
Abstract
The phosphorous fertilizers are a product of natural sedimentary phosphorite ores. Using this raw material to produce phosphoric acid and classic phosphorous fertilizers has generated well-known ecological problems. A new and perspective way to use the same materials is creating a new type of time-delayed fertilizers applying high-energy milling (HEM) activation method. The impact of the mechanical forces over the solids is mostly revealed through the changes of the quantities being related to the energetic stability and reactivity of the solid phase. The aim of this work is to report the results from the investigation on the chemical and thermal reactions in composites of natural apatite , which are HEM activated for different times and thermally treated, (from Tunisia) and ammonium sulphate. The Tunisian phosphorite belongs to the ‘basic’ apatites having a Ca/P ratio of 1.70–1.77 and is characterized by a complex mineral composition with major component carbonate-fluorapatite. The used ammonium sulphate—(NH4)2SO4 is obtained as a by-product from cleaning industrial waste gases, using e-beam technology. The composites of Tunisian phosphorite ores and ammonium sulphate, mixed in a mass ratio 1:1, were HEM activated during 10 min to 50 h with 20 mm Fe-milling bodies and temperature treated up to 1,100 °C. As a result, the chemical properties of the treated composites changed. Proofs were found for (i) formation of new phases during HEM activation such as NH4Ca(PO3)3 (NH4)2CaH4(P2O7)2, (NH4)2Ca3(P2O7)2.6H2O, CaH2P2O7 and α-Ca2P2O7; and (ii) decreasing of temperature intervals of phase changes in comparison to untreated composite.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The production of phosphoric acid and Phosphorous fertilizers from natural apatite by the classic acid-leaching method creates well-known ecological problems [1–3]. Using the method in addition to high-energy ball milling and thermal methods allows one to obtain new products (fertilizers) which can transform water-soluble and insoluble forms of P2O5 to citrate-soluble forms [4, 5]. A new possibility in this topic is the creation of ecologically safe complex fertilizer compounds and soil improvers by thermal and high-energy milling (HEM) methods from natural apatite mixed with industrial and semi-industrial wastes [6–8]. An important topic of interest during complex treatment of natural apatite is a possibility of solid-phase synthesis of the main (Ca, PO4, F and OH) and accessory (SiO2, Na2O, R2O3 - R = Al, Fe, etc.) components of the natural system.
In our previous works we investigated chemical and thermal reactions of
-
(i)
High energy activated and thermal treated composites of natural apatite from Tunisia and ammonium sulphate (pure for analysis—p. a.) grade in a mass ratio 1:1, where the HEM activation was carried out with planetary mill (Cr–Ni milling balls) and the thermal treatment—at temperatures up to 1,100 °C. The HEM activation was at wide range of times [4, 9].
-
(ii)
Thermal-treated composites of natural apatite from Tunisia and ammonium sulphate—by-product from the electron-beam waste gas cleaning system in the Thermal Power Plant Maritsa-East-2, Bulgaria in a mass ratio 1:1. The thermal treatment was accomplished at dynamic and isothermal heating conditions at temperatures up to 1,100 °C [4].
The obtained results show a promising mechanism of the thermal decomposition reactions requiring additional refinement.
In order to extend our earlier studies we investigated the chemical and thermal reactions in a new composite of natural apatite that is HEM activated for different times and thermally treated (from Tunisia) and ammonium sulphate, obtained as a by-product from cleaning industrial waste gases, using e-beam technology, in a mass ratio 1:1. The phase transformations of the composites after HEM and thermal treatment to solid products are confirmed using a complex of the different analyses: standard chemical analysis, X-ray powder diffraction, and infrared spectroscopy. A relationship was found between phase transformations in solids and the conditions of HEM activation.
Materials
Raw materials used for the new composite:
-
natural carbonate-fluorapatite Ca5F(PO4)3 (CFAp) [4, 7, 10] from Tunisian sedimentary phosphorite ore deposit with the following chemical composition: 29.5 % P2O total5 ; 6.9 % P2O ass5 (by 2 % citric acid); 3.2 % F; 46.5 % CaO; 0.55 % R2O3 (R = Al, Fe); 1.1 % SO3; 7.3 % SiO2; 0.35 % MgO; 0.05 % Cl; 6,2 % CO2; moisture content 3.14 % and a granulometric size of the particles of 0.8 mm.
