
Abstract
This study explores the comprehensive characterization of Y3+ and Co2+ co-substituted LaFeO3 nanoparticles synthesized via the sol-gel auto-combustion method. The synthesized samples, La1-xYxFe1-y CoyO3 (x = 0,0.10 and y = 0,0.03,0.05,0.07) were characterized by employing various techniques such as x-ray diffraction (XRD), Scanning Electron Microscopy (SEM) with EDX, Raman spectroscopy, UV-visible spectroscopy, and Vibrating Sample Magnetometry (VSM). The Raman and XRD analysis, supported by Rietveld refinement, provided conclusive evidence of a pure orthorhombic LaFeO3 phase. Microstructural studies unveiled an agglomerate-type, irregular particle distribution, while EDX analysis confirmed the elemental composition. The XPS study gives evidence about the presence of both Fe2+ and Fe3+ oxidation states, and Co has a Co3+ oxidation state. UV-vis spectroscopy demonstrated enhanced visible light absorption, revealing a reduced bandgap with increasing doping percentages. VSM measurements exhibited M-H loops, substantiating the weak ferromagnetic nature of the materials. Moreover, these nanoparticles exhibit dielectric constants and low dielectric losses, making them suitable for use in devices for communication. Overall, these findings may offer valuable contributions to the understanding of nanomaterial characteristics for potential applications in diverse fields.
Graphical Abstract

Research Highlights
-
Synthesis of La1-xYxFe1-y CoyO3 (x = 0,0.10 & y = 0,0.03,0.05,0.07) nanoparticles by sol-gel method.
-
Confirmation of orthorhombic structure with Pbnm space group from XRD, Rietveld and Raman analysis with a decrease in crystallite size from 48 to 12 nm.
-
Disclosement of the presence of both Fe2+ and Fe3+ oxidation states and Co doping has a Co2+ oxidation state from XPS analysis. The bandgap tuning from 2.09 to 1.56 eV with an increase in Co2+ doping concentrations.
-
Weak ferromagnetic behavior observed for all the NPs. Enhanced Ms, Mr, and Hc for the 7% Co2+ doped LFO.
-
The 7% Co2+ doped LFO exhibited the highest dielectric constant, lowest tangent loss and highest conductivity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
In recent years, Perovskite oxides have drawn a substantial attraction because of their favorable traits and unique chemical and physical features, making them useful for numerous technological applications in domains notably energy conversion, environmental remediation, electrochemical sensors, transducers, capacitors, dynamic random-access memory and non-volatile computer chips [1, 2]. These materials, which get their name from the mineral perovskites, are distinguished by their remarkable electrical and optical properties due to their distinct crystal structure [3]. Perovskite materials generally have the chemical formula ABO3, where A is mostly an alkali earth metal or lanthanide and B is a transition metal [4, 5].
Among the perovskites, LaFeO3 (LFO) has been raised as a most potential and promising candidate with exceptional characteristics, including high catalytic activity, electrical conductivity and stability under extreme conditions [6]. The structure of LFO is orthorhombic with space group Pbnm at ambient temperature, retaining up to ~957 °C. Furthermore, a first-order structural phase transition from orthorhombic to rhombohedral occurs between 960 °C-1005 °C and a second-order phase transition from rhombohedral to cubic at very high temperatures ~1867 °C, close to the melting temperature (~1890 °C) [7]. The presence of magnetic ordering in LFO is created by the antiferromagnetic superexchange interaction between two Fe3+ ions via oxygen anion, where the Fe-O-Fe bond angle is close to 180°. Each Fe3+ in LFO is surrounded by six anti-parallel Fe3+ nearest neighbors [8]. Moreover, LFO exhibits weak ferromagnetic ordering due to the partial ion Fe3+ tilt spin and anti-parallel interaction [9]. Furthermore, band gap tuning of perovskite nanoparticles proved to be efficient for various applications including solar cells, optoelectronic and optical devices, and photo-catalysts for water splitting [10].
Additionally, in thin film or heterostructure forms, LFO has fascinating features related to its electrical conductivity. Because of this ability, LFO possesses the potential to be used in a wider range of electrical and optoelectronic applications, such as photovoltaic cells, memristors, and resistive switching devices [11]. More opportunities to improve LFO’s performance in various applications arise from its electronic property controllable via interface effects, strain engineering, and controlled defect engineering [12]. The exceptional stability of LFO under harsh environmental conditions is another advantage, making it suitable for high-temperature applications. Its tolerance towards oxygen vacancies and thermal expansion mismatch has been extensively studied, highlighting its potential in solid oxide fuel cells, oxygen separation membranes, and thermal barrier coatings [13].
