensor (for dopamine and paracetamol) within a Cu-MOF (metal–organic framework), achieved by growing Cu-MOF in situ on HNTs/reduced graphene oxide surfaces [16]. In addition, because it has a hollow tubular structure and biocompatibility, HNT has substantial potential for various applications, including biomedical and packaging uses [13, 14, 17]. This potential stems from their tubular structure, which allows for effective transport of fluid medicines and antibacterial agents, for example [18, 19]. But this all depends on understanding the influence of the nanoparticle on the matrix. Nanoparticles must be well distributed and well dispersed for the performance of the nanocomposite to be optimized. The successful production of these nanocomposites is influenced by the behavior of the material observed when there is variation in the thermomechanical conditions. So, to ensure the final properties of the product, it is necessary to know the kinetics of crystallization of PLA/HNT [20,21,22,23].
The crystallinity of a polymeric material is one of the main characteristics that determine its properties and, consequently, its performance during application. High crystallinity may be required depending on the use, positively affecting properties such as elastic modulus, chemical resistance and barrier behavior to liquids and gases, among others. However, molecular ordering can negatively impact applications that depend on high transparency, high degradability and dimensional stability during processing. The crystallization process is influenced by intrinsic parameters of the polymer matrix (chemical structure, type of chain, molar mass), as well as by process conditions (time, temperature, pressure, deformation) and the presence of additives (nucleating agents, reinforcing fillers, functional fillers, etc.), and their understanding and control have great scientific and technological importance [24, 25].
Shear-induced and strain-induced crystallizations represent alternative methods to accelerate the crystallization kinetics, often occurring during processing through the orientation of the polymer chains. Bojda et al. [26] studied the shear flow effects on two distinct PLA grades (varying in d-isomer content) and noted that shear flow indeed accelerated crystallization, whereas a higher d-isomer content had the opposite effect, decelerating it. Similarly, Zin et al. [27] and Yin et al. [28] investigated the impact of strain on crystallization in PLA (with varying d-isomer content) and reported consistent findings with Bojda's research.
The main objective of this work was to analyze the influence of the d-isomer content and the presence and content of HNT on the non-isothermal crystallization kinetics, in quiescent and shear-induced conditions, of PLA/HNT nanocomposites. This aimed to observe which conditions increase the crystallization rate and crystallinity. Thermal and rheological characterizations of PLA/HNT nanocomposites were also conducted.
Experimental
Materials
Polylactic acid (PLA) used was the Ingeo Biopolymer 4032D (1.5% PLA d-isomer) and 4043D (5% PLA d-isomer), produced by NatureWorks. According to the manufacturer, the 4032D resin has a specific mass of 1.24 g cm−3 (ASTM D792), a melt flow index of 7 g 10−1 min−1 (210 °C 2.16−1 kg−1) and a melting range between 155 and 170 °C. The 4043D resin has a specific mass of 1.24 g cm−3 (ASTM D792), a melt flow index of 6 g 10−1 min−1 (210 °C 2.16−1 kg−1), a melting range between 145 and 160 °C, and glass transition temperature between 55 and 60 °C. Throughout this work, the 4032D and 4043D PLAs will be referred to as PLA1.5D and PLA5.0D, respectively, alluding to their d-isomer content. The halloysite nanotubes (HNT) were the Dragonite-XR grade, extracted by Applied Minerals Inc. According to the manufacturer, the material has a surface area of 64.66 m2 g−1, an average internal diameter of 15 nm, an average external diameter of 50 nm, an average length of 1.75 µm and a density of 2.52 g cm−3. Its cation exchange capacity (CEC) is 8 meq 100−1 g−1.
Filaments production
The two grades of PLA pellets were cryogenically grounded using a Mikro-Bantam® hammer mill (Hosokawa Micron Powder Systems). The filaments were produced by melt extrusion using both PLA grades and four different concentrations of HNT (0, 2.5, 5.0 and 10.0 mass percentage). The materials were mixed manually in plastic bags and dried for 12 h at 60 °C in a circulating air oven before being added to the extruder. The twin-screw extruder was a model MP19 from B&P Process Equipment Systems with a screw diameter of 19 mm and L/D of 25. The processing conditions were adjusted in order to obtain homogeneous filaments with an average diameter of 1.75 ± 0.10 mm. The temperature profile was between 155 and 175 °C, the screw speed was 60 rpm, and the feed speed was 1 kg h−1. The filaments were cooled in water at room temperature. Before characterization, the materials were dried for 12 h at 60 °C in a circulating air oven.
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was used to assess the thermal stability of PLA and its nanocomposites. The equipment used was the model Q50 from TA Instruments. The tests occurred under an inert atmosphere (N2), at a constant heating rate (20 °C min−1), from room temperature to 800 °C. Experimental data were analyzed with TA Universal Analysis 2000 software (TA Instruments).
Rheological properties
The influence of the presence and content of HNT on the rheological behavior of the nanocomposites was evaluated by parallel plate rheometry. Viscosity behavior as a function of shear rate and storage modulus (G′) and loss modulus (G″) as a function of frequency (oscillatory regime between 0.02 and 500 rad s−1) were obtained in a stress-controlled rheometer AR-G2, from TA Instruments. The main test parameters were plate diameter of 25 mm, gap of 1 mm, temperature of 200 °C and inert atmosphere of N2.
Crystallization kinetics by differential scanning calorimetry
The quiescent non-isothermal crystallization kinetics was analyzed by differential scanning calorimetry (DSC) using a TA Instruments equipment, model Q2000. The materials were subjected to four different cooling rates Φ: 1, 3, 5 and 10 °C min−1, adopting the following experimental procedure: (1) heating from 20 °C to 200 °C at 10 °C min−1; (2) isothermal stage at 200 °C for 5 min to balance the temperature; (3) cooling from 200 °C to 20 °C at a given Φ; (4) isothermal stage at 20 °C for 5 min to balance the temperature; and (5) second heating from 20 to 200 °C at 10 °C min−1.
The heat of crystallization ΔHc was obtained from the cooling steps of the non-isothermal analyses by the complete integration of the crystallization peak divided by the sample mass. The relative crystallinity as a function of temperature Xr(T) was calculated as the ratio between the integral of the area under the exothermic peak at a given temperature T (ΔHT) and the integral of the total area under the exothermic peak (ΔH0), according to Eq. 1 [12]:
where the numerator is the heat generated at a certain temperature T and the denominator is the total heat obtained during complete crystallization [12].
The crystallization temperature during cooling can be converted to the crystallization time according to Eq. 2 [12]:
where T is the temperature at time t and T0 is the initial temperature. The transformation from T to t can be done when a constant Φ is used. Thus, the time to obtain 50% relative crystallinity, t1/2, can be determined.
The degree of crystallinity of the samples was obtained by Eq. 3 [12]:
where ΔHf is the enthalpy of melting of the sample, ΔHcc is the enthalpy of cold crystallization occurring during heating, ΔH 0f is the equilibrium melting enthalpy (the theoretical value for 100% crystalline PLA is 93 J g−1) [29] and φ is the mass fraction of HNT.
