
Abstract
The influence of the branching polyol on the thermal behavior of phthalic anhydride (PA)–polyol polyesters was determined under non-isothermal conditions. The polyol was glycerol, trimethylolpropane, and pentaerythritol, used in a molar ratio of 1.5:1, 1.5:1, and 2:1 in respect of anhydride, respectively. The thermal behavior of the synthesized polyesters was qualitatively estimated by the thermoanalytical curves obtained at a heating rate of 10 °C min−1. Quantitatively, the thermal behavior of the prepared polyesters was studied by a kinetic analysis using three different methods: Friedman, Flynn–Wall–Ozawa, and modified nonparametric kinetic method. By all the samples, two thermodegradation steps were observed. The beginning of the first step of degradation was considered the qualitative criterion for the thermal stability estimation: The pentaerythritol-containing polyester was the most thermostable, until 195 °C. Also the kinetic analysis the same sample presented the highest activation energy, i.e., the lowest thermodegradation rate.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
One the most important characteristics of a polymeric material is its thermal behavior, including the thermal stability. This last one is usually determined by thermal analysis under non-isothermal conditions [1], when not only the mass loss, but also the kinetic of the thermally induced events can be established. However, the non-isothermal kinetics by polymer degradation is a subject of discussion [2].
By different applications of phthalic anhydride-based saturated polyesters, the thermomechanical properties can be modified by means of different polyols [3, 4]. The tri- or tetraols determine a branching of the polymeric chain, the consequence being a loss of elasticity and an increasing of mechanical and chemical resistance, leading to a prolonged working life.
In this paper, the influence of the branching polyol on the thermal behavior of phthalic-type saturated polyesters is described. Glycerol and trimethylolpropane were used as triols, without and with an ethyl side group, respectively; pentaerythritol was selected as tetrol. In order to avoid the hyperbranching [5], molar ratio of anhydride:polyol corresponding to their functionality was used for synthesis. The experimental and data processing strategy was similar with that in our previous paper [6–12].
Materials and methods
Materials
O-Phthalic anhydride (PA) used in this study was purchased from Merck (M = 148.1 g mol−1, 99 %, melting point 129–133 °C), glycerol (GLY) from ChimReactiv S.R.L. (M = 92 g mol−1, 99.5 %, d = 1.262 g cm−3), trimethylolpropane (TMP) from Merck (M = 134.17 g mol−1; 99 %; melting point 57–61 °C), and pentaerythritol (PE) from Merck (M = 136.14 g mol−1, d = 1.396 g cm−3, melting point 259–260 °C).
Synthesis
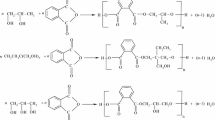
For a complete esterification without hyperbranching, the tri- and tetra-functionality of the polyols was taken into account, so that the anhydride:polyol molar was PA:GLY = 1.5:1, PA:TMP = 1.5:1 and PA:PE = 2:1, respectively.
The non-catalyzed reaction was performed at temperature until 220 °C for 6–7 h, up to acidity index less than 20 mg KOH g−1.
The reaction was carried out in an equipment for polyesterification, which was formed of a three-necked glass flask, equipped with a stirrer, a condenser, a Dean-Stark separator, and thermometer. The heating was achieved by means of an electric heating nest.
Phthalic anhydride and polyol were mixed and heated under continuous stirring. No solvents were used, and the formed water was continuously removed from the mixture by distillation.
FT-IR/UATR spectroscopy of polyesters was performed using a PerkinElmer Spectrum 100 FT-IR Spectrometer with UATR (see Fig. 1).

FT-IR spectra of synthesized polyesters
Thermal analysis was performed in dynamic synthetic air atmosphere (5.0 Linde Gas with a flow of 100 mL min−1) using a Diamond TG/DTA PerkinElmer. All the experiments were carried out from 25 °C up to 500 °C, at heating rates β = 7, 10, 12, 15, and 20 °C min−1.
The results of thermal analysis in dynamic synthetic air atmosphere are presented in Fig. 2.

TG/DTG/HF curves of synthesized sample for heating rate β = 10 °C min−1
Results and discussion
The spectra presented in Fig. 1 allow the following remarks:
-
No IR signals in the range 3670–3580 cm−1 indicate the absence of free hydroxyl groups by all the three prepared polyesters;
-
The peak at 1716–1712 cm−1 is characteristic for the νC=O vibration by aliphatic esters and appears by all the samples; also at 1256 cm−1, it is a characteristic signal for C–CO–O stretching, i.e., ester group;
-
In the range 2200–1960 cm−1, there are the benzene ring substitution patterns;
-
Between 1180 and 1020 cm−1, the fingerprint of secondary and tertiary alcohols, respectively, at 750–720 cm−1 that one of alkanes skeletal vibrations are present.