Selective doping, at A-site/B-site/both, is a process that is used to enhance and modify the properties of perovskite materials, making these useful for multifunctional applications such as electronic devices, memory devices, optoelectronic devices and energy-related technologies [14]. The doping with rare earth elements like cerium, praseodymium, neodymium, samarium and gadolinium ions at A site of LFO brings a noticeable change in its magnetic and optical properties [15]. An excellent improvement in the result of dye degradation has been reported on europium, gadolinium, dysprosium, and neodymium-doped LFO [16]. Yttrium-doped lanthanum ferrite also exhibited weak ferromagnetic properties along with remanent magnetization [5, 16]. Tb-doped LFO exhibited significant improvements in dielectric results [17]. Transition metal doping on the B-site of LFO has been studied extensively during the past two decades [18,19,20,21]. Since the transition metal cations have similar ionic radii with Fe, these are mostly preferred as dopants. However, the dopant concentration plays a significant role in the crystal structure formation. Rini et al. has also reported that beyond a certain dopant concentration (Co2+), the structure of LFO changed from orthorhombic to rhombohedral [22]. Gu et al., [23] have also studied the effect of cobalt doping in LFO and reported about sustainable evidence of the antiferromagnetic behavior and its use in n-butanol sensors [23, 24]. Manganese (Mn) is also a prominent doped material on the B-site that gives a new perspective for memory device creation [25]. It has also been published that the introduction of dopants like Mn, Co and Cu in LFO remarkably enhanced the photoelectrochemical performance [26]. Moreover, co-doping with divalent metal ions at A-site and transition metal ions at B-sites in LFO has shown a significant effect on its structural and electrical properties.
After a thorough study on doped LFO, it has been revealed that there are rarely any studies on the collective doping of yttrium and cobalt ions. In this work, we present a comprehensive investigation of the Y-Co ion-doped LFO synthesized by auto-combustion method. The effect of doping concentrations on the structural, microstructural, optical, magnetic and dielectric properties is thoroughly studied, exploring their application domain in multifunctional devices.
2 Experimental details
2.1 Synthesis method
The perovskite samples of La1-xYxFe1-yCoyO3 (x = 0,0.10 and y = 0,0.03,0.05,0.07) are prepared from nitrates precursor-based modified auto combustion method. The base materials are of analytic-grade lanthanum nitrate (LaN3O9.6H2O), ferric nitrate (Fe (NO3)3.9H2O) along with the dopants yttrium nitrate hexahydrate(Y(NO3)3.6H2O) and cobalt nitrate hexahydrate (Co(NO3)2.6H2O). Initially, the appropriate amount of raw materials were added to DI water to prepare 0.05 mol/L solution. After the proper dissolution of powders, the chelating agent citric acid monohydrate (C6H8O7.H2O) was added, maintaining a cation-to-anion ratio of 1:4. The polymerization process started by stirring the solution in a magnetic stirrer at 550 rpm for 3 hr. Subsequently, the sol was kept inside the hot air oven at 150 °C overnight to form the xerogel, followed by the auto-combustion method. Finally, the collected powder was grinded finely and calcined in a muffle furnace at 800 °C for 4 hr, where the heating and cooling rates were set at 5 °C per min. The final products with their code names are presented in Table 1.
2.2 Characterization techniques
All the X-ray diffraction (XRD) patterns were traced with the help of PANalytical X’pert PRO with Cu Kα radiation to determine the structure, phase and space group. The FULLPROOF SUITE software was used for the refinement of observed XRD peaks, which helped to determine the crystal structure. The 3D structure of all NPs are obtained using VESTA software. The visible Raman spectrometer (Renishaw), using an Argon gas LASER of wavelength 514.5 nm, was used to confirm the phase and chemical structure of all synthesized samples. The Field Emission Scanning Electron Microscope (FESEM) was used to get an insight visual of the samples to determine the size, shape and particle growth. The absorbance and band gap energy (Eg) of all the materials were analyzed by UV-vis spectrometer (Cecil CE3055). To measure the absorbance of the materials, the powder samples were compacted into pellets, and a standard reference in the range of 200–900 nm wavelength was used for proper absorbance data. After getting the values, we analyzed the band gap energy (Eg) by plotting the Tauc plot using the standard equation. The chemical state analysis of the samples were analyzed using X-ray photoelectron spectroscopy (XPS) (Physical Electronics, PHI 5000 VersaProbe III). All the peaks were analysed using XPEAK41 software. To obtain the impedance data, all the powder samples were pelletized with a diameter of ~12 mm and thickness of ~1 mm for dielectric measurements. The pellets were sintered at 500◦C for 2 hr. An impedance analyzer (HIOKI IM3536, Japan) was used to measure the dielectric properties of the sintered pellets at room temperature. Vibrating Sample Magnetometry (VSM) (Lake Shore 7410 Series VSM) was adopted for magnetic measurement at room temperature.
3 Results and discussion
3.1 (Micro)structural characterization
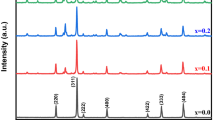
X-ray diffraction is a valuable technique used to get information regarding crystal structures, texture, orientation, phases, etc. Figure 1a–d shows the XRD patterns with the refinement of La1-xYxFe1-yCoyO3 (x = 0,0.10 and y = 0, 0.03, 0.05, 0.07), observed at room temperature. In Fig. 1a, all the peaks of pure LFO are distinguished, sharp, perfectly matched and indexed according to the standard JCPDS file 74-2203 [27]. The refinement of XRD patterns are done using FULLPROOF suite software. It gives an excellent agreement to the orthorhombic structure (space group Pbnm) of ferrite samples without changing the structural and lattice symmetry. The absence of extra peaks, other than lanthanum ferrite, in the observed XRD patterns indicated the phase purity of the samples. The different parameters like bond lengths, bond angles, Rwp, Rp and χ are observed from Rietveld refinement analysis and presented in Table 2. It is observed that there is a decrease in unit cell volume, which may be due to the substitution of Co over Fe for the cationic distribution at both sites. The 3D views of the crystal structures (shown inset to Fig. 1a–d) of all the NPs are obtained by using VESTA software and respective bond angles and bond lengths are calculated. The bond length enhanced, and the Fe-O-Fe angle reduced concentrations. The average crystallite size was calculated (using Scherrer’s equation [28]) as 42, 23,22, and 21 nm for LFO, LFO-1, LFO-2, and LFO-3, which gives a clear indication of gradual decrement in crystallite size for the synthesized perovskite materials & the interplanar spacing (d) are computed and listed in Table 2.

