
Abstract
We report the effect of lignin-coated cellulose nanocrystals (L-CNCs) on the crystallization behavior of poly(lactic acid) (PLA). PLA/L-CNC nanocomposites were prepared by melt mixing, and the crystallization behavior of PLA was investigated using differential scanning calorimetry. Isothermal crystallization data were analyzed using Avrami and Lauritzen–Hoffman secondary nucleation theory, while the equilibrium melting temperature was determined using the nonlinear Hoffman–Weeks method. The lignin-coated cellulose nanocrystals acted as a nucleating agent and significantly increased the rate of crystallization and degree of crystallinity of PLA in PLA/L-CNC nanocomposites. The Avrami exponent, n, increased in the presence of L-CNCs, displaying a conversion from lamellar morphology to two-dimensional crystal growth. PLA/L-CNC nanocomposites also gave lower values of the nucleation parameters, \(K_{\text{g}}\) and σ e, due to a reduction in the activation energy for nucleation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(lactic acid) (PLA) is considered to be a sustainable alternative to petroleum-based plastics due to its excellent physical properties, biodegradability, and thermal processability [1, 2]. PLA is used in a wide variety of applications such as packaging, automotive, and biomedical. However, it is highly brittle and prone to hydrolytic degradation which has limited its applications [1]. The incorporation of fillers as reinforcing agents has been shown to improve its physical properties and hydrolytic stability. However, the incorporation of non-biodegradable filler is undesirable for many applications, especially in biomedical. As amorphous regions of PLA are weaker and more susceptible to hydrolytic degradation than the crystalline regions, another approach to improve its hydrolytic stability and physical properties is to increase its degree of crystallinity. However, PLA has been found to undergo very slow crystallization, taking several minutes to crystallize completely at elevated temperature. Thus, normal industrial processing conditions result in low crystallinity. The incorporation of nucleating agents can significantly increase the rate of crystallization and can cause PLA to achieve a higher crystallinity during industrial processing [3–7]. Although there are several types of nucleating agents known for PLA the development of an environmentally friendly and a biocompatible, nucleating agent is highly desired to expand its applications.
Recently, cellulose nanofillers have generated a great deal of interest in the field of nanocomposites as they are renewable and biodegradable, have a high aspect ratio, are non-toxic, and have low density and excellent reinforcement properties [8, 9]. Cellulose is the most abundant natural biopolymer on earth. It is a homopolymer made up of repeating β(1,4)-d-glucopyranose units. The physical and chemical properties of cellulose greatly depend on its specific structure. The hydroxyl groups on cellulose contribute to the chemical reactivity of the glucopyranosyl ring and the tendency to form intra- and intermolecular hydrogen bonds [10]. Hydrogen bonding is responsible for the crystalline nature of cellulose fibers resulting in its high tensile strength and modulus [11]. Cellulose nanocrystals (CNCs) and nanofibers (CNFs) obtained by acid hydrolysis or TEMPO oxidation of cellulose fibers have been recognized as highly crystalline high aspect ratio fillers that have been widely proposed for incorporation of biodegradable polymer composites [12–14]. The incorporation of CNCs and CNFs in a PLA matrix has resulted in a moderate improvement in its crystallization characteristics, mechanical, and barrier properties [15–21]. However, poor dispersion of CNCs due to their high aspect ratio, intra-particle hydrogen bonding, and poor compatibility with hydrophobic polymers are major limitations in realizing the full potential of CNCs in a PLA matrix. Surface functionalization of CNCs has led to improvement in dispersion, compatibility, and thermal and mechanical properties of PLA [22, 23]. Surface coating can be an alternative approach to improving the dispersion and compatibility of CNCs with polymers without affecting their intrinsic properties.