Even if the FT-IR data have a qualitative character, they confirm that all the alcoholic hydroxyl groups were esterificated.
The thermoanalytical curves for a heating rate of β = 10 °C min−1 are presented in Fig. 2. The mass loss as a function of temperature is similar by all the three samples. There is an initial degradation, followed by severe thermo-oxidations. The mass loss is almost a total of over 400 °C. Since the determinations were performed in dynamic air, the main process is thermo-oxidation, the majority of the thermal processes is being exothermic. The few endothermic processes are probably due to the thermodegradation of some carboxyl end groups over 180 °C [1].
At the beginning of the oxidative thermodegradation, some differences by the DTG/HF curves appear. For instance, by PA:TMP sample, a significant mass loss of 24 % with an exothermic effect of −48 J·g−1 and a DTG maximum at 320 °C is in connection with the existence of the ethyl side group of the polyol. Also, by PA:PE sample, the thermodegradation begins at 195 °C, higher than by the other samples (170 °C by PA:GLY and 185 °C by PA:TMP, respectively), and this is probably in connection with the more branched structure determined by the four hydroxyl groups.
For the preservation of the working properties of any one material, also polyester resins, the thermal stability is a “sine qua non” condition. Therefore, we consider that the beginning of the first step of degradation is in one case a reasonable criterion for the estimation of the thermal stability, even it is a more qualitatively one. So the thermal stability of our prepared samples increases as follows:
Kinetic analysis
The kinetic study was performed on the well-defined first step of the thermodegradation, i.e., the water loss. It is generally accepted that the reaction rate can be expressed as a product of two separable functions: k(T) dependent only on the reaction temperature T and f(α) dependent on the reaction degree α, respectively,
where t is the reaction time. Under non-isothermal condition, usually the reaction temperature depends linearly on time, T = T i + βt, where β is the constant heating rate, so Eq. (1) becomes:
Equation (2) is the starting point for all the data processing methods used in kinetic analysis. The TG/DTG curves furnish direct the dα/dt, α, and T for different conversion degrees can be determined.
We used four different kinetic methods:
-
Differential isoconversional by Friedman (FR) [13]
$$\ln \left( {\beta \times {\text{d}}\alpha /{\text{d}}t} \right)_{\upalpha } = \ln \left[ {A \times f(\alpha )} \right]_{\upalpha } {-}E/RT$$(3) -
Integral isoconversional by Flynn–Wall [14] and Ozawa [15] (FWO)
$$\ln \beta = \ln A/\left[ {R \times g(\alpha )} \right]{-}5.331{-}1.052 \times E/R \times T$$(4)where \(g(\alpha ) = \int\nolimits_{0}^{\alpha } {{\text{d}}\alpha /f(\alpha )}\) is the integral form of the conversion function.
Plotting between the left member of Eqs. (3) or (4) and 1/T, the value of the activation energy will be obtained from the line slope. Both methods allow the explanation of the k(T) function. The f(α) remains implicit, so these methods are “model free”.
By plotting the values of the activation energy E versus the conversion degree, the diagrams in Fig. 3 were obtained. There are variations in E versus α which do not exceed 10 %, and these variations are non-monotonous. Therefore, a mean value of the activation energy is reasonable for comparison (see Table 2).

Dependence of the activation energy on the conversion degree
The modified NPK method [16–24]
The previous presented methods are “model free,” nothing being asserted about the conversion dependence of the reaction rate. The reaction model g(α) accounts for the dependence on the conversion degree and f(T) accounts for temperature dependence. The reaction rate (r) is considered to be given by:
The reaction rate βdα/dT, measured from several experiments at different heating rates, β, was interpolated as a surface in a 3D space (βdα/dT, α, T) (see Fig. 3) and interpolated in order to generate a continuous surface corresponding to Eq (3). This surface is discretized as a ixj matrix M.
The nonparametric kinetic (NPK) method uses the singular value decomposition (SVD) algorithm to decompose matrix M into the two vectors [25]. The matrix M is decomposed as follows:
and the first columns of U and V matrix, respectively, u 1 and v 1, are analyzed for determining the kinetic model, i.e.,
and a temperature dependence, i.e.,
For the temperature dependence, the Arrhenius equation is selected, whereas for the kinetic model, we suggest the use of Šesták–Berggren [26] equation:
where α m describes the influence of physical phenomenon related with the presence of reaction product and (1 − α)n describes the chemical phenomenon which involves the remainder reactant (1 − α).