-
ammonium sulphate (NH4)2SO4—obtained as a by-product from cleaning industrial waste gases, using e-beam technology. The parameters and the properties of ammonium sulphate are given elsewhere [11, 12].
The composite composition: CFAp and by-product of (NH4)2SO4 in mass ratio 1:1 (TS0).
The initial TS0 is HEM activated for different times: 5, 10, 30 and 60 min, as well as for 5, 10, 15, 30, 40 and 50 h. As a result, we obtained 10 activated samples, TS5 m, TS10 m, TS30 m, TS60 m, TS5 h, TS10 h, TS15 h, TS30 h, TS40 h and TS50 h, respectively.
Methods
The HEM activation was carried out in a planetary mill Pulverisette-5, Fritsch Co (Germany). The activation times were from 10 min to 50 h with Fe-milling bodies with diameter of the milling bodies of 20 mm and sample mass of 0.020 kg. The HEM activation was chosen from our previous work [13]. The selection was made in the order to satisfy earlier obtained results for optimal activation time and our next investigation for the linear dependence of activation time and particle agglomeration, whereas the increasing level of agglomeration depends contrariwise on the specific surface area [4, 14, 15]. Furthermore, in the experiment we used another type of milling balls, Fe, because of their well-known catalytic activity and ability of increasing the solubility of activated composites [14, 16].
We used standardized methods for determination of P2O sol5 defined from Bulgarian National Standard 14131-88, according to which P2O sol5 can be determined by direct extraction in solution of ammonium citrate with pH 7 or by 2 % citric acid. This method is relevant to the Instruction of EEO 77/535, p. 3.1.4 ‘Extraction of phosphorus soluble in neutral ammonium citrate’ and the dragging out of P2O sol5 is performed by direct extraction.
The powder X-ray diffraction (XRD) measurements were made through a DRON 3 M diffractometer, using a Fe-filtered Cu-Kα radiation in the range 8–60° 2-theta, at an accelerating voltage of 40 kV and a current of 25 mA.
The obtained Fourier transform infrared (FTIR) spectra were registered on Bruker Tensor 37 spectrometer in the range 400–4,000 cm−1, using KBr pellet technique. A resolution of 2 cm−1 was used for collecting 60 scans for each sample.
The thermal analyses (TG/DTG/DTA) were performed on a Stanton Redcroft thermal analyzer STA 780 (England) in the temperature range 20–1,100 °C, with a heating rate of 10 °C min−1, the purging gas—dry air and flow rate 50 mL min−1. α-Al2O3 was used as reference material [17, 18]. For the thermal analyses experiments the Zirconium open crucibles with diameter of 4.5 mm and the sample mass of ~10 mg were used.
Results and discussion
Chemical analysis
The most important chemical component for plants in phosphorus fertilizers is P2O5 and more specifically the quantity of soluble P2O5 (P2O sol5 ). The P2O sol5 is slightly soluble in water, so it is difficult to be washed out by the rain from the soils. Thus P2O sol5 stays for a long time in soils and feeds the plants for a longer time. The used method for chemical analysis presents the quantity of P2O sol5 in per cent as the ratio to the total P2O5 (P2O tot5 )−P2O tot5 /P2O sol 5 %. In our earlier investigation [5, 19] we did not measure the quantity of P2O sol5 .
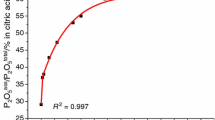
Figure 1 shows the dependence of P2O sol5 from HEM activation time for the non-activated sample (TS0) and the ten activated composites TS5 m, TS10 m, TS30 m, TS60 m, TS5 h, TS10 h, TS15 h, TS30 h, TS40 h and TS50 h. An activation time–exponential growth dependence of the P2O tot5 /P2O sol 5 % for the 10 samples (from 0 min to 40 h) was found. The experimentally measured values are shown in Fig. 1 by solid circles, while the solid line is the best fit to them, y = −18.05 exp(x/−7.7)−12(x/−798.66)+57.84 (R 2 = 0.992). A rapid increasing of the P2O sol5 amount from 0 to 60 min is followed by gradual increase—from 5 to 40 h activation. The sample TS50 h (presented by solid circle in the Figure) shows independent behaviour compared to other samples because of the strong influence of particle agglomeration [9, 15].