The following parameters were obtained from the second heating steps: glass transition temperature Tg; cold crystallization peak temperature T Pcc ; melting temperature Tm; enthalpy of cold crystallization ΔHcc; and enthalpy of fusion ΔHf.
Crystallization kinetics by rheometry
The shear flow-induced non-isothermal crystallization kinetics was evaluated on a TA Instruments AR-G2 rheometer, using the procedure described by Farah and Bretas [30] and a parallel plate geometry with a diameter of 25 mm and distance between plates of 1 mm. The material was melted at 200 °C and, after 3 min, was subjected to cooling at a given Φ (5 or 10 °C min−1) and shear rate (0.1 or 1.0 s−1). By monitoring the stress as a function of temperature, it was possible to obtain the temperature at which crystal structures began to grow.
Result and discussion
Thermogravimetric analysis
The main events of the thermogravimetry of pure halloysite are concentrated in: (1) between 50 and 150 °C, attributed to the release of adsorbed water on the surface and the beginning of the gradual decrease of water between the interlamellar layers of halloysite (2.0% reduction in mass); (2) between 100 and 400 °C, attributed to the total release of structural water (interlamellar water) and organic impurities (4.0% decrease in mass); and (3) between 400 and 600 °C, attributed to the dehydroxylation of the AlOH and SiOH groups (11.7% mass loss). Since samples only reached 800 °C, no event was attributed to the formation of new phases, reported to happen between 885 and 1000 °C [31, 32].
Table 1 shows the decomposition temperatures (onset Ton, peak Tp and end Tend) of each filament. At Tp and Tend, there is a small tendency for the temperature to decrease with increasing HNT concentration, that is, a tendency to reduce the temperature range in which decomposition occurs, shifting the maximum decomposition rate to lower temperature values. On the other hand, the Ton remains the same with the incorporation of HNT. Alakrach et al. [33] used different types of HNT in different mass percentages as nanofillers in the PLA. It is argued that these nanofillers may have a thermal barrier effect (stronger in clay, but HNT can have a limited effect), increasing thermal stability, but at the same time there is the opposite effect of water, which decreases thermal stability. The best thermal barrier effect is obtained with better dispersion, and in the work of Alakrach et al., this occurs with 5% HNT.
The thermal degradation of PLA, which occurs at T ≥ Tm, is mainly attributed to the random scission of the main chain and to the depolymerization reactions that release monomers as the final product (unzipping). If there is a decrease in thermal stability with the incorporation of HNT, there should be a decrease in Ton. However, this was not observed in this study.
Liu et al. [34], Wu et al. [13] and Kim et al. [35] investigated the effect on the thermal properties of PLA composites. Based on the TGA results, Liu et al. noticed that fillers caused a decrease in the matrix molar mass and, consequently, a drop in thermal stability during mixing and processing. The PLA degradation may be influenced by HNT, which can act as an accelerator for matrix decomposition at high temperatures. Wu et al. attributed the decrease in thermal stability to the release of water vapor from the HNT (both structural water and water adsorbed on the surface), accelerating the decomposition of the ester bonds of PLA. Kim et al. justified the decrease in thermal stability by the presence, on the surface of HNT, of Bronsted acid sites (Al–OH and Si–OH) that act as a catalyst to PLA pyrolysis [5, 6, 36,37,38,39,40,41,42,43]. A more intense change in thermal stability with the incorporation of HNT, especially in terms of Ton, may have been prevented in the current investigation due to the adequate drying process and adjustment of processing parameters.
Rheological properties
Figure 1 shows the curves of viscosity η as a function of the shear rate \(\dot{\gamma }\) obtained by parallel plate rheometry. The power-law model (η = m·γn−1) describes the experimental data collected, where m is the consistency index and n is the power-law index. Table 2 shows the values of initial viscosity η0 (Newtonian plateau) and n.

Viscosity versus shear rate curves of PLA and PLA/HNT: a PLA1.5D; b PLA5.0D
It was observed that the behaviors of all samples are practically Newtonian up to 10 s−1, as indicated by the plateau formed in Fig. 1 and by the values of n close to one in Table 2. No reduction in the viscosity was observed with the incorporation of HNT. On the contrary, there is a slight tendency toward an increase of η0 with increasing HNT content. This corroborates what was observed in the analysis of Ton based on TGA tests; that is, there is no evidence of relevant degradation in the PLA. For Kelnar et al. [44], the increase in viscosity in a PLA/HNT nanocomposite is given by the increase in particle anisometry (HNT), which leads to stronger interactions and short distances between particles (in a certain volumetric fraction of dispersed particles).
Cobos et al. [45] and Sharma et al. [46] studied the viscosity of the PLA/HNT nanocomposites and noticed a reduction in viscosity with the incorporation of HNT. For Sharma et al., the cause of the reduction in viscosity was the hydrolysis, while for Cobos et al., it was a plausible reduction in molecular mass. The presence of HNT justifies both phenomena.
Figure 2 shows the G′ and G″ curves (as a function of angular frequency) for all samples. All compositions showed typical behavior of molten thermoplastics in the terminal zone, with G″ greater than G′ at low oscillation frequencies. The results indicate that the behaviors of these materials are similar to that of a viscous liquid (G″ > G′) for the entire angular frequency range.

Storage modulus (G′) and loss modulus (G″) versus angular frequency of PLA and PLA/HNT: a PLA1.5D; b PLA5.0D
The degree of dispersion of HNT is related to the viscoelastic behavior of the nanocomposites, and the moduli and slopes of the curves in the low-frequency region (terminal zone) can be used to assess this dispersion and the formation of possible percolated structures [47]. Zhao et al. [48] demonstrated that the creation of a percolated network in nanocomposites could be rheologically visualized owing to the change in the behavior of the nanocomposite, which starts to present characteristics similar to those of a solid (with G′ > G″, and G′ and G″ ~ ω0, in the terminal zone, i.e., at low oscillation frequencies). Therefore, no signs of the formation of percolated networks were noticed in the current investigation.
Although there is an increase in G′ with the addition of HNT (a slight increase was also observed for η0), this did not change the predominantly viscous behavior of PLA in the terminal zone (but the behavior of PLA with an increase in HNT tends toward greater elasticity).
Crystallization kinetics by differential scanning calorimetry
Table 3 shows the results obtained with the cooling (at different Φ) and second heating steps of DSC tests. The results of Xc show that 5% of d-isomer in PLA is enough to inhibit the crystallization of the material at the Φ of 5 and 10 °C min−1, and in this case, the presence of the inorganic nanoparticles does not change the behavior of the matrix, and induces amorphous products. (With 4% of d-isomer, in the study by Díez-Rodriguez et al. [49], crystallization has already been restricted with a cooling/heating rate of 10 °C min−1.) The presence of 1.5% D does not completely prevent molecular ordering, as indicated by the values of Xc up to 49% found for the PLA1.5D samples. It is possible to observe that HNT acts as a nucleating agent for PLA since nanocomposites with higher nanofiller concentrations presented higher Xc. As Φ increased, a considerable reduction in Xc was observed, as expected [1]. Venkatesh et al. [50] concluded that HNT acts as a nucleating agent in PLA, as the PLA/HNT nanocomposite crystallizes faster than neat PLA.