a–d Refined XRD patterns of LFO, LFO-1, LFO-2, and LFO-3 NPs. The insets show the 3D view of the crystal structures using VESTA software
The Williamson-Hall method was also used to calculate the average crystallite size and microstrain of the NPs, shows in Fig. 2a–d [28]. The crystallite sizes can be obtained by using the relation βcosθ=4εsinθ + kλ/D where β is the FWHM, θ denotes the angle of diffraction, ε is lattice strain, k is a constant number 0.89, λ is 0.154 nm and D denotes the crystallite size. The crystallite sizes of the synthesized NPs were ranged from 12–48 nm. The Goldschmidt tolerance factor [29], \(T=\frac{\left(\left(1-x\right){R}_{{La}}+x{R}_{Y}\right)+{R}_{O}}{\surd 2[\{1-y\}{R}_{{Fe}}+y{R}_{{Co}}+{R}_{o}]}\), where R is the ionic radius of each constituent element, and x and y are the dopant concentration percentage, usually determines ion stability, distortion, and compatibility within a perovskite material. When the tolerance factor T was in the range of 0.71-0.9, the crystal structure would be orthorhombic/rhombohedral structure. In our case, the T value was observed to decrease with an increment of doping %, which gives an agreement for better ionic stability and less distortion. The microstrain (ε) indicates the degree of distortion in the crystalline lattice calculated by the W-H plot. The dislocation densities, δ, were obtained for these materials (Table 2), which confirmed the excellent crystallinity of La1-xYxFe1-yCoyO3 (x = 0.1, y = 0.03, 0.05, 0.07).

a–d Willamson-Hall (W-H) plots of LFO, LFO-1, LFO-2, and LFO-3 NPs
A non-destructive technique of chemical research, Raman spectroscopy yields much evidence on phase and polymorphy, molecular interactions, chemical structure, and crystallinity. The observed Raman shifts with respect to intensity is displayed in Fig. 3. The Theoretical analysis of orthorhombic structures using group theory yields an irreducible representation of the Pbnm space group, Г = 7Ag + 7B1g + 5B2g + 5B3g + 8Au + 10B1u + 8B2u + 10B3u [30]. Out of these modes, the Raman active modes corresponding to Pbnm space group are 7Ag + 7B1g + 5B2g + 5B3g [31].

Raman spectra of LFO, LFO-1, LFO-2 and LFO-3 NPs
Figure 3 shows the Raman spectra of LFO, LFO-1, LFO-2 and LFO-3 NPs, observed in the wavenumber range 200–2000 cm−1 at room temperature. All the detected Raman modes are presented in Table 3. Raman modes below 200 cm−1 in LFO can be attributed to the deformation caused by the displacement of La cations, whereas modes between 200–300 cm−1 are due to oxygen octahedral tilt in La. The 400–500 cm−1 mode corresponds to the oxygen octahedral bending vibrations. Modes above 500 cm−1 are ascribed to oxygen stretching vibrations [32, 33]. The Raman modes are shifted slightly towards the lower wave number, that could be attributed to the variation in the average ionic radii at A-site [34]. The comprehensive features witnessed above 1000 cm-1 are associated with second-order scattering [33].
The FESEM micrographs of LFO, LFO-1, LFO-2, and LFO-3 nanoparticles are depicted in Fig. 3a–d. The FESEM images significantly display the formation of irregular and inhomogeneous shapes of LFO. The FESEM image of LFO-2 shows well nanocrystalline grains and agglomerated random shapes, which is nearly similar to the longitudinal spherical signature of the particles. However, LFO nanoparticles were observed to be more porous and have greater grain size than the doped ones [18]. The typical EDS spectra for LFO & LFO-2 samples, shown in Fig. 3e, f, confirm the elemental composition and purity of these materials. The constituent elements of the samples, such as La, Y, Fe, Co, and O, are present in appropriate stoichiometric proportions (Fig. 4). The acquired results are corroborated with the observed uniform distribution of the constituent elements, displayed in color mapping images (shown in Fig. 5a–f).