The activation time dependence of measured P2O ass5 : /black circle/ samples with exponential growth dependence of the P2O tot5 /P2O ass 5 % and the best fit to these data /solid line//TS0, TS5 m, TS10 m, TS30 m, TS60 m, TS5 h, TS10 h, TS15 h, TS30 h and TS40 h/; /white circle/ sample outside exponential growth dependence/TS50 h/
Powder XRD
Figure 2 shows the powder XRD patterns for TS10 m, TS5 h, TS10 h and TS50 h HEM-activated samples together with the established crystalline phases. It is seen that the increase of the activation time leads to decrease in the peak intensity and to an increase in the peak widths. Furthermore, peaks of newly formed phases appear: Ca3(PO4)2.xH2O, (NH4)2SO4.2CaSO4, NH4Ca(PO3)3, (NH4)2CaH4(P2O7)2, (NH4)2Ca3(P2O7)2.6H2O, CaH2P2O7 and α-Ca2P2O7 together with phases of the raw materials Ca5F(PO4)3 и (NH4)2SO4. The existence of ammonium calcium pyrophosphates speaks in favour of solid-phase reactions between the components of the system during the activation. The phases (NH4)2SO4.2CaSO4 and α-Ca2P2O7 result from the thermal decomposition of non-activated composite and prove the identity of transformations during thermal treatment and HEM activation [4]. It is important to be noted that investigated samples indicate some differences in comparison with the early studied ones [8]: (i) the amount of newly formed crystalline phases is higher than that of the composite form CFAp and ammonium sulphate (p. a.), HEM activated with 20 mm Fe balls; (ii) there exist some new phases that were not proven in earlier powder XRD studies, which applies especially for the composites activated for 10 and 50 h; and (iii) for recently investigated samples the phase CaSO4.2H2O is missing.

Powder XRD patterns of composite of CFAp HEM activated for different times and by-product of ammonium sulphate in 1:1 mass ratio /samples TS10 m, TS5 h, TS10 h and TS50 h/
The formation of the new crystal phases is a result of the different milling balls used in this investigation. The new phases contain NH4 and OH− groups, increasing the sample solubility, a fact, confirming the results from chemical analysis (Fig. 1). Contrary, the obtained maximal amount of new phases of TS50 h does not match with the chemical analysis, because of the strong decreasing of specific surface area of the sample particles [9, 15].
FTIR measurements
The measured FTIR spectra show the influence of thermal treatment and HEM activation of composite on the formation of new solid phases. The obtained results are shown on Fig. 3.

FTIR spectra of non-activated (TS0) and composite/samples HEM activated for different times TS10 m, TS60 m and TS50 h/of CFAp and by-product of ammonium sulphate in 1:1 mass ratio
The raw CFAp and ammonium sulphate are proven for TS0 and all activated samples with the IR absorption bands of
-
PO4 3−-group in CFAp: symmetric P–O (ν1 and ν2) modes in the ranges 964–968 and 471–473 cm−1, respectively; asymmetric P–O (ν3) modes in the ranges 1,046–1,048, 1,103–1,106 and 1,186–1,188 cm−1; asymmetric P–O (ν4) mode in the ranges 570–575 and 616–620 cm−1.
-
SO4-group in (NH4)2SO4: asymmetric S–O (ν3) stretching mode in the range 1,103–1,106 cm−1.
-
NH4-group in (NH4)2SO4: asymmetric N–H (ν4) bending mode in the range 1,401–1,404 cm−1.
FTIR data confirm the results obtained by powder XRD measurements (Fig. 2). Furthermore, the IR measurements prove a new mineral phase (CaCO3) for all studied samples with symmetric O–C–O (ν2) bending mode (866–868 cm−1) of CO 2−3 in B-type CFAp and asymmetric O–C–O (ν3) stretching mode (1,433–1,450 cm−1) of CO 2−3 in CFAp. The CaCO3 phase is probably amorphous for proper investigation with powder XRD.
The IR measurements show the changes occurring after HEM activation of the samples due to the presence of
-
OH libration mode (in the range 654–656 cm−1) of CaH2P2O7—available for all HEM-activated samples.
-
Asymmetric P–O–P (ν3) stretching mode (730–738 cm−1 and 1,103–1,108 cm−1) of P2O7 4− in CaH2P2O7—available for all HEM-activated samples.