Both PLA1.5D and PLA5.0D samples do not have ΔHcc e T Pcc after cooling at a rate of 1 °C min−1 since the samples have enough time to crystallize at this rate. Pure PLA1.5D did not crystallize completely, on cooling, at 5 and 10 °C min−1, whereas its nanocomposites did not crystallize only at the cooling rate of 10 °C min−1, showing that the HNT helped in the crystallization, even at only 2.5% mass fraction. When comparing PLA1.5D and PLA5.0D, it is evident that the lower percentage of d-isomer in PLA1.5D allowed it to crystallize faster on cooling, indicating that D makes crystallization harder [1].
Table 3 also shows the results obtained from the second heating. It is known that Tg is a second-order transition that is manifested by the variation of the baseline of the DSC curve and that can be directly affected by crystallinity and molar mass. For the materials studied, the Tg does not seem to be influenced by changes in Φ or HNT content, at least in the ranges considered. The Tg values of PLA1.5D and its nanocomposites vary between 60 and 65 °C, and for PLA5.0D, they are around 60 and 61 °C. The d-isomer in this case can function as a defect in the polymer chain, causing a decrease in the crystal growth rate and an increase in the amorphous phase. As there is no significant change in Tg with the addition of HNT, there is a possibility that the nanofiller does not have much influence on the anchoring of the amorphous phase chains, and this could indicate that HNT may have low interaction with PLA or the HNT is in a small volume fraction [1, 51, 52].
The endothermic peak, present in the second heating, occurs at Tm. In samples that were subjected to Φ of 5 and 10 °C min−1, HNT did not cause a significant change in Tm, but the percentage of d-isomer is important, as PLA1.5D presented Tm between 169 and 170 °C and PLA5.0D had Tm between 151 and 153 °C [1, 2, 14, 53].
When exposed to a cooling rate of 3 °C min−1, PLA shows unique Tm values, similar to rates of 5 and 10 °C min−1, but for PLA1.5D with 5 and 10% HNT and PLA5.0D with 10% HNT, a second melting peak emerges leading to two Tm values (as shown in Table 3, where there are two values of Tm). This can be due to either the polymorphism of the PLA crystals or the melting of crystalline regions of varying sizes and perfections formed during cooling and crystallization. Most samples exposed to a cooling rate of 1 °C min−1 also show two endothermic peaks with the same explanation, except for PLA5.0D with 10% HNT, which only shows one peak (with a small shoulder), possibly due to the low rate or the influence of HNT in forming more homogeneous crystals [1, 2, 13, 53].
Dong et al. [54] concluded in their study that increasing the content of HNT in a PLA/HNT nanocomposite causes almost no change in Tg. However, increasing the percentage of HNT in the nanocomposites causes a decrease in Tm, as there are incomplete crystal structures formed by heterogeneous nucleation of HNT with thinner and/or less perfect crystalline lamellae (differently from what is proposed here, Dong et al. used electrospun mats).
Figures 3 and 4 are created based on Eqs. 1 and 2 (basically, the crystallization peak obtained at the cooling step was integrated with the baseline being tracked by the initial and final points, as the equations determine) and show a typical S shape for the curves of relative crystallinity Xr as a function of crystallization time. The shape obtained is associated with primary crystallization, the stage of crystallization where crystalline regions occupy the entire volume of the mass [20].

Relative crystallinity Xr curves of PLA1.5D as a function of crystallization time for different Φ: a 10 °C min−1; b 5 °C min−1; c 3 °C min−1; d 1 °C min.−1

Relative crystallinity Xr curves of PLA5.0D as a function of crystallization time for different Φ: a 3 °C min−1; b 1 °C min.−1
Observing the data in Table 3, Figs. 3 and 4, it can again be suggested that HNT influences the crystallization of PLA1.5D. The t1/2 increases with decreasing Φ, which indicates a lower crystallization rate (relative) and tends to decrease with an increasing percentage of HNT. The increase of d-isomer acts in a way that decreases the Φ necessary for crystallization to occur, as indicated by the fact that PLA5.0D could not be crystallized even during cooling at 5 °C min−1 [1].
Shi et al. [55, 56] studied PLA nanocomposites, and HNT worked as a nucleating agent. Halloysite particles increase the melt crystallization rate and shorten the crystallization time t1/2 by half. Wu et al. [13] studied the crystallization and melting behavior of the PLA (with 4% of d-isomer) nanocomposite with HNT from a DSC test with a Φ of 10 °C min−1 and explained that the lack of the crystallization peak of PLA and its nanocomposites was due to the rigid segments in the polymer backbone molecular chain. The study also cites that cold crystallization happens more easily with HNT, thus indicating that it can function as a nucleating agent, which was also observed in the current study.
Jeziorny’s model
Jeziorny [57] proposed a change for the Avrami model (Eq. 4) that can be used to study the non-isothermal crystallization kinetics. The model adopts the variable Zc, a modified crystallization rate constant that considers the Φ and is represented in Eq. 5.
where n is the Avrami constant, a function of the nucleation mechanism and the geometric shape of the growing crystal; Zt is the Avrami crystallization rate, constant that includes both nucleation and growth parameters; Xr is the relative crystallinity at an arbitrary time t [58]; and Zc is the modified crystallization rate constant [12, 57].
The graphs of log[− ln(1 − Xr)] versus log(t) were created using the DSC measurement data for the studied materials. The Xr were was transformed into log[− ln(1 − Xr)], and the crystallization time was converted using Eq. 2 for use in the Jeziorny model. The plotted data were then fitted with lines to determine the Avrami parameters and are displayed in the following table. It is possible to observe in Table 4 that the Zc values increase with the increase in the Φ, indicating an increase in the crystallization rate, something already expected by previous analyses. The rise of Zc means that at a higher Φ, there is a higher crystallization rate. Su et al. [59] studied PLA filled with carbon black, and the incorporation of filler had the same behavior on Zc.
The values of n are related to the processes of nucleation and growth. When n values are greater than three, they indicate three-dimensional growth of the formed crystals (spherulites), between two and three growth of disk-shaped crystals (two-dimensional) and values between one and two formation of crystals in a shape similar to rods [11, 12, 21, 57]. In the current investigation, most samples undergo three-dimensional growth (spherulite) [60].
Table 4 is used to calculate Xr of the Jeziorny model (using Eq. 4) for each sample and Φ. The model’s calculations were compared to the experimental Xr values, and the curves are displayed in Fig. 5. According to Li et al. [11, 12], the Jeziorny model is better suited for representing primary crystallization in PLA. As a result, the model and experimental data in Fig. 5 do not align, indicating secondary crystallization and spherulite impingement [59].