Recently, it has been reported that incorporation of lignin in CNCs/polymer composites can improve dispersion and compatibility [24, 25]. Lignin is an integral part of lignocellulose and has developed over time in close proximity with cellulose and it interacts with cellulose through electrostatic and van der Waal interactions. Thus, better dispersion of CNCs in the presence of lignin can be attributed to formation of a lignin layer on CNCs surfaces through electrostatic and van der Waal interactions which prevents re-aggregation of CNCs during processing and also improves interaction with the polymer matrix [24]. Considering better dispersion and compatibility of CNCs in the presence of lignin, it can be expected that a lignin-coated CNCs may act as an excellent nucleating agent and lead to higher rate of crystallization and degree of crystallinity of PLA. Lignin has been coated on cellulose nanocrystals produced by the AVAP process which minimally functionalizes the surface of the cellulose [26]. In the present study, lignin-coated cellulose nanocrystals (L-CNCs) were used to prepare PLA/L-CNC nanocomposites and their isothermal crystallization behavior was studied using differential scanning calorimetry (DSC). The crystallization kinetics data were analyzed using Avrami and Lauritzen–Hoffman secondary nucleation theory to understand the role of L-CNCs on the crystallization behavior of PLA. To the best of our knowledge, this is the first report on the effect of lignin-coated CNCs on the crystallization behavior of PLA.
Experimental
Materials
Commercial grade L-poly(lactic acid) (L-PLA) was procured from NatureWorks with trade name IngeoTM biopolymer 4043D. The lignin-coated cellulose nanocrystals (L-CNCs) having an average length and diameter of ~350 nm and ~5–7 nm, respectively, were produced from eucalyptus chips and provided by American Process Inc. (API). Before melt mixing, all the materials were dried in a vacuum oven at 90 °C for 2 h to avoid possible degradation due to moisture during processing.
Preparation of PLA/L-CNC nanocomposites
PLA/L-CNC nanocomposites were prepared by melt mixing using a HAAKE Rheocord 90 melt mixer. PLA and L-CNCs, 66.5 g, were mixed by shaking and added to the melt mixer portion wise over ~1 min at an initial temperature of 140 °C with a screw speed of 40 rpm. The temperature increased to 160 °C during mixing, and the mixing was continued for 11 min with an steady-state torque after the loading spikes of 14.7 N.m. A master-batch of 5 mass% L-CNCs was prepared and diluted in a second melt mixing step with the appropriate amount of neat PLA to prepare 0.3, 1.0, and 2.0 mass% samples of PLA/L-CNCs. The samples obtained after dilution were designated as PLA, PLA/L-CNCs-0.3 %, PLA/L-CNCs-1 %, and PLA/L-CNCs-2 %, respectively.
Characterization of PLA/L-CNC nanocomposites
Differential scanning calorimetry (DSC)
A TA Instruments Q2000 differential scanning calorimeter (DSC) was used for recording DSC scans under N2. To ensure reliability of the data obtained, heat flow and temperature were calibrated with indium and sapphire standards. For both isothermal and non-isothermal DSC experiments, 8–10 mg of samples in a closed aluminum pan were placed in the DSC. In the non-isothermal process, the samples were heated from room temperature to 200 °C and held for 5 min to remove thermal history and then cooled to 25 °C and reheated with ramp rates of 10 °C min−1. For isothermal crystallization studies, all samples were heated from room temperature to 200 °C and kept at 200 °C for 5 min. Then, the samples were quenched to the desired isothermal crystallization temperature (95, 100, 105, 110, and 115 °C) at a rate of 10 °C min−1 and held for sufficient time to allow complete crystallization from the quiescent melt. The exothermic curves of the heat flow as a function of time were recorded at different crystallization temperature for all the samples.
Results and discussion
Crystallization and melting behavior
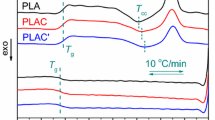
Crystallization and melting behavior of PLA in the presence of L-CNCs were investigated by analyzing non-isothermal DSC scans. Figure 1 shows the DSC cooling and second heating scans at 10 °C min−1 for PLA and PLA/L-CNC nanocomposites. The heating scans clearly show the presence of a glass transition (T g), crystallization exotherm defined as cold crystallization (T cc), and melting endotherm (T m) peaks. However, no exothermic peak was obtained during the cooling of PLA and the L-CNC nanocomposites. PLA crystallizes very slowly and usually takes long time to crystallize. The cooling cycle gives insufficient time to generate crystallinity; thus, it crystallizes during the second heating cycle resulting in a double melting peak. This double melting behavior is a common phenomenon in semicrystalline polymers, such as polyesters [27–30]. Possible causes of multiple melting behavior include different types of crystals having different morphologies (lamellar thickness and perfection), recrystallization, and re-melting processes during the subsequent heating [29, 30]. As expected, two melting peaks were observed in the heating scan of PLA and PLA/L-CNC nanocomposites. Incorporation of L-CNCs also reduced the melting temperature of PLA by ~6 °C. This decrease in melting temperature of PLA can be attributed to the formation of less stable random crystals during cold crystallization in the presence of L-CNCs. Furthermore, incorporation of L-CNCs showed no effect on the T g of PLA. However, cold crystallization of PLA was found to be significantly enhanced by the presence of L-CNCs. The crystallization temperature shifted to lower temperature from 129.3 to 112.5 °C, and the degree of crystallinity (X c) improved dramatically, on the addition of only 0.3 % of L-CNCs. The X c estimated from the melting endotherms was calculated using Eq. (1), and the results are summarized in Table 1.