FESEM images of the synthesized NPs: (a) LFO, (b) LFO-1, (c) LFO-2, (d) LFO-3 and EDX spectra for (e) LFO, (f) LFO-2 NPs

a–f Color mapping images of LFO-2 NPs and the constituent elements La, Y, Fe, Co, O, respectively
3.2 Chemical state investigation
The XPS technique was adopted to understand the surface chemistry of the synthesized NPs. The XPS analysis usually determines the elemental composition and chemical and electronic state of various elements within a material. The high-resolution XPS spectra of the La 3d core levels, along with the deconvoluted spectra for both LFO and LFO-2 samples, reveal distinctive doublets corresponding to La 3d3/2 and La 3d5/2 peaks, as illustrated in Fig. 6a. In the case of the LFO sample, the binding energies for the La 3d5/2 and La 3d3/2 peaks were observed at 836.1 and 853.2 eV, respectively, with accompanying shake-up peaks at 839.25 and 856.11 eV. However, for the LFO 2 sample, the peaks exhibited a slight shift towards lower binding energies compared to the LFO sample. The difference in binding energy between these two peaks is ∼ 16.8 eV for both LFO and LFO-2 samples, indicating the + 3 oxidation state of La ion in both samples [35]. The deconvoluted Fe 2p XPS spectra is depicted in Fig. 6b. For the LFO sample, distinct peaks were observed at 709.6 and 722.7 eV, corresponding to Fe2+ 2p3/2 and Fe2+ 2p1/2, respectively. Additionally, peaks at 711.8 and 725.1 eV, along with satellite peaks at 719.1 and 733.3 eV, were identified as Fe3+ 2p3/2 and Fe3+ 2p1/2, respectively. These findings suggested the coexistence of both +2 and +3 oxidation states of Fe ions within the prepared materials. Furthermore, a slight shift in the peak positions was observed for the LFO-2 sample compared to the LFO sample. This shift is indicative of perturbations in the Fe-O bonds, maybe resulting from the introduction of dopant atoms [26]. The deconvoluted Y 3d XPS spectrum reveals distinct peaks, with the Y 3d5/2 peak observed at 160.11 eV and the Y 3d3/2 peak at 158.24 eV, as depicted in Fig. 6e. The energy splitting of \(\sim\) 1.8 eV between the Y 3d5/2 and Y 3d3/2 peaks strongly indicates the presence of Y in the +3 oxidation state [36]. The high-resolution Co 2p XPS spectrum, in Fig. 6b, exhibits two prominent peaks at 781.85 and 794.11 eV, representing the Co 2p3/2 and Co 2p1/2 states, respectively. Additionally, two separate satellite peaks were also observed at approximately 787.20 and 803.49 eV. These findings strongly suggest a + 2 oxidation state for Co in the material [19, 37]. Figure 6c shows two O 1 s peaks observed at 529.44 eV and 531.3 eV, which may be attributed to oxygen lattice species (OL) and oxygen hydroxyl species (OH), respectively [38]. For LFO-2, these two peaks were observed at 529.35 eV and 531.28 eV. The peak at 529.35 eV is attributed to a metal oxygen bond, while the peak at 531.28 eV is associated to a low oxygen coordinated defect site and surface absorbed oxygen species [39].

XPS spectra of (a) La 3d, (b) Fe 2p, (c) O 1 s for LFO, (d) O 1 s for LFO-2, (e) Y 3d, (f) Co 2p for LFO and LFO-2 nanoparticles
3.3 Optical response
The optical response of the synthesized nanoparticles were investigated by recording the absorbance spectra using UV-vis spectroscopy in the wavelength range 200–800 nm displayed in Fig. 7a. A stronger absorbance is obtained in the wavelength range of 250–600 nm in case of LFO sample, showing the absorption edge at 597 nm. The inclusion of dopants slightly lowers the absorbance in this region. On the other hand, in the wavelength range 500–800 nm, it can be noticed that LFO-2 nanoparticles exhibited the highest absorbance among the doped samples. The absorption edges are found to be shifted slightly as a function of doping. The optical bandgaps of these samples were computed by plotting a graph between (αhν)2 and hν, where α denotes the absorbance. This is adopted in accordance to the Tauc’s model of energy bandgap determination for direct bandgap semiconductors, (αhυ)2 = A(Eg-hν) [18]. It can also be noted that the Tauc’s model for indirect bandgap semiconductors did not match well to our experimental results. The bandgap energies were estimated by extrapolating the linear region of the absorbance edge to the abscissa (Fig. 7b–e). Figure 7f shows the variation of bandgap energy with doping concentrations. It can be inferred that doping significantly influences in lowering the bandgap, Eg (for LFO-3) = 1.56 eV being the lowest one achieved. The results indicate that these doped nanoparticles can have potential use in solar cells and photocatalytic applications. The formation of sub-bandgap energy levels within the bandgap, caused by interface defects and cavities, is accountable for the minimal decrease in bandgap due to the transformation of energy levels [40].