Comparison between the relative crystallinity Xr as a function of the crystallization time of the Jeziorny model with the experimental data: a PLA1.5D 10 °C min−1; b PLA1.5D 5 °C min−1; c PLA1.5D 3 °C min−1; d PLA1.5D 1 °C min−1; e PLA5.0D 3 °C min−1; f PLA5.0D 1 °C min.−1
Mo’s model
Mo’s model relates the Φ to time at constant Xr, and Jeziorny’s correlates Xr with time at constant Φ. Mo’s method was developed by modifying the Avrami model, as expressed in Eqs. 6, 7 and 8 [11, 12, 61].
where K(T) is a function of cooling, which indicates the speed of the process (related to the global crystallization rate); m is the Ozawa exponent, which depends on the size of crystal growth; F(T) is the value of the Φ to be chosen in a unit crystallization time when the system has the same crystallinity; and α is the ratio between n and m, values linked to the growth dimension of the polymer crystals [11, 12, 61, 62].
Creating graphs of log(Φ) versus log(t) involved DSC data and Eqs. 2 and 6 for the studied materials. The plotted data were then fitted with lines using linear regression to determine the Mo parameters, with log(F(T)) being the constant and α being the slope of the line. There are two methods to obtain these points: The first is using the polynomial equation derived from the DSC data and Eq. 7 and finding the value of t by inserting Xr for a specific Φ in the equation; and the second method involves interpolating experimental data near the desired points. The behavior of log(F(T)) and α is shown in Fig. 6.

Dependence of the parameters log (F(T)) and α of the Mo model as a function of Xr: a PLA1.5D 0%HNT; b PLA1.5D 2.5%HNT; c PLA1.5D 5.0%HNT; d PLA1.5D 10.0%HNT; e PLA5.0D 10.0%HNT
According to the model, F(T) should depend on Xr, while α should be either independent or only slightly varying. The values of α in this study show minimal change with Xr due to three-dimensional crystal growth in crystallization [11, 12, 61, 62]. With increasing Xr, F(T) and α values also increase, indicating that the Xr of nanocomposites can be enhanced by speeding up the cooling process, and the refinement of crystals increases as crystallization progresses. Additionally, as noted by Liu et al. [11], F(T) values systematically increase with growing crystallinity, implying that a higher Φ should be used to attain a higher Xr. Thus, α can be considered to have a strong relationship with Φ.
The Mo parameters cannot be directly interpreted, but Fig. 6 shows growth in both log(F(T)) and α with increasing Xr for all samples. The results of neat PLA1.5D and its nanocomposites reveal that higher values of log(F(T)) and α occur when HNT is present. It is also evident that log(F(T)) values increase with increasing Xr, implying that higher Φ are required to achieve higher Xr with a given crystallization time, indicating that higher crystallization rates occur at higher Φ [11, 12, 63].
Estimations of the Xr of the Mo model were performed for each sample and available Φ on a point-by-point basis. The values calculated by the model were compared to the experimental results, which are shown in Figs. 7 to 10.

Comparison between the relative crystallinity Xr as a function of crystallization time of the Mo model with the experimental data at Φ of 10 °C min−1: a PLA1.5D 0%HNT; b PLA1.5D 2.5%HNT; c PLA1.5D 5.0%HNT; d PLA1.5D 10.0%HNT

Comparison between the relative crystallinity Xr as a function of crystallization time of the Mo model with the experimental data at Φ of 5 °C min−1: a PLA1.5D 0%HNT; b PLA1.5D 2.5%HNT; c PLA1.5D 5.0%HNT; d PLA1.5D 10.0%HNT

Comparison between the relative crystallinity Xr as a function of crystallization time of the Mo model with the experimental data at Φ of 3 °C min−1: a PLA1.5D 0%HNT; b PLA1.5D 2.5%HNT; c PLA1.5D 5.0%HNT; d PLA1.5D 10.0%HNT

Comparison between the relative crystallinity Xr as a function of crystallization time of the Mo model with the experimental data at Φ of 1 °C min−1: a PLA1.5D 0%HNT; b PLA1.5D 2.5%HNT; c PLA1.5D 5.0%HNT; d PLA1.5D 10.0%HNT
The Mo model has a better correlation between 10 and 90% Xr but does not fit well at the start and end of crystallization, resulting in a discrepancy with the experimental data. Nonetheless, compared to the Jeziorny model (which has a large number of data points), the Mo model provides results that more closely match the actual behavior of the materials, except for some samples at the cooling rate of 1 °C min−1 where Jeziorny performed better (such as the PLA5.0D with 10%HNT). Li et al. [11, 12, 64] found that the Mo model effectively describes the non-isothermal crystallization of PLAs with nucleating agents such as dilithium hexahydrophthalate, calcium phenylphosphonate or cadmium phenylmalonate.
Crystallization activation energy
Equation 8 is the Friedman method to determine the activation energy of crystallization [11, 12, 65,66,67]:
where dXr/dt is the instantaneous crystallization rate as a function of time for a given Xr conversion; R is the gas constant; ΔEx is the effective activation energy (the sum of the activation energies for the nucleation and crystal growth processes) for a given Xr conversion; and Tx is the set of temperatures related to a given Xr conversion at a different Φ [11, 12, 65,66,67].
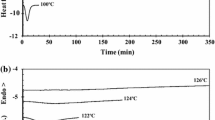
From Fig. 3 and Table 3, it is possible to obtain the instantaneous crystallization rate dXr/dt. By choosing different values of Xr (from 10 to 90%), related to a Tx, different values of dXr/dt are obtained, and it is possible to plot a graph ln(dXr/dt) versus 1000/Tx, where the slope of the linear regression will be equal to − ΔEx R−1. From the slopes, the values of the ΔEx can be calculated for the different Xr. These values are presented in Fig. 11. The data show that, in the case of the nanocomposites, ΔEx increases with Xr. Neat PLA1.5D has greater difficulty crystallizing than its nanocomposites under the conditions analyzed (especially at high crystallinity). Figure 11 shows that the activation energy is negative for all crystallizations. As the analysis is done on cooling, the temperature decreases and the process rate (Xr) increases, so ΔEx is negative and goes toward zero. On heating, increasing temperature usually brings higher reaction rates, so ΔEx becomes positive [68, 69].

Effective activation energy ΔEx as a function of relative crystallinity Xr
As the temperature drops below Tm and the melt converts to the crystalline phase, the ΔEx increases to zero. The point at which ΔEx changes to zero corresponds to the temperature of the maximum crystal growth rate. In simple terms, the negative sign of ΔEx means that the crystallization rate increases as the temperature decreases [69]. Adding more HNT led to a higher ΔEx (closer to zero) and increased the crystallization rate. The crystallization process is controlled by two processes: nucleation and spherulitic growth [70]. So, the nucleation effect seems to have been predominant, as the ΔEx values remained negative.