If the decomposition process is a result of two or more simultaneous steps, it means that r = Σr i, and consequently, the matrix M becomes:
The contribution of each step to the observed process is expressed by its explained variance λ, so that Σλ i = 100 %.
If the decomposition is a process with two simultaneous reactions, the matrix M became:
where u 1 and v 1 correspond to the first columns of matrix U and V, respectively, and u 2 and v 2 correspond to the secondary columns of matrix U and V, respectively.
It means that there are two elementary processes by the decomposition step, and the discrimination between them is possible by the values of the explained variance λ1 and λ2 (λ1 + λ2 ≈ 100 %).
In Table 1, the results by NPK method are systematized. All the three samples present two significant processes, the main with significance (explained variance) between 59 and 77 %, and the secondary one between 21 and 36 %, respectively. This is in agreement with the discussions above on the observed two steps from the DTG and HF maximums.
The dependence on conversion degree (Šesták–Berggren eq.) is rather different by samples/steps. But these equations were obtained using the “best fit” criterion, so they give a formal description, and any temptations for mechanistic speculations are risky.
Nevertheless, the NPK method gives a complete description of the reaction rate, without any approximations and with a natural (mathematical based) separation of the temperature, respective conversion dependence.
In Fig. 3, the variation in the activation energy values versus conversion degree obtained by the FR and FWO methods is presented. By inspecting the diagrams, it is observed that especially in case of PA:GLY and PA:PE, this variation do not exceed 10 %, so a mean value will be a reasonable solution for comparison purposes (see Table 2).
By inspecting the data in Table 2, a rather good agreement between the E mean values was obtained by the three kinetic methods. But most important is that each method indicates the same variation of the E value for the three polyesters, i.e.,
This relationship is somewhat different from the relationship (a), i.e., PA:GLY and PA:TMP changed reciprocal the places, respectively. But PA:PE is the most thermostable compound (according rel. a) and has the highest activation energy, i.e., the lowest decomposition rate. Certainly, the four methylol groups on the tertiary C atom have a positive effect on the stability and thermal lifetime of phthalic-based polyester.
Conclusions
Three branched polyols, i.e., glycerol, trimethylolpropane, and pentaerythritol, were used for preparing phthalic-based polyesters. The phthalic anhydride:polyol molar ratio was selected so that all the hydroxyl groups will be esterificated and no hyper branching occurs.
From both TG/DTG data (discussed qualitatively) and a kinetic analysis (quantitative evaluation), the pentaerythritol seems to have a positive effect on the thermal stability of the polymer. The arguments are a relative high-temperature range of the thermodegradation (begins around 200 °C) as well as a high value of the activation energy of the process. This observation is valid only in the studied series of polyols. An extension to the conclusion that all tetrols have a high thermostability by phthalic esters is risky.
For the kinetic analysis, three different methods were used. The modified NPK method allows complete description of the reaction rate (the temperature respective the conversion dependence) without any approximations.
References
Zhang T, Howell BA, Smith PB, Smith PS. Thermal degradation of glycerol/adipic acid hyperbranched poly(ester)s containing either hydroxyl or carboxyl end-groups. J Therm Anal Calorim. 2015;122:1221–9.
Howel BA. The utility of variable temperature techniques in the determination of kinetic parameters. Thermochim Acta. 2002;388:275–81.
Zhang P, et al. Thermal stability of aromatic polyesters prepared from di-phenolic acid and its esters. Polym Degrad Stab. 2009;94(8):1261–6.
Hansen D, et al. Synthesis and characterization of polyesters derived from glycerol and phthalic acid. Mater Res. 2007;10(3):257–60.
Zhang T, Howell BA, Dumitrascu A, Martin SJ. Synthesis and characterization of glycerol—adipic acid hyperbranched polyesters. Polymers. 2014;55:5062–72.
Bolcu C, Modra D, Vlase G, Doca N, Mihali C, Vlase T. Synthesis and thermal behaviour of some diisocyanate-silane compounds. J Therm Anal Calorim. 2014;115(1):489–94.
Bolcu C, Vlase G, Vlase T, Albu P, Doca N, Şisu E. Thermal behavior of some polyurethanes reticulated by aminated maltose. J Therm Anal Calorim. 2013;113(3):1409–14.