a UV-vis spectra, b–e estimation of bandgap energies using Tauc’s plots, f variation of energy bandgap with respect to Co2+ concentration of NPs
3.4 Magnetic response
Figure 8 exemplarily shows the M-H Loop obtained from the VSM measurements for La1-xYxFe1-yCoyO3 (x = 0.1, y = 0.03, 0.05, 0.07) NPs at room temperature (RT) with the applied magnetic field of 2 T. The LFO is usually known to exhibit weak ferromagnetic behavior because of the canted internal spin and the high percentage of uncompensated spins from the surfaces of the nanocrystals. The values of observed maximum magnetization (Ms), coercivity (Hc) and remanent magnetization (Mr) of the parent as well as doped samples are inscribed in Fig. 8. It is observed that for parent material (LFO), Ms and Hc values are 0.123 emu/g and 302 emu/g respectively, which is matched to the reported values by Sendil Kumar et al. [41]. The observation of weak ferromagnetism in cobalt-doped LFO may be a result of the substitution of Co ion in place of Fe at the B site of LFO [42]. Co substituted samples have a greater remanent magnetization (Mr) value than the parent material due to the enhancement of magneto crystalline energy of all the synthesized materials along with inclined angles modification of the Fe3+ moments [9, 43]. Amongst all, the LFO-3 exhibited the Ms of 0.136 emu/g and Hc as 337 emu/g. These results suggest that the Ms and Mr values increase with particle size, mainly due to spin non-collinearity at the surface [44]. The insets of Fig. 8a–d represent the rate of change of magnetization (dM/dH) with respect to the applied magnetic field. The loops exhibit differential magnetic susceptibilities (χ = dM/dH). All the observed loops show a typical butterfly shape, and the coercivity values can be calculated by obtaining the fullwidth at half maximum (FWHM), which is nearly equal to our observed values for all the samples [45].

a–d M-H curves at room temperature of LFO, LFO-1, LFO-2, and LFO-3 NPs. The insets show differential susceptibilities of respective NPs, e, f Variation of Ms, Hc, Mr and χ with respect to Co2+ concentrations
The squareness ratios of the M-H loops are computed by dividing the positive remanent magnetization to the maximum saturation magnetization. This parameter is highly significant in understanding the inter-grain magnetic interactions in the materials [45]. The nonzero values of these parameters confirm the presence of inter-grain magneto-static interaction in the materials. When S = 0.5, it confirms the existence of the randomly distributed non-interacting grains. Likewise, when S < 0.5, the magneto-static interactions between the grains may be allowed. The calculated squareness ratios of LFO, LFO-1, LFO-2 and LFO-3 are observed as 0.138, 0.16, 0.183 and 0.196, respectively. These values signify the possibility of magnetic interactions in our synthesized NPs. It may be one of the reasons for the enhanced magnetic parameters in the studied samples.
3.5 Dielectric response
The frequency-dependent dielectric responses of the sintered LFO, LFO-1, LFO-2, and LFO-3 pellets are evaluated at room temperature. Figure 9a depicts the monotonous decrease of dielectric constants (εr) with an increase in frequency for all the samples, indicating the usual dispersion behavior. A sharp rise in εr is quite evident for the samples with higher Co2+ dopant concentration: εr (LFO) = 348 and εr (LFO-3) = 1953 at a frequency of 3 Hz. It demonstrates the increase of exchange of Fe ions (Fe2+ ↔ Fe3+) due to partial Co2+ substitutions, thus stimulating the polarization effect, attributed to the Maxwell-Wagner type interfacial polarization, which is apparent in ferrites [46]. It is noteworthy to mention here that the XPS analysis on these samples also manifested the coexistence of Fe2+ and Fe3+ ions. The converged and flat lines at higher frequencies may be due to the effect of electronic polarization caused by the disorientation of the electric dipoles [47]. Figure 9b displays the frequency-dependent dielectric losses (tanδ) observed for the LFO, LFO-1, LFO-2, and LFO-3 pellets at room temperature. The tanδ values are quite small ( < 3) throughout the applied range of frequencies, reaching a minimal value (~0) at very high frequencies. The variation trend is similar in all the samples, showcasing the typical dielectric dispersion of tanδ [48].

Variation of (a) frequency-dependent dielectric response, and (b) dielectric tangent loss constant (tanδ) (c) Real (d) Imaginary (e) Cole-Cole plot of La1-xYxFe1-yCoyO3 (x = 0.1, y = 0.03, 0.05, 0.07) NPs
The electrical properties of these materials were investigated through complex impedance spectral analysis. Figure 9c, d displays the variation of the real and complex parts of the impedance (Z’, Z”) as a function of applied signal frequency, measured at room temperature. All the samples exhibited a decreasing trend in Z’, Z” up to 104 Hz, and thereafter converged and flat lines up to 106 Hz, following a similar trend to the dielectric constant graphs. This validates the dominance of space charge polarization in the low- frequency region, in line with the dielectric data presented above. Moreover, both impedance values were found to be highest for LFO and lowest for LFO-3 sample, indicating better conduction mechanism in LFO-3. Figure 9e shows the Nyquist plots (Z” vs. Z’) of the prepared samples. The observed semi-circular arcs indicate that the conduction mechanism in these samples is mainly dominated by the contributions from the movement of grain boundaries [49].