\(\Delta {\rm H}_{\text{m}}\) is the measured endothermic enthalpy of melting. The melting enthalpy, \(\Delta {\rm H}_{\text{f0}}\), of 100 % crystalline PLA was taken as 93.0 J g−1 from the literature [31]. The results presented in Table 1 clearly indicate that X c of PLA increased by almost sevenfold on the addition of only 0.3 % L-CNCs. This dramatic improvement in X c and T cc of PLA clearly shows that L-CNCs acted as nucleating sites and provided a surface to grow PLA crystals. Nucleating behavior of CNCs and CNFs in a PLA matrix has been reported, with most of the reports showing improvement in T cc with or without a significant change in X c [14, 20, 32–37]. In PLA/cellulose nanofiller nanocomposites, the maximum improvement in X c achieved so far is only two- to –threefold at nanofiller loading ranging from 1.0 to 7.5 mass%. It is worth to note that the X c of PLA/cellulose nanofiller nanocomposites reported in the literature was found to be sensitive to processing methods, loading of nanofiller, modification of PLA, and the cellulose nanofillers. The significant improvement in X c and T cc found in the present study can be attributed to the better dispersion and improved interfacial interaction between L-CNCs and PLA chains. Nucleating efficiency of nanofillers depends on their dispersion and surface properties. Better dispersion leads to more nucleating sites and thus higher rates of crystallization. The presence of the lignin coating on the L-CNCs reduced intra-particle H-bonding among CNCs and lead to better dispersion of CNCs in the PLA matrix. With increasing L-CNC loading, the drop in X c is possibly due to agglomeration of the L-CNCs or network formation above the percolation threshold. We have also investigated the effect of L-CNCs at lower loading, 0.1 mass% on the crystallization behavior of PLA; however, no significant effect was observed (data not shown).

DSC scans of neat PLA and PLA/L-CNC nanocomposites under nitrogen with a scan rate of 10 °C min−1
Isothermal crystallization behavior of PLA/L-CNC nanocomposites
Avrami analysis
In order to fully understand the effect of L-CNCs on the bulk crystallization rate of PLA, isothermal crystallization kinetics of PLA/L-CNC nanocomposites was studied. The isothermal crystallization exotherms of PLA and the nanocomposites (at different isothermal crystallization temperature, T c) are shown in Fig. 2. Figure 2 clearly shows that with increasing isothermal crystallization temperature the exotherms shift to lower time indicating that rate of crystallization is increasing with increasing temperature. This is a common phenomenon for semicrystalline polymers because polymeric chains have more mobility to arrange regularly at higher crystallization temperatures, whereas, at the same temperature, the presence of L-CNCs accelerated the rate of crystallization as compared to neat PLA, thus reducing the time required for complete crystallization.

DSC heating curves for a neat PLA and b PLA/L-CNC nanocomposites at different crystallization temperature
In general, evolution of crystallinity is linearly proportional to the evolution of heat released during the course of crystallization. The relative crystallinity (X t) as a function of crystallization time (t) can be calculated using Eq. 2:
where the numerator represents the heat generated at time t, while the denominator represents the total heat generated during the entire crystallization process. Using Eq. (2), the fraction of the relative crystalline phase, X t, for the sample at a particular time t was calculated and plotted in Fig. 3 with respect to t. Figure 3 shows that the rate of crystallization of PLA is very slow and both the nucleation and crystal growth processes are taking place simultaneously. However, the sigmoidal shape of X t vs. t plot of PLA/L-CNC nanocomposites clearly shows nucleation followed by crystal growth during crystallization.