Crystallization kinetics by rheometry
The orientation of polymer chains through shear flow affects the crystallization kinetics and morphology of polymers. Figures 12 and 13 show the results obtained by rheometry that elucidate the non-isothermal crystallization induced by shear flow using the higher cooling rates of this DSC analysis.

Viscosity curves as a function of temperature for the non-isothermal flow crystallization of PLA1.5D for different Φ and shear rates: a PLA1.5D 0%HNT; b PLA1.5D 2.5%HNT; c PLA1.5D 5.0%HNT; d PLA1.5D 10.0%HNT

Viscosity curves as a function of temperature for the non-isothermal flow crystallization of PLA5.0D for different Φ and shear rates: a PLA5.0D 0%HNT; b PLA5.0D 2.5%HNT; c PLA5.0D 5.0%HNT; d PLA5.0D 10.0%HNT
All samples analyzed had an increase in viscosity with a decrease in temperature in all situations observed. However, the test only ends after a sudden increase in viscosity occurs, as this characterizes the point where the polymer crystals reach a critical size, and from then on, the material starts to behave similarly to a solid. Table 5 presents the non-isothermal crystallization temperature TNIC for all the samples and conditions studied and a comparison with the final crystallization temperature found by DSC (without flow). The DSC results of PLA5.0D are not present since at Φ of 5 and 10 °C min−1 there was no crystallization on cooling during tests.
At TNIC, the viscous material starts to act similarly to a solid. The crystallization temperature without flow is lower, meaning that the crystallization time is higher and the crystallization rate is lower compared with the condition where the material suffers shear flow.
When comparing the TNIC for PLA5.0D and its nanocomposites, it is possible to observe that the values are very close even for the different Φ and shear rates, with a very slight increase in TNIC for the highest Φ, and a small drop in TNIC, in most samples, when there is an increase in the shear rate.
As for the PLA1.5D and its nanocomposites, a noticeable trend emerges. In most samples, except for pure PLA (where the values closely resemble those of PLA5.0D), there is a notable rise in TNIC as the shear rate increases and Φ decreases. Notably, this variation is more pronounced than in PLA5.0D, potentially exceeding 10 °C with increasing shear rate.
The results of PLA5.0D and its nanocomposites and neat PLA1.5D may indicate that neither the change in the Φ nor the change in the shear rate allows for great variation in crystallization, as the TNIC values are very close. Now for the PLA1.5D nanocomposites, it is possible to observe that a higher shear rate decreases the crystallization time (higher TNIC temperatures indicate that the crystals reached a critical size in shorter times), leading to an increase in the crystallization rate. Thus, it is acceptable to say that the increase in the shear rate favors crystallization, probably due to the orientation of the molecules, which allows the formation of crystalline nuclei more easily compared to cases where the melt is not submitted to shear forces. The higher percentage of d-isomer in PLA5.0D seems to act as a physical heterogeneity that prevents the total orientation of the polymeric chains of the crystalline domains under stress, hindering the crystallization of the material [28].
Using Table 5, it is possible to observe the crystallization temperatures data from DSC (quiescent) and the rheometer (shear-induced). Quiescent non-isothermal crystallization occurs more favorably in this study for the PLA1.5D, which has a lower d-isomer content, as in higher quantities, d-isomer tends to hinder PLA organization and make crystallization more challenging [26,27,28]. Studies by Han et al. [7], Xu et al. [8], Leoné et al. [9], Kovalcik et al. [10], Li et al. [11, 12] investigated the use of various nucleating agents, different from HNT, in PLA and reported an increase in crystallinity and acceleration of crystallization in the presence of these nucleating agents (at most cooling rates). Wu et al. [13] and Liu et al. [14] studied PLA/HNT nanocomposites and concluded that HNT worked as a nucleating agent for the PLA. Therefore, the effect observed in this study, where HNT assisted in the crystallization of the PLA and increased its crystallinity, can be replicated. The effect of HNT is more pronounced in PLA1.5D, with its influence reduced in PLA5.0D due to the high quantity of d-isomer.
The experimental data obtained through DSC analysis served as the basis for evaluating the congruence of various models with the empirical findings. According to Li et al. [11, 12], the Jeziorny model is conventionally considered an appropriate representation of primary crystallization. However, it falls short in closely aligning with the experimental data from this study, suggesting the presence of secondary crystallization and spherulite impingement [59]. In contrast, the Mo model, grounded in more intricate assumptions including the mechanisms of secondary crystallization, the influence of crystallization temperature on lamellar thickness and the treatment of cooling rate as a variable throughout the entire crystallization process, demonstrates a stronger correlation between 10 and 90% crystallinity (Xr). Nevertheless, it does exhibit limitations at the beginning and end of the crystallization process.
When compared to the Jeziorny model, which incorporates a greater number of data points, the Mo model yields results that closely mimic the actual behavior of the material. Exceptions arise, notably when the cooling rate is set at 1 °C min−1, where the Jeziorny model outperforms, particularly in cases such as PLA5.0D with 10% HNT. It is essential to emphasize that Li et al. [11, 12, 64] noted the effectiveness of the Mo model in describing non-isothermal crystallization in PLAs when nucleating agents such as dilithium hexahydrophthalate, calcium phenylphosphonate or cadmium phenylmalonate are introduced.
Shear-induced non-isothermal crystallization was explored by Bojda et al. [26], who investigated PLA with differing d-isomer mass percentages (1.8% and 2.5%) and found that a lower d-isomer content intensified the impact of shear flow on crystallization. Similarly, Tang et al. [71] examined non-isothermal crystallization in PLA/carbon nanotube nanocomposites, affirming that shear flow boosts crystallization in both pure PLA and nanocomposites. Meanwhile, Zin et al. [27] studied crystallization under strain-induced conditions in PLA with different d-isomer levels (2%, 4% and 12% in mass percentage). Their results, consistent with this study, indicated that a higher d-isomer content hindered crystallization, even in the presence of flow/strain, leading to reduced crystallinity in PLA—contrary to the usual promotion of crystallization by flow/strain. Indeed, these studies show evidence that the shear flow facilitates crystallization, whereas an increased d-isomer content exerts the opposite effect, thus reinforcing the results presented in Table 5 within this study.
A direct comparison from Table 5 reveals that the values of flow-induced crystallization temperatures (ranging between 110 to 130 °C) are higher than those of quiescent crystallization obtained via DSC (ranging between 104 and 109 °C). This demonstrates PLA's enhanced crystallization tendency in the presence of flow, despite the influence of a higher d-isomer content. By exclusively analyzing the crystallization temperatures, it is not possible to discern the effect of HNT; one can merely observe a slight trend of increased crystallization temperature in its presence. This could be attributed to the flow effect being more predominant in these data.