-
Asymmetric P–O (ν3) stretching mode (1,046–1,048 and 1,186–1,188 cm−1) of P2O7 4− in CaH2P2O7—available for all HEM-activated samples.
-
Asymmetric P–O–P (ν3) stretching mode (774–780 cm−1) of PO3 2− in NH4CaP3O9—available for samples TS60 m, TS5 h, TS10 h and TS50 h.
-
Asymmetric O–C–O (ν3) stretching mode (1,500–1,506 cm−1) of CO 2−3 in A/B-type CFAp—available for all HEM-activated samples. This band is a result from CO3 incorporation in the CFAp structure [19, 20].
-
Asymmetric (ν4) bending mode (1,401–1,404 cm−1) of NH +4 in NH4CaP3O9.
The IR measurements show the following solid-phase composition: CFAp, (NH4)2SO4, CaH2P2O7 (with structure incorporated H2O) and NH4CaP3O9. Table 1.
Thermal analysis
The results from thermal analysis are shown on Fig. 4a–c and Table 2.

Thermal decomposition of non-activated/TS0/and HEM-activated composite/samples TS10 m, TS60 m, TS5 h, TS10 h and TS50 h/of CFAp and by-product of ammonium sulphate in 1:1 mass ratio. a TG curves, b DTA curves and c DTG curves
The results show that thermal decomposition of the samples takes place during the following temperature stages: 310–380 °C, 380–450 °C, 610–770 °C and 770–1,100 °C, accompanied by mass losses from 39 to 44 %. The identified stages, together with the measured mass losses, are close to our previous measurements where we used Cr–Ni balls for HEM activation of the composite of CFAp and ammonium sulphate—p. a. grade in a mass ratio 1:1 [4].
TG,/DTG/DTA measurements show the decomposition dependencies of TS0. The analysis of these dependencies proves that thermal instability of (NH4)2SO4 controls the mechanism of chemical reaction between CFAp and (NH4)2SO4. As a result, from chemical interactions in the composite, the new phases are formed in different temperature stages. In the 310–380 °C range, (NH4)2SO4.2CaSO4, CaHPO4 and NH4HSO4 are formed as products from interaction of the two raw materials. In the 380–420 °C range, CaSO4 and α-Ca2P2O7 are formed as products from interaction between newly formed CaHPO4 and NH4HSO4 and raw Ca5F(PO4)3. Additionally, from ammonium calcium phosphate phases NH4CaP3O9 is formed. This phase is slowly soluble in ammonium citrate, a liquid, close to soil solutions. In the 420–450 °C range new quantity of α-Ca2P2O7 is formed, because of already possible interaction between CFAp and NH4CaP3O9 in this temperature range. In the 610–770 °C range a decarbonization of CaCO3 occurs (CaCO3 is presented as impurity in raw phosphorite ore). In the 770–1,100 °C range runs formation of Ca3(PO4)2 due to interaction between α-Ca2P2O7 and CaSO4 [4, 21].
The thermal decomposition experiments (Fig. 4) show 6–9 % decreasing of mass losses in case of TS10 m. For sample TS50 h 6 % decreasing of mass losses was found in comparison with TS0 and 4 % increasing of mass losses compared TS10 m. At all HEM-activated samples a general trend of decomposition temperatures decreasing is measured, best presented by TS10 m.
The mechanism of thermal chemical reactions of HEM-activated composite (Table 2) is defined from the main reactions of decomposition and interaction of raw CFAp and ammonium sulphate, namely formation of (NH4)2SO4.2CaSO4 and Ca3(PO4)2. At increasing temperature (380–420 °C range) the formation of NH4CaP3O9 and CaH2P2O7 occurs. The formation of CaH2P2O7 is damaged from the general catenation rule of P-atom (the ability of the atoms of phosphorus to form the branched and unbranched chains), according to which pyro- and poly-phosphates can be obtained only after the formation of the hydrogen phosphates [9, 22]. All thermal reactions are two-staged for the TS0 and one-staged for the HEM-activated samples. A trend of the decreasing of temperatures of the thermal decomposition of HEM-activated samples is also established.
The new crystalline phases are formed during interaction between (i) raw materials and (ii) hydrogen-, ammonium calcium-ortho-, and pyro-phosphates. The latter phases are a product of temperature treatment and HEM activation, which complicate the reaction mechanisms additionally. In the 420–450 °C range there are overlaid reactions of decomposition/dehydration with formation of insoluble calcium pyrophosphates.