Plot of relative percent crystallinity versus time for a neat PLA and b PLA/L-CNC nanocomposites
In order to quantitatively describe the macroscopic evolution of crystallinity during primary crystallization under quiescent isothermal conditions, classical Avrami theory was used [38, 39]. The Avrami equation is as follows:
Equation (3) can be rewritten in double logarithmic form as follows:
where k is the rate constant which depends on the geometry of the growing crystalline phase and n is the Avrami exponent which represent the nucleation and growth processes. The values of ln[-ln(1-X t)] were plotted against ln t (Fig. 4), and the value of n and k were calculated from the slope and intercept of the linear regression analysis. The crystallization process usually has two stages, namely the primary crystallization stage (linear portion) and the secondary crystallization stage (nonlinear portion). The Avrami theory does not describe the complete crystallization process and is usually applicable at the primary crystallization stage [33]. Thus, n and k values were calculated from the linear portions of the plots and results are summarized in Table 2. Figure 4 also shows the existence of secondary crystallization because at low X t values, the plots are linear, whereas at higher X t values, the curves deviate from linearity for all the samples [40].

Plots of ln[-ln(1−X t)] versus ln t for a neat PLA and b PLA/L-CNC nanocomposite
The k values of the PLA/L-CNC nanocomposites are higher than those of neat PLA and increase with increasing crystallization temperature for all the L-CNC nanocomposites indicating faster crystallization. Surprisingly, the rate of crystallization has almost doubled at 105 °C for all the nanocomposite samples. According to crystallization theory, crystal growth rate of semicrystalline polymers shows a bell-shaped temperature dependence [1]. PLA, however, shows a discontinuity in the crystal growth rate as a function of isothermal crystallization temperature. This unusual behavior of PLA has been reported by several researchers for intermediate molecular weight PLA [31, 41–44]. Kawai et al. [41] found that instead of displaying one maximum, the growth rate vs. temperature curve exhibits two maxima. The first maximum was found at 105 °C, while a second maximum was found at 130 °C showing a clear discontinuity at around 105–130 °C. This anomalous behavior of PLA was found to be dependent on molecular weight [45], tacticity, species of co-monomer [46], and change in regime from regime II to regime III [1, 47]. The n value relates to the nature of nucleation and the dimensionality of the growing crystals and usually takes an integer number between 1 and 4. It has been reported for various polymers that the n value adopts fractional numbers due to secondary crystallization. The value of n was found to be close to 1 for neat PLA, suggesting formation of lamellae, whereas n value close to 2 was obtained for PLA/L-CNC nanocomposites suggesting a two-dimensional crystal growth. The n value obtained in the present study is similar to those reported for the neat PLA and PLA/clay nanocomposites [21, 48].
Crystallization half time, t 1/2, is another important crystallization kinetics parameter; it is defined as the time needed to complete half of the isothermal crystallization and calculated as follows:
Usually, t 1/2 is employed to characterize the crystallization rate directly since it includes contributions from primary nucleation and growth processes. The growth rate, G, can be defined as the reciprocal of t 1/2, as follows:
As shown in Table 2, for PLA/L-CNC nanocomposites t 1/2 values are lower than that of neat PLA, indicating the nucleating property of L-CNCs. The t 1/2 values, for all samples, were found to decrease with increasing isothermal crystallization temperature due to the higher chain mobility at high temperature which led to faster crystallization.
Lauritzen–Hoffman secondary nucleation theory
Lauritzen–Hoffman theory [49] was used to further analyze the nucleating effect of L-CNCs on the crystallization process of PLA. According to Lauritzen–Hoffman theory, the temperature dependence of the linear growth rate of PLA can be represented by Eq. 7.