Conclusions
In this paper, we set out to investigate the non-isothermal crystallization kinetics of PLA/HNT nanocomposites under both quiescent and shear-induced conditions. The experimental data were analyzed using Jeziorny and Mo models, and the crystallization activation energies were determined by Friedman method. Additionally, this research encompassed thermal and rheological characterizations of PLA/HNT nanocomposites. The Mo model presented the best fit to describe the non-isothermal crystallization of PLA, possibly due to the fact that the method is based on more complex assumptions, such as how secondary crystallization occurs, the dependence of lamellar thickness on the crystallization temperature and the rate of cooling treated as a variable throughout the entire crystallization process. Even though it is not the best model, Jeziorny was able to indicate that the growth of PLA crystals is mostly three-dimensional, which is corroborated by Mo’s model. The evaluation of activation energy showed that HNT favors an increase in the crystallization rate and that nucleation was the predominant process. The flow-induced crystallization allowed the conclusion that the shear action could lead to crystallization at higher temperatures, which suggests that in these cases, there is the orientation of polymeric chains which induces molecular ordering. The percentage of d-isomer greatly affects crystallization to the point that it can prevent crystallization under certain conditions. It was noticed that the HNT nanofiller nearly loses its function as a nucleating agent in PLA5.0D samples, primarily attributed to the higher d-isomer percentage in this material.
Through the TGA analysis the results showed that the presence of HNT did not cause a sudden change in thermal stability. Furthermore, there was no evidence of degradation provoked by the addition of HNT. Meanwhile, the rheological studies of these PLA nanocomposites in steady and dynamic states showed that all samples presented a viscous behavior and that HNT did not lead to major changes in rheological properties in steady state. The results indicated the absence of percolated networks too.
Finally, the DSC results showed that the smaller amount of d-isomer in PLA1.5D does not hinder the organization of the molecular chains, and the addition of HNT acts as a nucleating agent, favoring crystallization. On the other hand, the resulting morphology of processing a nanocomposite of PLA5.0D should be an amorphous material, only showing higher crystallinity in the lower cooling rate (the addiction of HNT show signs of increasing crystallinity in the 3 °C min−1 cooling rate). The crystallization half-time followed the same tendency. The neat PLA1.5D can have 43% of crystallinity against 32% of the neat PLA5.0D at 1 °C min−1 cooling rate (which have more time to crystallize), and the difference changes to 49% against 36% when 10%HNT is added (at 1 °C min−1 cooling rate). At 10 °C min−1 cooling rate, PLA1.5D 10%HNT crystalize and the PLA5.0D is amorphous.
References
Androsch R, Schick C, Di Lorenzo ML. Kinetics of nucleation and growth of crystals of poly(l-lactic acid). In: Synthesis, structure and properties of poly(lactic acid). Cham: Springer; 2018. p. 235–72. https://doi.org/10.1007/12_2016_13.
Puchalski M, Kwolek S, Szparaga G, Chrzanowski M, Krucińska I. Investigation of the influence of PLA molecular structure on the crystalline forms (α′ and α) and mechanical properties of wet spinning fibres. Polymers. 2017. https://doi.org/10.3390/polym9010018.
Harris AM, Lee EC. Improving mechanical performance of injection molded PLA by controlling crystallinity. J Appl Polym Sci. 2008. https://doi.org/10.1002/app.27261.
Moldovan A, Cuc S, Prodan D, Rusu M, Popa D, Taut AC, Petean I, Bomboş D, Doukeh R, Nemes O. Development and characterization of polylactic acid (PLA)-based nanocomposites used for food packaging. Polymers. 2023. https://doi.org/10.3390/polym15132855.
Li G, Zhao M, Xu F, Yang B, Li X, Meng X, Teng L, Sun F, Li Y. Synthesis and biological application of polylactic acid. Molecules. 2020. https://doi.org/10.3390/molecules25215023.
Joseph TM, Kallingal A, Suresh AM, Mahapatra DK, Hasanin MS, Haponiuk J, Thomas S. 3D printing of polylactic acid: recent advances and opportunities. Int J Adv Manuf Technol. 2023. https://doi.org/10.1007/s00170-022-10795-y.
Han Q, Wang Y, Shao C, Zheng G, Li Q, Shen C. Nonisothermal crystallization kinetics of biodegradable poly(lactic acid)/zinc phenylphosphonate composites. J Compos Mater. 2014. https://doi.org/10.1177/0021998313502064.
Xu T, Wang Y, Han Q, He D, Li Q, Shen C. Nonisothermal crystallization kinetics of poly(lactic acid) nucleated with a multiamide nucleating agent. J Macromol Sci B. 2014;6:66. https://doi.org/10.1080/00222348.2014.910049.
Leoné N, Roy M, Saidi S, Kort G, Hermida-Merino D, Wilsens CHR. Improving processing, crystallization, and performance of poly-l-lactide with an amide-based organic compound as both plasticizer and nucleating agent. ACS Omega. 2019. https://doi.org/10.1021/acsomega.9b00848.
Kovalcik A, Pérez-Camargo RA, Fürst C, Kucharczyk P, Müller AJ. Nucleating efficiency and thermal stability of industrial non-purified lignins and ultrafine talc in poly(lactic acid) (PLA). Polym Degrad Stab. 2017. https://doi.org/10.1016/j.polymdegradstab.2017.07.009.
Li C, Dou Q. Non-isothermal crystallization kinetics and spherulitic morphology of nucleated poly(lactic acid): effect of dilithium hexahydrophthalate as a novel nucleating agent. Thermochim Acta. 2014. https://doi.org/10.1016/j.tca.2014.08.036.
Li C, Dou Q, Bai Z, Lu Q. Non-isothermal crystallization behaviors and spherulitic morphology of poly(lactic acid) nucleated by a novel nucleating agent. J Therm Anal Calorim. 2015. https://doi.org/10.1007/s10973-015-4677-y.
Wu W, Cao X, Zhang Y, He G. Polylactide/halloysite nanotube nanocomposites: thermal, mechanical properties, and foam processing. J Appl Polym Sci. 2013. https://doi.org/10.1002/app.39179.
Liu M, Zhang Y, Zhou C. Nanocomposites of halloysite and polylactide. Appl Clay Sci. 2013. https://doi.org/10.1016/j.clay.2013.02.019.
Jin J, Zhang Y, Ouyang J, Yang H. Halloysite nanotubes as hydrogen storage materials. Phys Chem Miner. 2014. https://doi.org/10.1007/s00269-013-0651-z.
Manoj D, Rajendran S, Hoang TKA, Ansar S, Joo SW, Vasseghian Y, Soto-Moscoso M. In-situ growth of 3D Cu-MOF on 1D halloysite nanotubes/reduced graphene oxide nanocomposite for simultaneous sensing of dopamine and paracetamol. J Ind Eng Chem. 2022. https://doi.org/10.1016/j.jiec.2022.05.022.
Yuan P, Tan D, Annabi-Bergaya F. Properties and applications of halloysite nanotubes: recent research advances and future prospects. Appl Clay Sci. 2015. https://doi.org/10.1016/j.clay.2015.05.001.
Machowska A, Klara J, Ledwójcik G, Wójcik K, Dulińska-Litewka J, Karewicz A. Clindamycin-loaded halloysite nanotubes as the antibacterial component of composite hydrogel for bone repair. Polymers. 2022. https://doi.org/10.3390/polym14235151.