At higher temperatures of treatment the chemical reactions of HEM-activated samples and TS0 are similar: decarbonization of CaCO3 and interaction of Ca2P2O7 with CaSO4 [23].
It is important to note the registration of exothermal effect of TS10 h, which results probably from HEM activation (the exothermal effect missing in case of TF0) (Fig. 4). The effect probably is a product from deformation of CFAp crystal structure and accumulation of mechanical energy during HEM activation [1]. Simultaneously, the spontaneous reconstruction of crystal phases occurs during relaxation of accumulated energy. All these lead to formation of stress fields—energy-unstable state for the system. The accumulated mechanical energy relaxes and causes chemical reactions with reduced heat energy consumption [24, 25]. With increasing of activation time the force of exothermal effect decreases probably due to increasing agglomeration effect.
Figure 5 presents the shifting of sample decomposition temperature depending on HEM activation time of composite for the different temperature stages. All shown dependences are not functional because of strong particle agglomeration influence [9, 15]. Nevertheless, it is possible to note the following trends:

Shifting of decomposition temperatures depending of time for HEM activation /samples TS0, TS60 m, TS5 h, TS10 h and TS50 h/: a temperature stage I: 20–195 °C, b temperature stage II: 310–380 °C, temperature stage III: 410–450 °C and stage IV: 620–700 °C, c temperature stage V: 820–1,020 °C
-
Stage I (Fig. 5a)—there exist two peaks of thermal decomposition situated near 41 and 181 °C. The shift of the peaks is with trend of increasing the temperature of decomposition by increasing the time of HEM activation.
-
Stages II, III and IV (Fig. 5b) are with peaks situated near 330, 420 and 693 °C, respectively. The observed trend is opposite to the previously described one: decreasing of the decomposition temperature with increasing the time of HEM activation. It is important to note that the minimum decomposition temperature for Stage II is measured for TS60; for Stage III—sample TS5 h and for Stage IV—sample TS50 h.
-
Stage V (Fig. 5c) with peaks near 857 and 927 °C shows trend similar to the previous one (for the HEM-activated samples over 60 min).
The obtained trends indicate that thermal treatment up to 190 °C is not sufficient to decrease the temperature of thermal decomposition in HEM-activated samples. Contrarily, the clear decreasing of temperature decomposition is marked at Stage II (temperature near 330 °C) for the sample TS60 m. The effects for the HEM-activated samples over 60 min and for samples threaded at temperature over 330 °C stay practically unchanged in comparison to the last described one. Therefore, it is not necessary to heat the samples at temperature over 330 °C and high energy activate for over 60 min. These results confirm well the results from the chemical and FTIR analyses.
As a result of the applied high-energy ball milling, the crystal structure of apatite accumulates mechanical energy and passes into a metastable energy state. A change of this type increases the apatite reactivity, which is accompanied with the formation of ammonium calcium phosphates. The appearance of an exothermal effect on the DTA curve probably is a function of the sample activation (the effect is missing on the DTA curve of the non-activated sample) [4]. The accumulated mechanical energy relaxes at higher temperatures, which is indicated by the exothermal effect on the DTA curve—the effect is best expressed as a transformation of CaH2P2O7 into α-Ca2P2O7 for the sample TS10 h. On increasing the activation time the intensity of the exothermal effect decreases probably due to a stronger particle agglomeration. Under subsequent thermal treatment of the activated samples, the following reactions occur: (i) reaction between activated apatite and ammonium sulphate with the formation of ammonium calcium hydrogen ortho- and pyro-phosphates; (ii) decomposition of previously formed new phases and (iii) decomposition of ammonium calcium hydrogen ortho- and pyro-phosphates. In addition, a reduction of the reaction temperature is observed.
After comparison between results of composite samples, HEM activated with different milling balls, some interesting facts are found. At HEM activation with 20 mm with Fe balls: (i) the temperature ranges with most intensive mass losses stay the same with the ones obtained at activation with Cr–Ni balls (380–420 and 420–450 °C) and (ii) one-stage reactions take place, unlike during activation with Cr–Ni balls, where ammonium- and ammonium calcium-pyrophosphates together with NH4CaP3O9 are obtained. All these results prove the catalytic and intensification effect of Fe-milling balls on the investigated composite [26]. The Fe balls are lighter than the Cr–Ni ones but their use does not lead to formation of larger diversity of ammonium salts.