The first term represents the diffusion of chains to the growth front, while the second is related to the secondary nucleation barrier. G represents the linear growth rate, G o is a pre-exponential factor, U* is the activation energy for transport of PLA chains to the crystallization site, K g is the nucleation parameter, R is the gas constant, T c is the isothermal crystallization temperature, T ∞ is the hypothetical temperature where crystallization process completed and is defined as T ∞ = T g–30 °C, f is a corrective factor that takes into account the variation of the equilibrium melting enthalpy with temperature, defined as \(f = 2 \, T_{\text{c}}^{{}} / \, (T_{\text{c}} - T_{\text{m}}^{0} )\), and ΔT is the degree of supercooling defined by \(\Delta T = T_{\text{m}}^{0} - T_{\text{c}}\) where \(T_{\text{m}}^{0}\) is the equilibrium melting temperature. The value of \(T_{\text{m}}^{0}\) for PLA and PLA/L-CNC nanocomposites (Fig. 5) was obtained from nonlinear Hoffmann–Week extrapolation [50], and the results are reported in Table 3. A plot of \(T_{\text{m}}^{0} /T_{\text{m}}^{0} - T_{\text{m}}\) vs. \(T_{\text{m}}^{0} /T_{\text{m}}^{0} - T_{\text{c}}\) yielding a straight line with slope of unity, i.e., (r = 1.00) gives the true \(T_{\text{m}}^{0}\) value. The value of \(T_{\text{m}}^{0}\) for neat PLA obtained in the present study is close to the values obtained using nonlinear Hoffmann–Week extrapolation for PLA having different molecular weights [5]. The nucleation constant, K g, is the energy required for the formation of nuclei of critical size and is defined by Eq. 8:
where n is a constant value that depends on the crystallization regime (i.e., it is equal to 2 in regime II and 4 for both regime I & III), b is the single layer thickness, σ is the lateral surface free energy, σ e is the free energy of folding which reflects the work required to create a new surface, \(\Delta {\rm H}_{\text{f}}\) is the heat of fusion per unit volume; \(T_{\text{m}}^{0}\) is the equilibrium melting temperature, and k B is the Boltzmann’s constant. It is often most convenient to rearrange Eq. 8 as:

Determination of equilibrium melting temperature for PLA using nonlinear Hoffman–Weeks equation
By plotting the log \(G + U*/2.303(T_{\text{c}}^{{}} - T_{\infty } )\) versus \(1/2.303 \, T_{\text{c}} (\Delta T)f\), the value of K g [i.e., K g = −(slope) (Fig. 6)] can be determined. For the calculation purpose, values of \(T_{\text{m}}^{0}\)(given in Table 3), T g = 56 °C, and U* = 1500 cal mol−1 were used and G was calculated using Eq. 6.

Plot of log G + U*/2.303 R (T c–T ∞ ) as a function of 1/2.303 T c f ΔT × 105
The lateral surface free energy, σ, is estimated by the Thomas–Stavely relationship (Eq. 10) [49]:
where \(\alpha\) is an empirical constant, which is usually assumed to be ~0.25. Taking \(\alpha\) = 0.25, molecular width (a) = 5.97 Å, molecular layer thickness (b) = 5.17 Å, and \(\Delta {\rm H}_{\text{f}}\) = 1.11 × 108 J m−3, σ can be calculated to be 1.54 × 10−2 J m−2. Using the values obtained for K g and σ, the folding surface free energy σ e can be calculated using Eq. 8.
The values of K g and \(\sigma_{\text{e}}\) obtained for PLA and L-CNC nanocomposites are tabulated in Table 3. The values of K g and \(\sigma_{\text{e}}\) are lower for the nanocomposite as compared to neat PLA which shows that the presence of L-CNCs decreased the activation energy of nucleation and the energy required to fold PLA chains to create a new surface. It is well known that addition of foreign particles provide a new surface to grow crystals on it, thus increasing the heterogeneity of the system. During heterogeneous nucleation, polymeric chains start growing on these preexisting surfaces and reduce the energy required for primary nucleation. The presence of L-CNCs in the PLA matrix reduced the work required to create a new surface for crystallization and catalyzed the crystallization process.
Conclusions
The L-CNCs acted as a nucleating agent and catalyzed the bulk crystallization process of PLA resulting in an about sevenfold increase in the degree of crystallinity. Crystallization and nucleation parameters for PLA/L-CNC nanocomposites were determined using Avrami and Lauritzen–Hoffman theory. Higher value of the Avrami exponent, n, and overall rate of crystallization, k, for PLA/L-CNC nanocomposites as compared to neat PLA showed the nucleating behavior of L-CNCs. Moreover, the n value for neat PLA changed from 1 to 2 in the presence of L-CNCs showing gradual growth from lamella to two-dimensional morphology. The nucleation parameters, K g and σ e, calculated from secondary nucleation theory for PLA/L-CNC nanocomposites showed lower values for the nanocomposites suggesting that addition of L-CNCs lowered the energy requirement for chain folding for PLA chains. The present study indicates that lignin-coated cellulose nanocrystals are an excellent nucleating agent for PLA and can help to achieve optimum physical properties and hydrolytic stability of PLA.