4 Conclusions
Yttrium and cobalt-doped Lanthanum Ferrites were successfully synthesized using the auto-combustion method. The XRD study, along with Rietveld refinement and Raman spectroscopy, provided a good agreement on a pure phase orthorhombic structure with pbnm space group in all the synthesized NPs. As the doping concentration increased, a change in bond length and bond angles and a decrease in crystallite size from 48 to 12 nm was observed. The microstructural studies using FESEM and EDX demonstrated the agglomerated structure and proper elemental composition. The chemical state analysis provided evidence of the presence of both the Fe2+ and Fe3+ ions. The UV-vis spectroscopy technique was adopted to study the absorbance and bandgap of the prepared NPs. The bandgap could be tuned from 2.09 to 1.56 eV with the increase of Co doping concentrations. The VSM study for all NPs showed weak ferromagnetic behavior in the parent material as well as in doped ones. The value of Ms for all the NPs increased from 0.123 emu/g to 0.136 emu/g upon doping. The frequency-dependent dielectric constant and loss followed the same trend of all the samples. LFO-3 attained the highest dielectric constant value (≈ 1953 at 3 Hz) compared to other materials. Y and Co doped LFO-3 exhibited the lowest ( ≈ 1.79 at 3 Hz). The impedance study explained that LFO-3 exhibited the highest conductivity. The materials with improved dielectric constant and low dielectric loss can be beneficial in memory storage devices and Bluetooth devices.
References
Suresh Kumar N, Chandra Babu Naidu K (2021) A review on perovskite solar cells (PSCs), materials and applications. J Materiomics 7:940–956. https://doi.org/10.1016/j.jmat.2021.04.002
Goel P, Sundriyal S, Shrivastav V et al. (2021) Perovskite materials as superior and powerful platforms for energy conversion and storage applications. Nano Energy 80:105552. https://doi.org/10.1016/j.nanoen.2020.105552
Matin MA, Hossain MN, Rhaman MM, et al. (2019) Dielectric and optical properties of Ni-doped LaFeO3 nanoparticles. SN Appl Sci 1: https://doi.org/10.1007/s42452-019-1453-9
Simoes AZ, Ortega PP, Ramirez MA et al. (2023) Magnetoelectric coupling at room temperature in LaTiO3/SrTiO3 heterojunctions. Mater Res Bull 162:112169. https://doi.org/10.1016/j.materresbull.2023.112169
Tolentino Cabral AC, Tafur Tanta UM, Simões AZ et al. (2023) Unveiling the metal-insulator transition at YTiO3/LaTiO3 interfaces grown by the soft chemical method. Mater Chem Phys 302:127709. https://doi.org/10.1016/j.matchemphys.2023.127709
Chertkova VP, Iskortseva AN, Pazhetnov EM et al. (2022) Evaluation of the Efficiency of Photoelectrochemical Activity Enhancement for the Nanostructured LaFeO3 Photocathode by Surface Passivation and Co-Catalyst Deposition. Nanomaterials (Basel) 12:4327. https://doi.org/10.3390/nano12234327
Fossdal A, Menon M, Waernhus I, et al. Crystal Structure and Thermal Expansion of La1xSrxFeO3 Materials Journal of American Ceramics 87(10):1952–1958. https://doi.org/10.1111/j.1151-2916.2004.tb06346.x
Mitra A, Shaw A, Chakrabarti PK (2020) Structural transformation induced enhanced multiferroicity in Al3+ and Ti4+ co-doped LaFeO3. Adv Powder Technol 31:2469–2479. https://doi.org/10.1016/j.apt.2020.04.013
Lin Q, Yang X, Lin J et al. (2018) The Structure and Magnetic properties of Magnesium-substituted LaFeO3 Perovskite negative electrode material by Citrate sol-gel. Int J Hydrog Energy 43:12720–12729. https://doi.org/10.1016/j.ijhydene.2018.03.156
Sitt A, Hadar I, Banin U (2013) Band-gap engineering, optoelectronic properties and applications of colloidal heterostructured semiconductor nanorods. Nano Today 8:494–513. https://doi.org/10.1016/j.nantod.2013.08.002
Rana DK, Mehta V, Kundu SK, Basu S (2020) Development of organic-inorganic flexible PVDF-LaFeO3 nanocomposites for the enhancement of electrical, ferroelectric and magnetic properties. Mater Chem Phys 242:122491. https://doi.org/10.1016/j.matchemphys.2019.122491
Shringi AK, Betal A, Sahu S et al. (2022) Resistive Switching and Synaptic Behavior of Perovskite Lanthanum Orthoferrite Thin Film for Neuromorphic Computing. IEEE Trans Electron Devices 69:6465–6470. https://doi.org/10.1109/TED.2022.3209957
Nga Phan TT, My Dinh TT, Duc Nguyen M et al. (2022) Hierarchically structured LaFeO3 with hollow core and porous shell as efficient sensing material for ethanol detection. Sens Actuators B Chem 354:131195. https://doi.org/10.1016/j.snb.2021.131195
Ranieri MGA, Ortega PP, Moreno H et al. (2021) Resistive switching and multiferroic behavior of La0.5Pr0.5FeO3 ferrite thin films. J Alloy Compd 851:156936. https://doi.org/10.1016/j.jallcom.2020.156936