Yousefi P, Hamedi S, Garmaroody ER, Koosha M. Antibacterial nanobiocomposite based on halloysite nanotubes and extracted xylan from bagasse pith. Int J Biol Macromol. 2020. https://doi.org/10.1016/j.ijbiomac.2020.05.209.
Hsiao B. Polymer crystallization: the development of crystalline order in thermoplastic polymers by Jerold M. Schultz (University of Delaware). J Am Chem Soc. 2002;6:66. https://doi.org/10.1021/ja015359h.
Giammona G, Craparo EF. Biomedical applications of polylactide (pla) and its copolymers. Molecules. 2018. https://doi.org/10.3390/molecules23040980.
Saeidlou S, Huneault MA, Li H, Park CB. Poly(lactic acid) crystallization. Prog Polym Sci. 2012. https://doi.org/10.1016/j.progpolymsci.2012.07.005.
Noether H. Structure of crystalline polymers. Hiroyuki Tadokoro, Wiley‐Interscience, New York, 1979, 465 pp. Journal of Polymer Science: Polymer Letters Editio; 1980. https://doi.org/10.1002/pol.1980.130180220.
Long Y, Shanks RA, Stachurski ZH. Kinetics of polymer crystallisation. Prog Polym Sci. 1995. https://doi.org/10.1016/0079-6700(95)00002-W.
Alexis F. Factors affecting the degradation and drug-release mechanism of poly(lactic acid) and poly[(lactic acid)-co-(glycolic acid)]. Polym Int. 2005. https://doi.org/10.1002/pi.1697.
Bojda J, Piorkowska E. Shear-induced nonisothermal crystallization of two grades of PLA. Polym Test. 2016. https://doi.org/10.1016/j.polymertesting.2016.01.006.
Zin MRM, Mahendrasingam A, Konkel C, Narayanan T. Effect of d-isomer content on strain-induced crystallization behaviour of poly(lactic acid) polymer under high speed uniaxial drawing. Polymer. 2021. https://doi.org/10.1016/j.polymer.2021.123422.
Yin Y, Zhang X, Song Y, Vos S, Wang R, Joziasse CAP, Liu G, Wang D. Effect of nucleating agents on the strain-induced crystallization of poly(l lactide). Polymer. 2015. https://doi.org/10.1016/j.polymer.2015.03.061.
Gao W, Zhang Y, Ramanujan D, Ramani K, Chen Y, Williams CB, Wang CCL, Shin YC, Zhang S, Zavattieri PD. The status, challenges, and future of additive manufacturing in engineering. Comput Aided Des. 2015. https://doi.org/10.1016/j.cad.2015.04.001.
Farah M, Bretas RES. Characterization of i-PP shear induced crystallization layers developed in a slit die. J Appl Polym Sci. 2004. https://doi.org/10.1002/app.13576.
Joussein E, Petit S, Churchman J, Theng B, Righi D, Delvaux B. Halloysite clay minerals—a review. Clay Miner. 2005. https://doi.org/10.1180/0009855054040180.
Polanský R, Kadlec P, Kolská Z, Švorčík V. Influence of dehydration on the dielectric and structural properties of organically modified montmorillonite and halloysite nanotubes. Appl Clay Sci. 2017. https://doi.org/10.1016/j.clay.2017.07.027.
Alakrach AM, Noriman NZ, Alsaadi MA, Sam ST, Pasbakhsh P, Dahham OS, Shayfull Z. Thermal properties of PLA/HNTs composites: Effect of different halloysite nanotube. In: Proceeding of AIP conference proceedings 2030, USA; 2018. https://doi.org/10.1063/1.5066693.
Liu X, Wang T, Chow L, Yang M, Mitchell J. Effects of inorganic fillers on the thermal and mechanical properties of poly(lactic acid). Int J Polym Sci. 2014. https://doi.org/10.1155/2014/827028.
Kim YH, Kwon SH, Choi HJ, Choi K, Kao N, Bhattacharya SN, Gupta RK. Thermal, mechanical, and rheological characterization of polylactic acid/halloysite nanotube nanocomposites. J Macromol Sci. 2016. https://doi.org/10.1080/00222348.2016.1187054.
Zhou Q, Xanthos M. Nanoclay and crystallinity effects on the hydrolytic degradation of polylactides. Polym Degrad Stab. 2008. https://doi.org/10.1016/j.polymdegradstab.2008.05.014.
Södergard A, Stolt M. Properties of lactic acid based polymers and their correlation with composition. Prog Polym Sci. 2002. https://doi.org/10.1016/S0079-6700(02)00012-6.
Kopinke FD, Remmler M, Mackenzie K, Möder M, Wachsen O. Thermal decomposition of biodegradable polyesters—II poly(lactic acid). Polym Degrad Stab. 1996. https://doi.org/10.1016/0141-3910(96)00102-4.
McNeill IC, Leiper HA. Degradation studies of some polyesters and polycarbonates—1. polylactide: general features of the degradation under programmed heating conditions. Polym Degrad Stab. 1985. https://doi.org/10.1016/0141-3910(85)90050-3.
McNeill IC, Leiper HA. Degradation studies of some polyesters and polycarbonates—2. polylactide: degradation under isothermal conditions, thermal degradation mechanism and photolysis of the polymer. Polym Degrad Stab. 1985. https://doi.org/10.1016/0141-3910(85)90035-7.
Tsuji H, Fukui I, Daimon H, Fujie K. Poly(l-lactide) xi. lactide formation by thermal depolymerisation of poly(l-lactide) in a closed system. Polym Degrad Stab. 2003;6:66. https://doi.org/10.1016/S0141-3910(03)00150-2.
Lim LT, Auras R, Rubino M. Processing technologies for poly(lactic acid). Prog Polym Sci. 2008. https://doi.org/10.1016/j.progpolymsci.2008.05.004.
Nishida H. Thermal degradation. In: Auras RA, Lim LT, Selke SEM, Tsuji H, editors. Poly(lactic acid): synthesis, structures, properties, processing, and applications. Wiley; 2022. pp. 455–66. https://doi.org/10.1002/9781119767480.ch20.
Kelnar I. Effect of halloysite on structure and properties of melt-drawn PCL/PLA microfibrillar composites. Express Polym Lett. 2016. https://doi.org/10.3144/expresspolymlett.2016.36.
Sharma S, Singh A, Majumdar A, Butola B. Harnessing the ductility of polylactic acid/halloysite nanocomposites by synergistic effects of impact modifier and plasticiser. Compos B Eng. 2020. https://doi.org/10.1016/j.compositesb.2020.107845.
Cobos CM, Garzón L, López J, Fenollar O, Ferrándiz S. Study of thermal and rheological properties of PLA loaded with carbon and halloysite nanotubes for additive manufacturing. Rapid Prototyp J. 2019. https://doi.org/10.1108/RPJ-11-2018-0289.