References
Jiménez A, Peltzer M, Ruseckaite R. Poly (lactic acid) science and technology: processing, properties, additives and applications. R Soc Chem. 2014.
Garlotta D. A literature review of poly(lactic acid). J Polym Environ. 2001;9(2):63–84. doi:10.1023/a:1020200822435.
Vasanthakumari R, Pennings AJ. Crystallization kinetics of poly(l-lactic acid). Polymer. 1983;24(2):175–8. doi:10.1016/0032-3861(83)90129-5.
Saeidlou S, Huneault MA, Li H, Park CB. Poly(lactic acid) crystallization. Prog Polym Sci. 2012;37(12):1657–77. doi:10.1016/j.progpolymsci.2012.07.005.
He Y, Fan Z, Hu Y, Wu T, Wei J, Li S. DSC analysis of isothermal melt-crystallization, glass transition and melting behavior of poly(l-lactide) with different molecular weights. Eur Polymer J. 2007;43(10):4431–9. doi:10.1016/j.eurpolymj.2007.07.007.
Li C, Dou Q, Bai Z, Lu Q. Non-isothermal crystallization behaviors and spherulitic morphology of poly(lactic acid) nucleated by a novel nucleating agent. J Therm Anal Calorim. 2015;122(1):407–17. doi:10.1007/s10973-015-4677-y.
Jandas PJ, Mohanty S, Nayak SK. Thermal properties and cold crystallization kinetics of surface-treated banana fiber (BF)-reinforced poly(lactic acid) (PLA) nanocomposites. J Therm Anal Calorim. 2013;114(3):1265–78. doi:10.1007/s10973-013-3102-7.
Lin N, Huang J, Dufresne A. Preparation, properties and applications of polysaccharide nanocrystals in advanced functional nanomaterials: a review. Nanoscale. 2012;4(11):3274–94. doi:10.1039/c2nr30260h.
Eichhorn SJ, Dufresne A, Aranguren M, Marcovich NE, Capadona JR, Rowan SJ, et al. Review: current international research into cellulose nanofibres and nanocomposites. J Mater Sci. 2010;45(1):1–33. doi:10.1007/s10853-009-3874-0.
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J. Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev. 2011;40(7):3941–94. doi:10.1039/c0cs00108b.
Nishino T, Matsuda I, Hirao K. All-cellulose composite. Macromolecules. 2004;37(20):7683–7. doi:10.1021/ma049300h.
Abdul Khalil HPS, Bhat AH, Ireana Yusra AF. Green composites from sustainable cellulose nanofibrils: a review. Carbohydr Polym. 2012;87(2):963–79. doi:10.1016/j.carbpol.2011.08.078.
Takagi H, Asano A. Effects of processing conditions on flexural properties of cellulose nanofiber reinforced “green” composites. Compos A Appl Sci Manuf. 2008;39(4):685–9. doi:10.1016/j.compositesa.2007.08.019.
Liu D, Yuan X, Bhattacharyya D. The effects of cellulose nanowhiskers on electrospun poly (lactic acid) nanofibres. J Mater Sci. 2012;47(7):3159–65. doi:10.1007/s10853-011-6150-z.
Kamal MR, Khoshkava V. Effect of cellulose nanocrystals (CNC) on rheological and mechanical properties and crystallization behavior of PLA/CNC nanocomposites. Carbohydr Polym. 2015;123:105–14. doi:10.1016/j.carbpol.2015.01.012.
Luo H, Xiong G, Li Q, Ma C, Zhu Y, Guo R, et al. Preparation and properties of a novel porous poly(lactic acid) composite reinforced with bacterial cellulose nanowhiskers. Fibers Polym. 2014;15(12):2591–6. doi:10.1007/s12221-014-2591-8.
Jonoobi M, Harun J, Mathew AP, Oksman K. Mechanical properties of cellulose nanofiber (CNF) reinforced polylactic acid (PLA) prepared by twin screw extrusion. Compos Sci Technol. 2010;70(12):1742–7. doi:10.1016/j.compscitech.2010.07.005.