Ateia EE, Ismail H, Elshimy H, Abdelmaksoud MK (2021) Structural and Magnetic Tuning of LaFeO3 Orthoferrite Substituted Different Rare Earth Elements to Optimize Their Technological Applications. J Inorg Organomet Polym Mater 31:1713–1725. https://doi.org/10.1007/s10904-021-01887-5
Gao TT, Zhu XN, Chen J et al. (2019) Conductive, dielectric and magnetic properties of Y-substituted LaFeO3 ceramics. J Alloy Compd 792:665–672. https://doi.org/10.1016/j.jallcom.2019.04.069
Bashir B, Warsi MF, Khan MA et al. (2015) Rare earth Tb3+ doped LaFeO3 nanoparticles: New materials for high frequency devices fabrication. Ceram Int 41:9199–9202. https://doi.org/10.1016/j.ceramint.2015.03.235
Subudhi S, Mahapatra A, Mandal M et al. (2020) Effect of Co Doping in Tuning the Band Gap of LaFeO3. Integr Ferroelectr 205:61–65. https://doi.org/10.1080/10584587.2019.1674998
Kalasina S, Kongsawatvoragul K, Phattharasupakun N et al. (2020) Cobalt oxysulphide/hydroxide nanosheets with dual properties based on electrochromism and a charge storage mechanism. RSC Adv 10:14154–14160. https://doi.org/10.1039/d0ra01714k
Triyono D, Hanifah U, Laysandra H (2020) Structural and optical properties of Mg-substituted LaFeO3 nanoparticles prepared by a sol-gel method. Results Phys 16:102995. https://doi.org/10.1016/j.rinp.2020.102995
Lin Q, Lin J, Yang X et al. (2019) The effects of Mg2+ and Ba2+ dopants on the microstructure and magnetic properties of doubly-doped LaFeO3 perovskite catalytic nanocrystals. Ceram Int 45:3333–3340. https://doi.org/10.1016/j.ceramint.2018.10.246
Rini EG, Gupta MK, Mittal R et al. (2021) Structural change from Pbnm to R3̅c phase with varying Fe/Mn content in (1-x) LaFeO3.xLaMnO3 solid solution leading to modifications in octahedral tilt and valence states. J Alloy Compd 883:160761. https://doi.org/10.1016/j.jallcom.2021.160761
Gu J, Zhang B, Li Y et al. (2021) Synthesis of spindle-like Co-doped LaFeO3 porous microstructure for high performance n-butanol sensor. Sens Actuators B Chem 343:130125. https://doi.org/10.1016/j.snb.2021.130125
Arman MM, Ahmed MA (2022) Effects of vacancy co-doping on the structure, magnetic and dielectric properties of LaFeO3 perovskite nanoparticles. Appl Phys A Mater Sci Process 128:554. https://doi.org/10.1007/s00339-022-05623-9
Raji RK, Ramachandran T, Muralidharan M et al. (2021) Twitching the inherent properties: the impact of transition metal Mn-doped on LaFeO3-based perovskite materials. J Mater Sci: Mater Electron 32:25528–25544. https://doi.org/10.1007/s10854-021-07018-7
Peng Q, Shan B, Wen Y, Chen R (2015) Enhanced charge transport of LaFeO3 via transition metal (Mn, Co, Cu) doping for visible light photoelectrochemical water oxidation. Int J Hydrog Energy 40:15423–15431. https://doi.org/10.1016/j.ijhydene.2015.09.072
Kum−onsa P, Thongbai P (2021) Dielectric properties of poly(vinylidene fluoride)-based nanocomposites containing a LaFeO3 nanoparticle filler. J Mater Sci: Mater Electron 32:13985–13993. https://doi.org/10.1007/s10854-021-05974-8
Indriyani A, Yulizar Y, Tri Yunarti R et al. (2021) One-pot green fabrication of BiFeO3 nanoparticles via Abelmoschus esculentus L. leaves extracts for photocatalytic dye degradation. Appl Surf Sci 563:150113. https://doi.org/10.1016/j.apsusc.2021.150113
Ateia EE, Abdelamksoud MK, Rizk MA (2017) Improvement of the physical properties of novel (1 − x) CoFe2O4+(x) LaFeO3 nanocomposites for technological applications. J Mater Sci: Mater Electron 28:16547–16553. https://doi.org/10.1007/s10854-017-7567-1
Chu X, Zhou S, Zhang W, Shui H (2009) Trimethylamine sensing properties of nano-LaFeO3 prepared using solid-state reaction in the presence of PEG400. Mater Sci Eng: B 164:65–69. https://doi.org/10.1016/j.mseb.2009.06.014
Martín-Carrón L, De Andrés A (2001) Melting of the cooperative Jahn-Teller distortion in LaMnO3 single crystal studied by Raman spectroscopy. Eur Phys J B 22:11–16. https://doi.org/10.1007/PL00011129
Martín-Carrón L, de Andrés A, Martínez-Lope MJ et al. (2002) Raman phonons as a probe of disorder, fluctuations, and local structure in doped and undoped orthorhombic and rhombohedral manganites. Phys Rev B 66:174303. https://doi.org/10.1103/PhysRevB.66.174303
Manchón-Gordón AF, Sánchez-Jiménez PE, Blázquez JS et al. (2023) Structural, Vibrational, and Magnetic Characterization of Orthoferrite LaFeO3 Ceramic Prepared by Reaction Flash Sintering. Materials 16:1019. https://doi.org/10.3390/ma16031019