Conclusions
The high-energy ball milling activation and the thermal decomposition (up to 1,100 °C) experiments of a composite from natural apatite and ammonium sulphate prove an increased activity of the samples with increasing of activation degree. We observed evidences for
-
(i)
Solid-phase synthesis of (NH4)2H2P2O7, CaH2P2O7, α-Ca2P2O7, NH4CaP3O9 and Ca3(PO4)2.
-
(ii)
Optimal time for HEM activation—up to 60 min because of particle agglomeration process due to longer times of HEM activation (5 and 10 h), in spite of maximum number of ammonium calcium phosphate phases formation during these activation times. The optimal HEM activation time of 60 min is confirmed as well as of rapid increasing of P2O sol5 concentration.
-
(iii)
Optimal temperature for thermal activation is up to 450 °C because of beginning of dehydration process and decomposition of ammonium calcium phosphates with formation of insoluble pyrophosphates.
-
(iv)
Use of by-product of (NH4)2SO4 did not influence negatively on the chemical reactions and can be used successfully in obtaining fertilizer components and soil conditioner.
-
(v)
To avoid losses of NH3 and SO2 (useful and expensive industrial raw materials as well as atmospheric pollutants) during HEM and thermal activations, the raw material for the composite needs to be HEM-activated apatite and non-activated ammonium sulphate.
-
(vi)
Longer activation time (up to 50 h) results due to agglomeration effects and diffusion problems arising in chemical reactions between the components of the system.
The value of this paper is that the new data obtained confirm the possibility to recover partially ammonia and the same time to obtain new fertilizer product with the content of 3 nutrition elements as N, P and S. A next step of experimental work is needed to confirm the efficiency of such new method.
Abbreviations
- HEM:
-
High-energy milling
- CFAp:
-
Natural carbonate fluorapatite Ca5F(PO4)3
- TS0:
-
Non-activated mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1
- TS5m:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 5 min
- TS10m:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 10 min
- TS30m:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 30 min
- TS60m:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 60 min
- TS5h:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 5 h
- TS10h:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 10 h
- TS15h:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 15 h
- TS30h:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 30 h
- TS40h:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 40 h
- TS50h:
-
Mixture Ca5F(PO4)3 + (NH4)2SO4 – mass ratio 1:1, HEM activated 50 h
- XRD:
-
Powder X-ray diffraction
- FTIR:
-
Furies transform infrared
- TG/DTG/DTA TA:
-
Thermal analyses
- P2O sol5 :
-
Soluble P2O5
- P2O tot5 :
-
Total P2O5
References
Pelovski Y, Petkova V, Dombalov I. Thermotribochemical treatment of low grade natural phosphates. J Therm Anal Cal. 2007;88:207–12.
Chaikina MV. Mechanochemistry of natural and synthetic apatites. In: Avvakumov EG, editors. Novosibirsk, Publishing house of SB RAS, Branch “GEO”, 2002, p. 11–15; 105–107; 114–115; 139.
Wieczorek-Ciurowa Kr, Gamrat K. Mechanochemical syntheses as an example of green processes. J Therm Anal Calorim. 2007;88:213–7.
Petkova V, Pelovski Y, Dombalov I, Tonsuaadu K. Thermochemical investigations of natural phosphate with ammonium sulphate additive. J Therm Anal Cal. 2005;80:701–8.
Yaneva V, Petrov O, Petkova V. Structural and spectroscopic studies of the Nanosize Appatite (Syrian). Mat Res Bull. 2009;44:693–9.
Ivanova V, Petkova V, Pelovski Y. Thermal analysis of new soil sorption regulators. J Therm Anal Cal. 2003;74:387–94.
Petkova V, Serafimova E, Petrova N, Pelovski Y. Thermochemistry of triboactivated natural and NH4-exchanged Clinoptilolite mixed with Tunisian Apatite. J Therm Anal Cal. 2011;105(2):535–44.
Petrova N, Petkova V. Structural changes in the system natural apatite - NH(4) clinoptilolite during triboactivation. Bulg Chem Commun. 2011;43(2):301–7.