Wang T, Drzal LT. Cellulose-Nanofiber-Reinforced Poly(lactic acid) composites prepared by a water-based approach. ACS Appl Mater Interfaces. 2012;4(10):5079–85. doi:10.1021/am301438g.
Dhar P, Tarafder D, Kumar A, Katiyar V. Effect of cellulose nanocrystal polymorphs on mechanical, barrier and thermal properties of poly(lactic acid) based bionanocomposites. RSC Adv. 2015;5(74):60426–40. doi:10.1039/c5ra06840a.
Cho S, Park H, Yun Y, Jin H-J. Cellulose nanowhisker-incorporated poly(lactic acid) composites for high thermal stability. Fibers Polym. 2013;14(6):1001–5. doi:10.1007/s12221-013-1001-y.
Liu Y, Wang L, He Y, Fan Z, Li S. Non-isothermal crystallization kinetics of poly(L-lactide). Polym Int. 2010;59(12):1616–21. doi:10.1002/pi.2894.
Eyley S, Thielemans W. Surface modification of cellulose nanocrystals. Nanoscale. 2014;6(14):7764–79. doi:10.1039/c4nr01756k.
Raquez JM, Murena Y, Goffin AL, Habibi Y, Ruelle B, DeBuyl F, et al. Surface-modification of cellulose nanowhiskers and their use as nanoreinforcers into polylactide: a sustainably-integrated approach. Compos Sci Technol. 2012;72(5):544–9. doi:10.1016/j.compscitech.2011.11.017.
Hambardzumyan A, Foulon L, Chabbert B, Aguie-Beghin V. Natural organic UV-absorbent coatings based on cellulose and lignin: designed effects on spectroscopic properties. Biomacromolecules. 2012;13(12):4081–8. doi:10.1021/bm301373b.
Ago M, Jakes JE, Johansson L-S, Park S, Rojas OJ. Interfacial properties of lignin-based electrospun nanofibers and films reinforced with cellulose nanocrystals. ACS Appl Mater Interfaces. 2012;4(12):6849–56. doi:10.1021/am302008p.
Nelson K, Retsina T. Innovative nanocellulose process breaks the cost barrier. TAPPI. 2014;13:19–23.
Ge C, Ding P, Shi L, Fu J. Isothermal crystallization kinetics and melting behavior of poly(ethylene terephthalate)/barite nanocomposites. J Polym Sci Part B Polym Phys. 2009;47(7):655–68. doi:10.1002/polb.21669.
Gupta A, Choudhary V. Thermal and mechanical properties of poly(trimethylene terephthalate)/acid-treated multiwalled carbon nanotube composites. J Mater Sci. 2013;48(20):7063–70.
Yasuniwa M, Iura K, Dan Y. Melting behavior of poly(l-lactic acid): effects of crystallization temperature and time. Polymer. 2007;48(18):5398–407. doi:10.1016/j.polymer.2007.07.012.
Song P, Chen G, Wei Z, Zhang W, Liang J. Calorimetric analysis of the multiple melting behavior of melt-crystallized poly(l-lactic acid) with a low optical purity. J Therm Anal Calorim. 2013;111(2):1507–14. doi:10.1007/s10973-012-2502-4.
Yasuniwa M, Tsubakihara S, Iura K, Ono Y, Dan Y, Takahashi K. Crystallization behavior of poly(l-lactic acid). Polymer. 2006;47(21):7554–63. doi:10.1016/j.polymer.2006.08.054.
Pei A, Zhou Q, Berglund LA. Functionalized cellulose nanocrystals as biobased nucleation agents in poly(l-lactide) (PLLA)/crystallization and mechanical property effects. Compos Sci Technol. 2010;70(5):815–21. doi:10.1016/j.compscitech.2010.01.018.
Robles E, Urruzola I, Labidi J, Serrano L. Surface-modified nano-cellulose as reinforcement in poly(lactic acid) to conform new composites. Ind Crops Prod. 2015;71:44–53. doi:10.1016/j.indcrop.2015.03.075.
Pracella M, Haque MMU, Puglia D. Morphology and properties tuning of PLA/cellulose nanocrystals bio-nanocomposites by means of reactive functionalization and blending with PVAc. Polymer. 2014;55:3720–8. doi:10.1016/j.polymer.2014.06.071.