Vlase T, Bolcu C, Vlase G, Mogos A, Doca N. Thermooxidative stabilization of a MDI polyol polyisocyanate. J Therm Anal Calorim. 2010;99:973–9.
Albu P, Bolcu C, Vlase G, Doca N, Vlase T. Kinetics of degradation under non-isothermal conditions of a thermooxidative stabilized polyurethane. J Therm Anal Calorim. 2011;105(2):685–9.
Vlase T, Vlase G, Doca N, Iliescu S, Ilia G. Thermo-oxidative degradation of polymers containing phosphorus in the main chain. High Perform Polym. 2010;22(7):863–75.
Vlase T, Doca N, Vlase G, Bolcu C, Borcan F. Kinetics of non-isothermal decomposition of three IRGANOX-type antioxidants. J Therm Anal Calorim. 2008;92:15–8.
Albu P, Bolcu C, Vlase G, Doca N, Vlase T. Kinetics of degradation under nonisothermal conditions of a thermooxidative stabilized polyurethane. J Therm Anal Calorim. 2011;105:685–9.
Friedman HL. Kinetics of thermal degradation of char-foaming plastics from thermogravimetry: application to a phenolic resin. J Polym Sci. 1965;6C:183–95.
Flynn JH, Wall LA. A quick, direct method for the determination of activation energy from thermogravimetric data. Polym Lett. 1966;4:323–8.
Ozawa T. A new method of analyzing thermogravimetric data. Bull Chem Soc Jpn. 1965;38:1881–6.
Birta N, Doca N, Vlase G, Vlase T. Kinetic of sorbitol decomposition under non-isothermal conditions. J Therm Anal Calorim. 2008;92:35–638.
Anghel M, Vlase G, Bilanin M, Vlase T, Albu P, Fuliaş A, Tolan I, Doca N. Comparative study on the thermal behavior of two similar triterpenes from birch. J Therm Anal Calorim. 2013;113(3):1379–85.
Fuliaş A, Vlase G, Vlase T, Soica C, Heghes A, Craina M, Ledeti I. Comparative kinetic analysis on thermal degradation of some cephalosporins using TG and DSC data. Chem Centr J. 2013;7(1):70.
Fulias A, Vlase G, Grigorie C, Ledeţi I, Albu P, Bilanin M, Vlase T. Thermal behaviour studies of procaine and benzocaine: Part 1. Kinetic analysis of the active substances under non-isothermal conditions. J Therm Anal Calorim. 2013;113(1):265–71.
Ledeti I, Vlase G, Vlase T, Bercean V, Fulias A. Kinetic of solid state degradation of transitional coordinative compounds containing functionalized 1,2,4-triazolic ligand. J Therm Anal Calorim. 2015;121(3):1049–57.
Patrutescu C, Vlase G, Turcus V, Ardelean D, Vlase T, Albu P. TG/DTG/DTA data used for determining the kinetic parameters of the thermal degradation process of an immunosuppressive agent: mycophenolate mofetil. J Therm Anal Calorim. 2015;121(3):983–8.
Ledeti I, Fulias A, Vlase G, Vlase T, Doca N. Novel triazolic copper(II) complex: synthesis, thermal behaviour and kinetic study. Rev Roum Chim. 2013;58(4–5):441–50.
Ledeti I, Vlase G, Vlase T, Doca N, Bercean V, Fulias A. Thermal decomposition, kinetic study and evolved gas analysis of 1,3,5-triazine-2,4,6-triamine. J Therm Anal Calorim. 2014;118(2):1057–63.
Fulias A, Vlase G, Vlase T, Onetiu D, Doca N, Ledeti I. Thermal degradation of B-group vitamins: B-1, B-2 and B-6. J Therm Anal Calorim. 2014;118(2):1033–8.
Wall ME. Singular value decomposition and principal component analysis. In: Berrar DP, Dubitzky W, Granzow M, editors. A practical approach to microarray data analysis, vol. 9. Norwel, MA: Kluwer; 2003. p. 91–109 LANL LA-UR-02.
Šesták J, Berggren G. Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures. Thermochim Acta. 1971;3:1–12.
Acknowledgements
This work was supported by POSCCE Grant No. 12PO102418/5124/22.05.2014, SMIS 50328: “New energetic efficient technology for synthesis of polyester copolymers.”
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vlase, G., Modra, D., Albu, P. et al. Thermal behavior of saturated phthalic-type polyesters. Influence of the branching polyol. J Therm Anal Calorim 127, 409–414 (2017). https://doi.org/10.1007/s10973-016-5667-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5667-4