Arasheva M, Dombalov Jv. Investigation on thermal stability and phase transformations in the system Marocco phosphorite - (NH4)3H(SO4)2 - NH4HSO4. J Therm Anal. 1995;43:359–68.
Koleva V, Petkova V. IR spectroscopic study of high energy activated Tunisian phosphorite. Vib Spectrosc. 2012;58:125–32.
Petkova V. Investigation of Investigation of the thermal decomposition of triboactivated samples of ammonium sulphate. Int J Bal Trib Assoc. 2004;10(3):344–54.
Petkova V, Pelovski Y, Hristova V. Thermal analysis for identification of E-beam nanosize Ammonium Sulfate. J Therm Anal Cal. 2005;82:813–7.
Pelovski Y, Petkova V, Dombalov I. Thermal analysis of mechanoactivated mixtures of tunisia phosphorite and ammonium sulfate. J Therm Anal Cal. 2003;72:967–80.
Petkova V, Yaneva V. The effect of mechano-chemical activation on the chemical activity, structural and thermal properties of carbonate substituted apatite from Syria. Part I. Chemical, structural, and spectroscopic investigations. J Environ Prot Ecol. 2012;13(2A):979–94.
Tõnsuaadu K, Kaljuvee T, Petkova V, Traksmaa R, Bender V, Kirsimäe K. Impact of mechanical activation on physical and chemical properties of phosphorite concentrates. Int J Min Pro. 2011;100(100):104–9.
Petkova V, Pelovski Y, Dombalov I, Kostadinova P. Influence of triboactivation conditions on the synthesis in natural phosphate. Ammonium sulphate system. J Therm Anal Cal. 2005;80:709–14.
Šulcová P. Thermal stability and colour properties of new pigments based on BiREO3. J Therm Anal Calorim. 2012;109:639–42.
Šulcová P, Večeřa J, Strnadlová L. Study of doped CeO2 prepared by different synthesis. J Therm Anal Calorim. 2012;108(2):519–23.
Tõnsuaadu K, Gross KA, Plūduma L, Veiderma M. A review on the thermal stability of calcium apatites. J Therm Anal Calorim. 2012;110(2):647–59.
Jebri S, Boughzala H, Bechrifa A, Jemal M. Structural analysis and thermochemistry of “A” type phosphostrontium carbonate hydroxyapatites. J Therm Anal Calorim. 2012;107(3):963–72.
Kostova B, Petrova N, Petkova V. The high energy milling effect on position of CO3-ions in the structure of sedimentary apatite. Bul Chem Commun. 2013;45(4):601–6.
Gorodylova N, Dohnalová Z, Šulcová P. Effect of the synthesis conditions on the formation of MgSrP2O7 and its characterisation for pigmentary application. J Therm Anal. 2013;113(1):147–55.
Larson PR, Madden AS, Tas A. Cuneyt, non-stirred synthesis of Na- and Mg-doped, carbonated apatitic calcium phosphate. Ceram Int. 2013;39:1485–93.
Boldyrev BB. Mechanochemistry of inorganic solids. Proc Indian Natl Sci Acad. 1986;52A:400–17.
Balaz P. Mechanochemistry in nanoscience and minerals engineering. Springer-Verlag Berlin Heidelberg, ISBN: 978-3-540-74854-0 e-ISBN: 978-3-540-74855-7, 2008.
Baláž P, Achimovičová M, Baláž M, Billik P, Cherkezova-Zheleva Z, Criado JM, Francesco D, Dutková E, Gaffet E, Gotor FJ, Kumar R, Mitov I, Rojac T, Senna M, Streletskii A, Wieczorek-Ciurowa K, Hallmarks of mechanochemistry: from nanoparticles to technology. Chem Soc Rev. 2013;42:7571–637.
Acknowledgements
Authors gratefully acknowledge the financial support of this work by the Central Fund of Strategic Development of New Bulgarian University. The Slovak agency ‘Grant’ and the Ministry of Education and Sciences of Bulgaria are gratefully acknowledged also for the financial support granted under contracts Nos. VEGA 2/0009/11 and DNTS/Slovakia/01/3.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kostova, B., Petkova, V. Effect of high-energy milling and thermal treatment on the solid-phase reactions in apatite–ammonium sulphate system. J Therm Anal Calorim 116, 737–746 (2014). https://doi.org/10.1007/s10973-014-3747-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-014-3747-x