Benali EM, Benali A, Bejar M et al. (2020) Effect of synthesis route on structural, morphological, Raman, dielectric, and electric properties of La0.8Ba0.1Bi0.1FeO3. J Mater Sci: Mater Electron 31:3197–3214. https://doi.org/10.1007/s10854-020-02867-0
Vijayaraghavan T, Sivasubramanian R, Hussain S, Ashok A (2017) A Facile Synthesis of LaFeO3-Based Perovskites and Their Application towards Sensing of Neurotransmitters. ChemistrySelect 2:5570–5577. https://doi.org/10.1002/slct.201700723
Wu Y, Zhou X, Li M et al. (2019) 2D/3D interface engineering: direct Z-scheme g-C3N4/YMnO3 heterojunction for reinforced visible-light photocatalytic oxidation. J Mater Sci: Mater Electron 30:17601–17611. https://doi.org/10.1007/s10854-019-02109-y
Jena A, Penki TR, Munichandraiah N, Shivashankar SA (2016) Flower-like porous cobalt(II) monoxide nanostructures as anode material for Li-ion batteries. J Electroanalytical Chem 761:21–27. https://doi.org/10.1016/j.jelechem.2015.11.003
Munisha B, Mishra B, Nanda J et al. (2023) Enhanced photocatalytic degradation of 4-nitrophenol using polyacrylamide assisted Ce-doped YMnO3 nanoparticles. J Rare Earths 41:1541–1550. https://doi.org/10.1016/j.jre.2022.11.022
Liu Y, Sun T, Dong G et al. (2019) Dependence on sintering temperature of structure, optical and magnetic properties of La0.625Ca0.315Sr0.06MnO3 perovskite nanoparticles. Ceram Int 45:17467–17475. https://doi.org/10.1016/j.ceramint.2019.05.308
Ansari AA, Abushad M, Arshad M et al. (2021) Microstructure, optical and dielectric properties of cobalt-doped zinc ferrite nanostructures. J Mater Sci: Mater Electron 32:21988–22002. https://doi.org/10.1007/s10854-021-06647-2
Kumar AS, Raja MM, Bhatnagar AK (2014) Surface driven effects on magnetic properties of antiferromagnetic LaFeO3 nanocrystalline ferrite. J Appl Phys 116:113912. https://doi.org/10.1063/1.4896191
Mahapatra AS, Mitra A, Mallick A et al. (2018) Modulation of magnetic and dielectric property of LaFeO3 by simultaneous doping with Ca2+ and Co2+-ions. J Alloys Compd 743:274–282. https://doi.org/10.1016/j.jallcom.2018.01.207
Polat O, Coskun M, Yildirim Y et al. (2023) Variation in the dielectric and magnetic characteristics of multiferroic LuFeO3 as a result of cobalt substitution at Fe sites. J Alloys Compd 963:170939. https://doi.org/10.1016/j.jallcom.2023.170939
Gharagozlou M, Bayati R (2015) Low temperature processing and magnetic properties of zinc ferrite nanoparticles. Superlattices Microstruct 78:190–200. https://doi.org/10.1016/j.spmi.2014.12.004
Sharma S, Kumar M, Srinet G et al. (2021) Structural, Raman analysis and exchange bias effects in Mn doped multiferroic Bi0.80La0.10Ca0.10Fe1-xMnxO3 ceramics. Ceram Int 47:6834–6841. https://doi.org/10.1016/j.ceramint.2020.11.026
Sebastian RM, Xavier S, Mohammed EM (2015) Dielectric Behavior and AC Conductivity of Mg 2+ Doped Zinc Ferrite Nanoparticles Synthesized by Sol-Gel Technique. Ferroelectrics 481:48–56. https://doi.org/10.1080/00150193.2015.1049491
Shukla J, Mishra A Influence of Ba2+ Doping on Structural and Electrical Transport Properties of YMnO3 Ceramics. Journal of Superconductivity and Novel Magnetism 34: 451–459. https://doi.org/10.1007/s10948-020-05693-x
Mishra B, Munisha B, Nanda J et al. (2022) Hydrothermally Synthesized Magnesium doped Zinc Ferrite Nanoparticles: An extensive study on structural, optical, magnetic, and dielectric properties. Mater Chem Phys 292:126791. https://doi.org/10.1016/j.matchemphys.2022.126791
Heiba ZK, Altowairqi Y (2022) Modifying the structure and dielectric characteristics of MnFe2O4/ZnMn2O4 heterostructure nanocomposites via changing the composition. J Solgel Sci Technol 101:323–334. https://doi.org/10.1007/s10971-021-05711-x
Acknowledgements
The authors would like to thank Dr. Chhatrapati Parida, Dept. of Physics, OUAT, BBSR, for providing the LCR meter facility.
Author information
Authors and Affiliations
Contributions
JN and DM: Formulation of Research Problem and Methodology. DM and SP: Synthesis of Materials. DM: Characterization and data collection using XRD, SEM, XPS, VSM and LCR meter. DM, KJS and SG: Characterization and data collection using Raman and UV-visible spectroscopy. DM: Data analysis and original draft preparation. JN and DM: Reviewing and Editing the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mishra, D., Nanda, J., Parida, S. et al. Effect of Y3+ and Co2+ co-doping on the structural, optical, magnetic and dielectric properties of LaFeO3 nanoparticles. J Sol-Gel Sci Technol 111, 381–394 (2024). https://doi.org/10.1007/s10971-024-06452-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-024-06452-3