Marini J, Bretas R. Influence of shape and surface modification of nanoparticle on the rheological and dynamic-mechanical properties of polyamide 6 nanocomposites. Polym Eng Sci. 2013. https://doi.org/10.1002/pen.23405.
Zhao J, Morgan AB, Harris JD. Rheological characterization of polystyrene–clay nanocomposites to compare the degree of exfoliation and dispersion. Polymer. 2005. https://doi.org/10.1016/j.polymer.2005.04.038.
Díez-Rodríguez T, Blázquez-Blázquez E, Pérez E, Cerrada M. influence of content in d isomer and incorporation of sba-15 silica on the crystallization ability and mechanical properties in plla based materials. Polymers. 2022. https://doi.org/10.3390/polym14061237.
Venkatesh C, Fuenmayor E, Doran P, Major I, Lyons J, Devine D. Additive manufacturing of PLA/HNT nanocomposites for biomedical applications. Procedia Manuf. 2019. https://doi.org/10.1016/j.promfg.2020.01.003.
Sin LT, Rahmat A, Rahman WAWA. 3—Thermal properties of poly(lactic acid). In: Sin LT, Rahmat A, Rahman WAWA, editors. Polylactic Acid. William Andrew Publishing; 2013. pp. 109–41. https://doi.org/10.1016/B978-1-4377-4459-0.00003-2.
Righetti MC. Amorphous fractions of poly(lactic acid). In: Di Lorenzo M, Androsch R, editors. Synthesis, structure and properties of poly(lactic acid). Springer, Cham; 2017. pp. 195–234. https://doi.org/10.1007/12_2016_14.
Murariu M, Dechief AL, Paint Y, Peeterbroeck S, Bonnaud L, Dubois P. Polylactide (PLA)-halloysite nanocomposites: production, morphology and key-properties. J Polym Environ. 2012. https://doi.org/10.1007/s10924-012-0488-4.
Dong Y, Marshall J, Haroosh HJ, Mohammadzadehmoghadam S, Liu D, Qi X, Lau K. Polylactic acid (PLA)/halloysite nanotube (HNT) composite mats: influence of HNT content and modification. Compos A Appl Sci Manuf. 2015. https://doi.org/10.1016/j.compositesa.2015.05.011.
Shi X, Zhang G, Phuong V, Lazzeri A. Synergistic effects of nucleating agents and plasticizers on the crystallization behavior of poly(lactic acid). Molecules. 2015. https://doi.org/10.3390/molecules20011579.
Shi X, Zhang G, Siligardi C, Ori G, Lazzeri A. Comparison of precipitated calcium carbonate/polylactic acid and halloysite/polylactic acid nanocomposites. J Nanomater. 2015. https://doi.org/10.1155/2015/905210.
Jeziorny A. Parameters characterizing the kinetics of the nonisothermal crystallization of poly(ethylene terephthalate) determined by d.s.c. Polymer. 1978. https://doi.org/10.1016/0032-3861(78)90060-5.
Sperling LH. The crystalline state. In: Sperling LH, editor. Introduction to physical polymer science. New York: Wiley; 2005. pp. 239–23. https://doi.org/10.1002/0471757128.ch6.
Su Z, Liu Y, Guo W, Li Q, Wu C. Crystallization behavior of poly(lactic acid) filled with modified carbon black. J Macromol Sci Part B. 2009. https://doi.org/10.1080/00222340902837238.
Hinrichs V, Kalinka G, Hinrichsen G. An Avrami-based model for the description of the secondary crystallization of polymers. J Macromol Sci B. 1996. https://doi.org/10.1080/00222349608220382.
Liu T, Mo Z, Zhang H. Nonisothermal crystallization behavior of a novel poly(aryl ether ketone): PEDEKmK. J Appl Polym Sci. 1998. https://doi.org/10.1002/(SICI)1097-4628(19980131)67:5%3c815::AID-APP6%3e3.0.CO;2-W.
Liu T, Mo Z, Wang S, Zhang H. Nonisothermal melt and cold crystallization kinetics of poly(aryl ether ether ketone ketone). Polym Eng Sci. 1997. https://doi.org/10.1002/pen.11700.
Vyazovkin S. Nonisothermal crystallization of polymers: getting more out of kinetic analysis of differential scanning calorimetry data. Polym Cryst. 2018. https://doi.org/10.1002/pcr2.10003.
Li Y, Han C, Yancun Y, Xiao L, Shao Y. Crystallization behaviors of poly(lactic acid) composites fabricated using functionalized eggshell powder and poly(ethylene glycol). Thermochim Acta. 2018. https://doi.org/10.1016/j.tca.2018.03.011.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry application to a phenolic resin. J Polym Sci C Polym Symp. 1964. https://doi.org/10.1002/polc.5070060121.
Vyazovkin S. Evaluation of activation energy of thermally stimulated solidstate reactions under arbitrary variation of temperature. J Comput Chem. 1997. https://doi.org/10.1002/(SICI)1096-987X(199702)18:3%3C393::AID-JCC9%3E3.0.CO;2-P.
Vyazovkin S. Modification of the integral isoconversional method to account for variation in the activation energy. J Comput Chem. 2001. https://doi.org/10.1002/1096-987X(20010130)22:2%3C178::AID-JCC5%3E3.0.CO;2-%23.
Revell LE, Williamson BE. Why are some reactions slower at higher temperatures? J Chem Educ. 2013. https://doi.org/10.1021/ed400086w.
Vyazovkin S. A time to search: finding the meaning of variable activation energy. Phys Chem Chem Phys. 2016. https://doi.org/10.1039/C6CP02491B.
Mucha M, Królikowski Z. Application of dsc to study crystallization kinetics of polypropylene containing fillers. J Therm Anal Calorim. 2003. https://doi.org/10.1023/B:JTAN.0000005193.66789.ea.
Tang H, Chen J, Wang Y, Xu J, Hsiao B, Zhong G, Zhong G. Shear flow and carbon nanotubes synergistically induced nonisothermal crystallization of poly(lactic acid) and its application in injection molding. Biomacromol. 2012. https://doi.org/10.1021/bm3013617.
Acknowledgements
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. The authors would like to thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Process Number: 88887.351910/2019-00) for the financial aid.
Author information
Authors and Affiliations
Contributions
Conceptualization was contributed by G.G.B., L.C.C. and J.M.; methodology was contributed by G.G.B., L.C.C. and J.M.; formal analysis was contributed by G.G.B.; investigation was contributed by G.G.B.; writing—original draft preparation was contributed G.G.B.; writing—reviewing and editing was contributed by G.G.B., C.A.G.B., T.A.A., L.C.C. and J.M.; supervision was contributed by L.C.C. and J.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Biazin, G.G., Beatrice, C.A.G., Augusto, T.A. et al. Quiescent and shear-induced non-isothermal crystallization kinetics of PLA/HNT nanocomposites. J Therm Anal Calorim 148, 13463–13485 (2023). https://doi.org/10.1007/s10973-023-12648-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-023-12648-6