Sanchez-Garcia M, Lagaron J. On the use of plant cellulose nanowhiskers to enhance the barrier properties of polylactic acid. Cellulose. 2010;17(5):987–1004. doi:10.1007/s10570-010-9430-x.
Herrera N, Mathew AP, Oksman K. Plasticized polylactic acid/cellulose nanocomposites prepared using melt-extrusion and liquid feeding: mechanical, thermal and optical properties. Compos Sci Technol. 2015;106:149–55. doi:10.1016/j.compscitech.2014.11.012.
Martinez-Sanz M, Abdelwahab MA, Lopez-Rubio A, Lagaron JM, Chiellini E, Williams TG, et al. Incorporation of poly(glycidylmethacrylate) grafted bacterial cellulose nanowhiskers in poly(lactic acid) nanocomposites: improved barrier and mechanical properties. Eur Polymer J. 2013;49:2062–72. doi:10.1016/j.eurpolymj.2013.04.035.
Avrami M. Kinetics of phase change. I. General theory. J Chem Phys. 1939;7:1103–12.
Avrami M. Kinetics of phase change. II. Transformation-time relations for random distribution of nuclei. J Chem Phys. 1940;8:212–24.
Lambrigger M. Non-isothermal polymer crystallization kinetics and Avrami master curves. Polym Eng Sci. 1998;38(4):610–5.
Kawai T, Rahman N, Matsuba G, Nishida K, Kanaya T, Nakano M, et al. Crystallization and melting behavior of poly (l-lactic acid). Macromolecules. 2007;40(26):9463–9. doi:10.1021/ma070082c.
Tsuji H, Takai H, Saha SK. Isothermal and non-isothermal crystallization behavior of poly(l-lactic acid): effects of stereocomplex as nucleating agent. Polymer. 2006;47(11):3826–37. doi:10.1016/j.polymer.2006.03.074.
Tsuji H, Miyase T, Tezuka Y, Saha SK. Physical properties, crystallization, and spherulite growth of linear and 3-arm poly(l-lactide)s. Biomacromolecules. 2005;6(1):244–54. doi:10.1021/bm049552q.
Di Lorenzo ML. Crystallization behavior of poly(l-lactic acid). Eur Polym J. 2005;41(3):569–75. doi:10.1016/j.eurpolymj.2004.10.020.
Abe H, Kikkawa Y, Inoue Y, Doi Y. Morphological and kinetic analyses of regime transition for poly[(S)-lactide] crystal growth. Biomacromolecules. 2001;2(3):1007–14. doi:10.1021/bm015543v.
Tsuji H, Tezuka Y, Saha SK, Suzuki M, Itsuno S. Spherulite growth of l-lactide copolymers: effects of tacticity and comonomers. Polymer. 2005;46(13):4917–27. doi:10.1016/j.polymer.2005.03.069.
Di Lorenzo ML. Determination of spherulite growth rates of poly(l-lactic acid) using combined isothermal and non-isothermal procedures. Polymer. 2001;42(23):9441–6. doi:10.1016/S0032-3861(01)00499-2.
Vasanthan N, Ly H, Ghosh S. Impact of nanoclay on isothermal cold crystallization kinetics and polymorphism of poly(l-Lactic Acid) nanocomposites. J Phys Chem B. 2011;115(31):9556–63. doi:10.1021/jp203322d.
Hoffman JD, Miller RL. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: theory and experiment. Polymer. 1997;38(13):3151–212. doi:10.1016/S0032-3861(97)00071-2.
Marand H, Xu J, Srinivas S. Determination of the equilibrium melting temperature of polymer crystals: linear and nonlinear Hoffman-Weeks extrapolations. Macromolecules. 1998;31(23):8219–29. doi:10.1021/ma980747y.
Acknowledgements
The authors would like to gratefully acknowledge partial support of this work by U.S. Forest Service Grant 11-JV-11111101-050.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gupta, A., Simmons, W., Schueneman, G.T. et al. Lignin-coated cellulose nanocrystals as promising nucleating agent for poly(lactic acid). J Therm Anal Calorim 126, 1243–1251 (2016). https://doi.org/10.1007/s10973-016-5657-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5657-6